
Abstract
The vertebrate neuromuscular junction (NMJ) is a specialised chemical synapse involved in the transmission of bioelectric signals between a motor neuron and a skeletal muscle fiber, leading to muscle contraction. Typically, the NMJ is a tripartite synapse comprising (a) a presynaptic region represented by the motor nerve ending, (b) a postsynaptic skeletal motor endplate area, and (c) perisynaptic Schwann cells (PSCs) that shield the motor nerve terminal. Increasing evidence points towards the role of PSCs in the maintenance and control of neuromuscular integrity, transmission, and plasticity. Acetylcholine (ACh) is the main neurotransmitter at the vertebrate skeletal NMJ, and its role is fine-tuned by co-released purinergic neuromodulators, like adenosine 5′-triphosphate (ATP) and its metabolite adenosine (ADO). Adenine nucleotides modulate transmitter release and expression of postsynaptic ACh receptors at motor synapses via the activation of P2Y and P2X receptors. Endogenously generated ADO modulates ACh release by acting via co-localised inhibitory A1 and facilitatory A2A receptors on motor nerve terminals, whose tonic activation depends on the neuronal firing pattern and their interplay with cholinergic receptors and neuropeptides. Thus, the concerted action of adenine nucleotides, ADO, and ACh/neuropeptide co-transmitters is paramount to adapting the neuromuscular transmission to the working load under pathological conditions, like Myasthenia gravis. Unravelling these functional complexities prompted us to review our knowledge about the way purines orchestrate neuromuscular transmission and plasticity in light of the tripartite synapse concept, emphasising the often-forgotten role of PSCs in this context.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The neuromuscular junction (NMJ) is a specialised chemical synapse involved in the transmission of electric signals between motor neurons and skeletal muscle fibres that are necessary for muscle contraction. The presynaptic region of the NMJ comprises the demyelinated part of the motoneuron axon terminal, which is normally shielded by perisynaptic Schwann cells (PSCs) [1, 2]. Unlike Schwann cells (SCs) that wrap the axons of myelinated motor neurons, PSCs are non-myelinating cells that play a fundamental role in the formation, maintenance, and regulation of skeletal NMJs [2, 3]. The synaptic cleft is a gap that separates the pre- and postsynaptic regions and contains a basal lamina of specialised extracellular matrix [1]. The postsynaptic motor endplate consists of deep plasma membrane invaginations (synaptic folds) and crests, where muscle-type α1 subunit-containing nicotinic receptors (nAChRs) are clustered. In vertebrates, the main neurotransmitter released by motor nerve terminals is acetylcholine (ACh), which accumulates only very briefly in the synaptic cleft during neuronal firing due to the presence of highly active acetylcholinesterase (AChE) enzymes. Activation of α1-containing nicotinic receptors by ACh released from motor nerve terminals triggers small depolarisations of the motor endplate region. Synchronisation of plasma membrane depolarisations results in the generation of an action potential, leading to subsequent skeletal muscle contraction [3].
Besides its role in muscle contraction, ACh regulates its release through the activation of presynaptic nAChRs and muscarinic receptors (mAChRs). In addition to facilitatory nAChRs containing α3β2 subunits [4, 5], nerve terminals are also endowed with M1 (facilitatory) and M2 (inhibitory) mAChRs [6, 7]. It has also been demonstrated that ACh spillover from the neuromuscular synapse participates in a negative feedback loop to restrain its release through the activation of nAChRs containing α7 subunits (α7 nAChRs) located on the plasma membrane of PSCs [8, 9]. Thus, increasing evidence suggests that PSCs can sense and tune neuromuscular transmission efficacy [10,11,12,13]. Besides ACh, other released signalling molecules (e.g. glutamate, purine nucleotides or nucleosides, and neuropeptides) play important roles in neuromuscular transmission regulation [14,15,16], namely by interfering with the activity of PSCs [8, 10,11,12,13]. In this review, we will briefly summarise the modulatory roles of adenosine (ADO) and adenine nucleotides on neuromuscular transmission; emphasis will be given to the role of distinct purinoceptor subtypes and their interplay with other neuromodulatory signalling pathways at both synaptic and perisynaptic levels. Understanding the crosstalk between cholinergic and purinergic cascades that regulate neurotransmitter release given the tripartite synapse concept may be relevant to guide the rationale of therapeutic approaches for diseases affecting the safety margin of neuromuscular transmission, such as myasthenia gravis and other myasthenic syndromes [17].
Pre- and Postsynaptic Signalling by Adenine Nucleotides
ATP is Co-released with ACh: Co-transmission and Neuromodulation by Purines
Considered the “molecular currency unit” found in all forms of life, adenosine 5′-triphosphate (ATP) is also known for its role as a neurotransmitter and neuromodulator both in central and peripheral synapses [18]. Unlike other nucleotides or their metabolites, ATP is stored in synaptic vesicles together with other neurotransmitters, such as ACh in cholinergic neurons. The ACh:ATP ratio in synaptic vesicles has been estimated to be between 1:1 to 10:1, depending on the technique and preparation used [19,20,21,22]. As such, ATP is co-released with ACh during neuromuscular transmission, and it accumulates at the synaptic cleft in a frequency-dependent manner during neuronal activity [16, 23, 24].
ATP can also originate from twitching skeletal muscle fibres in amphibian and rodent NMJs [25,26,27,28]. Pharmacological evidence suggests that pannexin-1 hemichannels mediate the release of ATP from skeletal myotubes [29]. Estimates about the proportion of ATP directly released from contracting muscle fibres vis a vis that of neuronal origin range between 15 and 80%; this contention depends on experimental settings concerning the use of low- vs high-frequency nerve stimulation patterns and the pre- vs post-junctional selectivity of the neuromuscular blockers used to paralyse skeletal nerve fibres [25,26,27,28].
Under appropriate conditions (e.g. hypotonicity, stimulation with uracil nucleotides), cultured SCs are also able to release ATP; yet, the relevance of this phenomenon in the context of PSC signalling and neuromuscular transmission remains to be elucidated in situ [30,31,32]. PSCs are also able to release ATP via a yet undisclosed mechanism that may be involved in the modulation of Ach release via the activation of presynaptic purinoceptors (see below) [10].
Neuromodulation by adenine nucleotides occurs either directly, via the activation of ionotropic P2X and metabotropic P2Y receptors (P2XRs and P2YRs, respectively) [18], or indirectly, after their extracellular breakdown into ADO and subsequent activation of P1 receptors, which include four distinct subtypes (A1, A2A, A2B, and A3; see below) [33]. Nerve terminals, skeletal muscle fibres, and PSCs express various subtypes of P2XRs and P2YRs (see Table 1) [34,35,36]. Yet, the pathophysiological relevance of ATP and its metabolite adenosine 5′-diphosphate (ADP) in the control of neuromuscular transmission is far from being consensual, mostly because research has been conducted under distinct experimental conditions, which include the use of different preparations from diverse animal species and developmental stages. Most often, no attempt has been made to separate P2- from P1-mediated effects [16].
P2Y Receptor-mediated Signalling
Attenuation of quantal ACh release by ATP may involve the activation of one or more types of pertussis-toxin-sensitive Gi/o-coupled P2Y (most likely the P2Y12R and/or P2Y13R subtypes), but not ionotropic P2XRs, at the NMJ of amphibia [37, 38] and rodents [35]. Immunohistochemistry and pharmacological data point towards the involvement of the ADP-sensitive P2Y13R as the main purinoceptor mediating the inhibitory effect of ATP on ACh release at the mouse neuromuscular synapse. However, this is not consensual given to the fact that ATP-induced decreases in non-quantal transmitter release at mammalian NMJs may also involve other P2YR subtypes coupled to different intracellular signalling pathways [39, 40]. Data in amphibia considerably differ from that obtained in mammals. The mechanism underlying ATP-mediated inhibition of ACh release in frogs involves ADP-sensitive P2Y12Rs, but not P2Y13Rs [41], via an intracellular mechanism linking the production of reactive oxygen species (ROS; including H2O2), oxidation of the synaptosomal-associated protein 25 (SNAP25), and decrease in synaptic vesicle release probability [42, 43]. The coupling of inhibitory P2YRs to a second ROS-independent signalling pathway involving PLC/PKC/PLA2/COX activation has also been proven [37, 42, 44]. In support of this theory, byproducts of PLA2 and COX activity, like arachidonic acid and prostaglandin E2, are known to decrease ACh release by suppressing Ca2+ influx into frog motor nerve endings [45].
It is also worth noting that modulation of evoked transmitter release by ATP is more often observed under low neuronal stimulation frequencies (≤ 1 Hz). It remains to be elucidated if the inhibitory role of ATP is maintained under more physiological nerve-firing conditions (e.g. 50–100 Hz bursts), which are known to affect neuromodulation by other purines, such as ADO. Previous studies from our group using selective receptor agonists and antagonists showed that ADO (and its analogues) could activate both presynaptic A1 inhibitory/A2A facilitatory receptors depending on the motor nerve stimulation paradigm and, thus, on the amount of ADO accumulated at the rat NMJ (see below) [6, 46]. Contrariwise, the P2 purinoceptor(s) involved in the aforementioned ATP-mediated effects require comprehensive investigations.
P2X Receptor-mediated Signalling
Besides the predominant inhibitory effect of P2YRs at the NMJ, the net effect of ATP may be balanced via the activation of Ca2+-permeable facilitatory P2XRs [47,48,49]. Yet, independent studies have shown that non-hydrolysable ATP analogues can either facilitate or inhibit ACh release from motor nerve terminals [38, 50, 51]. ATP significantly decreases the amplitude of nerve-evoked muscle twitches of rat extensor digitorum longus (EDL) and soleus muscles while increasing the twitch amplitude at the mouse EDL, diaphragm, and soleus muscles [52,53,54]. These findings confirm our contention that the effect of ATP on neuromuscular transmission significantly differs among species [52]. It is also interesting to note that the facilitation of ACh release by P2 receptors is normally coupled to the activation of presynaptic facilitatory nAChRs [50].
The presence of the P2X7R was detected on mammalian motor nerve terminals by immunofluorescence and electron microscopy, where it acts to increase synaptic vesicle exocytosis [48, 49, 55]. The P2X7R exhibits low desensitisation probability and high conductance to monovalent and divalent cations, like Ca2+, which might explain its ability to increase ACh exocytosis [33, 56, 57]. Nevertheless, the true relevance of P2X7R activation on neuromuscular transmission is still a matter of debate because some authors failed to reproduce the facilitation of transmitter exocytosis using P2X7R agonists in mammals and amphibia NMJs [37]. Downstream activation of pannexin-1 hemichannels may partially occlude the facilitatory effect of the P2X7R given that the formation of large membrane pores may allow the release of inhibitory adenine nucleotides among other substances with molecular weight up to 900 Da [55, 58]. The low affinity of the P2X7R for ATP suggests that it might have negligible activity under physiological conditions, even considering that this receptor is sensitised by transient decreases in extracellular Ca2+ levels that are normally observed during high-frequency stimulation trains [33]. Thus, one may speculate that activation of P2X7Rs on mammalian motor nerve terminals might only occur under pathological conditions (e.g. ischaemia or muscle damage) when extracellular ATP levels increase considerably [49].
Extracellular Catabolism of Adenine Nucleotides and ADO Formation at the NMJ
ATP is sequentially hydrolysed into ADP, adenosine 5′- monophosphate (AMP), and ADO by extracellular ectonucleotidases at mammalian NMJs [24, 26, 59, 60]. However, the characterisation of the enzymes responsible for the extracellular ATP breakdown at neuromuscular synapses is still lacking. It has been demonstrated that nucleoside triphosphate diphosphohydrolase 2 (NTPDase2 or ATPase) is located in the vicinity of mammalian motor endplates [61], which may contribute to the preferential accumulation of ADP compared to that of AMP when exogenous ATP is used as substrate [24]. So, ATP and/or ADP may transiently accumulate at the NMJ during high-frequency neuronal firing or under pathological conditions. High extracellular ATP and/or ADP levels feed-forwardly inhibit AMP dephosphorylation to ADO by ecto-5′-nucleotidase/CD73, thus partially preventing its formation [46, 62]. Under such conditions (e.g. continuous stimulation with 50 Hz trains), neuromodulation through ATP/ADP-sensitive P2-mediated purinoceptors is likely to prevail over ADO-mediated signals operated by P1 receptors.
The rat NMJ is also equipped with an ecto‐AMP deaminase pathway that metabolises adenine nucleotides to inosine 5′-monophosphate (IMP), bypassing ADO formation [24]. Thus, while the ecto‐5′‐nucleotidase/CD73 pathway modulates the rate of ADO formation from released nucleotides, alternative AMP deamination to IMP controls the amount of AMP available for ADO formation. In the innervated frog sartorius muscle, extracellular endogenous amounts of ADO are also balanced by exo-AMP deaminase and ecto-5'-nucleotidase activity. When only one of these enzymes is inhibited, the evoked release of adenine nucleotides becomes undetectable. Data strongly suggest that each of these enzymes is able, on its own, to break down whole AMP resulting from the catabolism of released adenine nucleotides upon stimulation [25].
ADO: Modulator of Neuromodulators at the NMJ
ADO Formation and Inactivation at the NMJ
ADO is a purine nucleoside ubiquitously found in most synapses, where it exerts neuromodulatory roles via the activation of P1 receptors. Unlike neurotransmitters, ADO is not stored in synaptic vesicles nor released by exocytosis [18, 63]. The nucleoside is released as such from intracellular compartments via equilibrative nucleoside transporters (ENTs); ADO may also originate from the extracellular catabolism of ATP co-released with other neurotransmitters, including ACh (see above) [63, 64]. Intracellularly there are two major pathways leading to ADO formation: AMP hydrolysis catalysed by the cytosolic 5′-nucleotidase or hydrolysis of S-(5′-adenosyl)-L-homocysteine (SAH) by SAH hydrolase. Intracellular ADO accumulation is balanced by its formation and/or uptake from the extracellular milieu (via ENTs) and the activity of two intracellular enzymes, adenosine kinase (ADK, Km for ADO in the nanomolar range) and adenosine deaminase (ADA, Km for ADO in the micromolar range), converting the nucleoside into AMP and inosine, respectively [63, 65]. Since the Km value of ADA was found to be at least ninefold higher than that of ADK, the preferential phosphorylation of ADO to AMP (a less membrane-permeable compound) constitutes the main driving force to take up ADO from the extracellular milieu while keeping low cytosolic levels of the nucleoside, both in peripheral and central nervous system synapses [66].
Contracting muscle fibres were also identified as a major source of ADO at several NMJs [22, 26, 28, 60]. ADO progressively accumulates when ecto-5′-nucleotidase/CD73 and ecto-ADA are inhibited in the innervated frog sartorius, thus suggesting that the nucleoside is released as such even in the absence of nerve stimulation. Using the muscle-type nAChR antagonist, d-tubocurarine, authors estimated that extracellular ADO amounts originated from 0.2 Hz twitching skeletal muscle fibres were roughly identical to the nucleoside released from stimulated motor nerve terminals [25]. These findings challenged a previously published paper showing that ADO release was essentially abolished in the presence of d-tubocurarine, providing that ecto-5′-nucleotidase/CD73 was also inhibited, hence suggesting that contracting muscle fibres contribute to most extracellular ADO released as such [22]. A major limitation of both studies’ estimations relies on the fact that d-tubocurarine abrogates the ACh/ATP release facilitatory drive due to the activation of α3β2-containing nicotinic autoreceptors on mammalian motor nerve terminals besides its ability to block α1-containing nAChRs on skeletal muscle fibres [4, 14, 67].
Using an HPLC technique, we showed that selective muscle paralysis with μ‐conotoxin GIIIB, a toxin that blocks muscle‐specific voltage‐gated Na+ channels without affecting neuronal function [4, 68], significantly (> 90%) decreased nerve‐evoked ADO outflow without much affecting (∼ 15%) the release of ATP (and related nucleotides) [28, 69]. Data also showed that endogenous ADO generated during high-frequency nerve stimulation of motor endplates of myasthenia gravis patients was insufficient to maintain transmitter release demand through activation of the A2A receptor (A2AR)-mediated presynaptic facilitatory drive [28]. Interestingly, myasthenic patients have impaired oxidative metabolism and a noticeable shift to glycolytic metabolism in skeletal muscles during exercise, which yields to higher‐end Pi/ATP ratio and reduced levels of synaptic ADO levels [70]. Taken together, these findings strengthen the idea that deficient ADO release from myasthenic skeletal muscle fibres may contribute to explaining the neuromuscular deficits observed in patients, which cannot be compensated by the smaller ADO amounts formed from released adenine nucleotides. Curiously, both neuromuscular impairments and ADO neuromodulation deficits in myasthenic motor endplates could be rehabilitated using the nucleoside precursor, AMP, or methylprednisolone; the latter is a synthetic glucocorticoid immunosuppressant that is widely used to prevent myasthenic crisis because it also amplifies neuromuscular transmission by increasing endogenous ADO availability and activation of facilitatory A2ARs, which promotes synaptic vesicle recycling and release [51, 69, 70].
More recently, increasing evidence has strengthened the idea that PSCs, along with motor nerve terminals and skeletal muscle fibres, are also able to release ADO [8, 10, 71,72,73]. We provided recent data suggesting that activation of α7 nAChRs on PSCs controls ACh spillover from motor endplates by promoting ADO outflow via ENT1 and retrograde activation of presynaptic inhibitory A1 (A1Rs) [8]. However, the mechanism(s) underlying ADO release from PSCs still need to be elucidated in the future (see discussion below).
Extracellular ADO accumulation is also fine-tuning regulated by the nucleoside inactivation mechanisms, both cellular uptake and deamination [60, 74, 75]. The presence of ecto-ADA at both mammalian and amphibian NMJs is responsible for the conversion of ADO into inosine in the synaptic cleft [24, 28, 60]. Motor synapses are also endowed with the ENT1 [75, 76]. Both uptake and deamination coordinate to remove extracellular ADO, thus contributing to fine-tuning regulate activation of P1 receptor subtypes on motor nerve terminals [46, 75, 77, 78].
ADO-mediated Signalling via P1 Purinoceptors
Neuromodulation by ADO depends on the activation of metabotropic P1 receptors, which are sub-classified in A1, A2A, A2B, and A3 (Table 1). In rodents, A1R and A2AR display high affinity for ADO, while A2BR and A3R are low-affinity receptors. Most commonly, A1R and A3Rs couple to Gi/o proteins, while A2AR and A2BR are preferentially coupled to Gs protein subunits. As such, activation of A2AR and A2BR normally results in stimulation of adenylyl cyclase (AC), resulting in increases in intracellular 3′-5′-cyclic adenosine monophosphate (cAMP) and subsequent protein kinase A (PKA) activation. Conversely, activation of A1R and A3R subtypes results in the suppression of AC activity, thus decreasing cAMP levels and PKA activation [79]. It is noteworthy that these receptors may also couple to non-canonical pathways, thus regulating intracellular concentrations of Ca2+ and/or K+ through the activation of Gq protein-coupled mechanisms; others include a direct interference with ion currents across the plasma membrane via the βγ subunit of G proteins [63, 80], although some controversy still exists on this matter.
A1 Receptors
ADO released as such or formed from the extracellular breakdown of released ATP reaches very small amounts in non-stimulated motor endplates. ADO exerts a predominantly inhibitory effect on the release of ACh via A1R activation [46, 75, 78] when motor nerves fire at low activation frequencies [38, 74, 81]. The mechanism by which the A1R inhibits ACh release from motor nerve terminals is still poorly understood, but it is thought to involve direct inhibition of the secretory apparatus or inhibition of Ca2+ currents. At the frog NMJ, activation of A1R inhibits ACh release through a Ca2+-independent mechanism, suggesting a direct interference with proteins involved in vesicular exocytosis [82,83,84]. At mammalian NMJs, the A1R-mediated inhibition of ACh release depends on the inhibition of Ca2+ currents via N (Cav2.2) [85] and P/Q (Cav2.1) voltage-sensitive channels [86,87,88]. Since inhibition of N-type (Cav2.2) Ca2+ currents is insufficient to affect both spontaneous and evoked ACh release from mammalian NMJ under resting conditions [86, 87, 89], reduction of transmitter release by A1Rs may predominantly rely on the suppression of Ca2+ influx via P/Q-type (Cav2.1) channels (Fig. 1a), even though cross-talk between A1Rs and proteins of the exocytosis apparatus (such as Rab3A) via G protein βγ subunits has also been reported [90, 91].

Crosstalk between purinergic and cholinergic auto-receptors to control nerve-evoked ACh release at the mammalian neuromuscular junction A under resting conditions and B during intense neuronal activity. Bold arrows indicate an increase in the strength of the proposed pathways. Predominant receptors operating in each condition appear as fully opaque, while non-predominant receptors appear as semi-transparent. See text for details. Abbreviations: ACh, acetylcholine; ADO, adenosine; AMP, adenosine 5′-monophosphate; ATP, adenosine 5′-triphosphate; cAMP, 3′-5′-cyclic adenosine monophosphate; ENT1, equilibrative nucleoside transporter 1; EPP, endplate potential; IP3, inositol 1,4,5-triphosphate; PKA, protein kinase A; PKC, protein kinase C
The A1R located on motor nerve terminals may also be the effector of synaptic depression caused by other inhibitory signalling molecules, like the non-proteinogenic amino acid L-citrulline [72, 76]. This byproduct of nitric oxide (NO) resulting from the breakdown of L-arginine by nitric oxide synthase (NOS) [92, 93] inhibits nerve-evoked ACh release by increasing ADO outflow at the neuromuscular synapse via ENT1 transporters, fostering activation of the inhibitory A1R. In contrast to that observed with L-citrulline, ADO does not seem to play a role in NO-induced inhibition of ACh release from motor nerve terminals [72, 76].
A2A Receptors
In 1991, previous results from Correia-de-Sá et al. demonstrated by electrophysiological and neurochemical methods that ADO A1-inhibitory and A2-excitatory receptors coexist at rat phrenic nerve terminals to fine-tune regulate evoked ACh release [78]. This seminal paper unravelled for the first time the neuronal activity of excitatory A2ARs outside basal ganglia in the CNS.
Activation of A2ARs at the mammalian NMJ is revealed when motor neurons fire at high-frequency rates and/or when nerve terminals are focally depolarised [46]. Under such conditions that roughly mimic the firing rate of motor neurons during initiation of normal respiratory or voluntary motion, ATP co-released with ACh reaches levels that are beyond the critical level required for ADO formation [19, 94]. Experimental data from our group showed that bypass of the initial feedforward inhibition of ecto-5′-nucleotidase/CD73 transiently caused by high ATP levels is required before ADO boosts at the motor endplate to allow subsequent activation of facilitatory A2ARs [16, 46, 75].
The opposing effects of ADO at the mammalian NMJ may not be exclusively dependent on the nerve stimulation paradigm nor on the nucleoside concentration in the synaptic cleft, but it is also determined by its source. While ADO activates both A1R and A2AR depending on the extracellular concentration of the nucleoside, ADO resulting from the extracellular catabolism of ATP co-released with ACh preferentially activates facilitatory A2ARs [46, 75, 94]. This feature, which is commonly observed in other peripheral and central synapses [95,96,97,98], may be attributable to the proximity of ecto-5′-nucleotidase/CD73 and A2ARs, thus favouring the activation of this receptor subtype following ADO formation by AMP breakdown [95, 97].
The A2AR-mediated facilitation of ACh release from motor nerve endings is thought to be mainly caused by normally “quiescent” high-capacity L-type (Cav1) VGCCs, which allow the influx of Ca2+ when “predominant” P/Q (Cav2.1) channels are desensitised, such as during high-frequency neuronal firing (Fig. 1b) [28, 77, 86]. The mechanism downstream activation of the A2AR on motor nerve terminals is dependent on cAMP formation by AC [99, 100], leading to stimulation of PKA activity and subsequent Ca2+ influx through “quiescent” L-type (Cav1) VGCCs (Fig. 1b) [101, 102]. Involvement of Ca2+ release from intracellular stores may also concur to facilitate ACh release from motor nerve terminals during intense neuronal activation [86, 102]. Phosphorylation of synapsin I by PKA may account to facilitate the mobilisation of synaptic vesicles to the active release zones following A2AR activation [51, 103], but this mechanism has never been directly addressed at vertebrate NMJs.
A2B and A3 Receptors
Scarce evidence shows the presence and function of A2BR and A3R at the NMJ. Localisation of A2BR in presynaptic nerve terminals, as well as at the motor endplate region, has been observed at the mouse NMJ, but their roles are largely undefined [104]. Using high-resolution confocal microscopy, these authors also identified the A1R in PSCs and nerve terminals and the A2AR in the postsynaptic skeletal muscle, as well as in axon nerve terminals of the mouse NMJ. Despite this, the AC/cAMP/PKA pathway may be involved in the trafficking and clustering of nAChRs on the postsynaptic membrane, meaning that postjunctional A2BRs are important for neuromuscular transmission efficacy and the long-term stability of the NMJ [105]. In the mouse diaphragm, activation A2BRs with either adenosine or a synthetic ADO analogue increases the opening probability of nAChRs on skeletal muscle fibres. This effect was abolished in the presence of an AC inhibitor, thus confirming the involvement of the AC/cAMP/PKA pathway [106]. The PKA-mediated phosphorylation of the skeletal-type nAChR favours its opening probability and duration [107], prompting a putative synergistic interplay between postsynaptic A2BRs and nAChRs on neuromuscular transmission, besides the role of this low-affinity ADO receptor subtype in NMJ stability promotion. The physiological significance of the A2BR activation on neuromuscular transmission remains to be elucidated given the fact that it requires high extracellular ADO amounts, which are normally unavailable at this particular synapse unless cellular damage due to ischaemia or inflammation is also present.
A3Rs have also been localised at presynaptic nerve terminals (and possibly the postsynaptic membrane) of mammalian NMJs. Activation of the A3R downregulates both spontaneous and evoked ACh release [104, 108]. This putative inhibitory effect on neuromuscular transmission may, indeed, be mediated by inosine, the ADO metabolite resulting from ADA activity [109]. Inosine decreases the frequency of MEPPs as well as the amplitude of nerve-evoked endplate potentials (EPPs) in mammalian but not in frog NMJs [74, 104, 108]. Inosine may decrease the release probability either directly, by activating low-affinity inhibitory A3Rs, or indirectly, through inhibition of ADO cellular uptake by ENTs [104, 108].
Pathophysiological Implications of P1 Receptor Activation Deficits
The loss of function of A2AR seems to be a hallmark of ageing and some neuromuscular disorders (Table 1). While tonic activation of inhibitory A1R is largely preserved, deficits in the activity of excitatory A2AR were observed in diaphragm motor endplates of aged rats, thus contributing to the characteristic age-related neuromuscular transmission impairment [110]. Loss of the A2AR tone may also play a role in neuromuscular diseases, like myasthenia gravis and amyotrophic lateral sclerosis (ALS; see discussion below) [28, 111]. However, changes in the protein density of A2AR predominantly located on nerve axon terminals do not account for the functional deficits observed in the autoimmune myasthenia gravis (EAMG) rat model compared with naïve animals, as demonstrated by immunofluorescence confocal microscopy [69]. Myasthenia gravis is a B-cell-mediated and T-cell-dependent autoimmune disease characterised by the production of antibodies directed against muscle-type nAChRs containing α1 subunits; this attack reduces the number of effective nAChRs at motor endplates to nearly one-third of the normal amount, at least partially explaining the chronic muscle weakness that is characteristic of this disease [112]. Using rats with toxin-induced myasthenia gravis (TIMG), our group demonstrated that the A2AR-mediated facilitation of ACh release during high-frequency stimulation bursts is significantly impaired due to insufficient ADO outflow from myasthenic skeletal muscle fibres. Consequently, the necessary shift from fast desensitising P/Q (Cav2.1) to facilitatory high-conductance L-type (Cav1) VGCCs is compromised, which contributes to tetanic failure and reduced vesicular exocytosis [28]. Likewise, the endogenous levels of ADO were below those required to activate A2AR at both the TReg/THelper immunological and the neuromuscular synapse in the EAMG rat model [69]. As such, this animal model presents deficits in sustaining muscle contraction during high-frequency neuronal firing (tetanic failure) and exaggerated T- and B-cell-mediated immune responses, leading to unrestrained production of antibodies against the muscle-type nAChR.
A strategy to experimentally overcome both immunological and neuromuscular transmission deficits in myasthenic animals was the rehabilitation of the A2AR tonus through in vitro application of AMP, which is rapidly converted into ADO by ecto-5′-nucleotidase/CD73 [28, 69]. Whether this strategy works in vivo remains to be elucidated, for instance using newly developed A2AR agonists (prodrugs) consisting of 2-substituted AMP derivatives that are locally activated by ecto-5′-nucleotidase/CD73 [113]. Selective activation of A2ARs using CGS 21680C also specifically reduced the production of antibodies against the muscle-type α1-containing nAChRs by B-cells and increased the proliferation of Treg cells in EAMG rats. Intraperitoneal treatment of these rats with CGS 21680C also decreased the myasthenic symptoms [114].
Besides the immunosuppressive effect, rehabilitation of the facilitatory A2AR-mediated tonus may also afford an explanation for glucocorticoid improvements of the neuromuscular transmission deficits in myasthenic patients. At the rat NMJ, methylprednisolone facilitates neurotransmitter release during high-frequency bursts; the effect was potentiated by blocking inhibitory muscarinic M2 and A1Rs, but it was prevented by blockage of facilitatory M1 and A2ARs [51, 101]. Methylprednisolone favours ATP release under resting conditions, thus fostering ADO formation from ATP breakdown and, thereby, A2AR activation, allowing these receptors to play a facilitatory role from the beginning of nerve stimulation onwards. Phosphorylation of synapsin I clustered with vesicles in the reserve pool causes the priming of mature vesicles to the readily releasable pool, allowing maintenance of transmitter exocytosis during high-frequency stimulation [103, 115]. Taking this into consideration, together with the fact that activation of M1 and A2ARs cooperate downstream to activate PKA, one may speculate that methylprednisolone facilitates ACh release by increasing the readily releasable pool during high-frequency stimuli through the prevention of vesicle clustering in the reserve pool [51, 115].
The putative involvement of adenosinergic signalling in the pathophysiology of ALS has also been reported [116]. ALS is a neurodegenerative disease leading to motor neuron dysfunction, resulting in impairment of neuromuscular transmission [117]. Using electrophysiology, Nascimento et al. demonstrated the role of A2AR on neuromuscular transmission in the ALS SOD1(G93A) mouse model, as well as its nuances during disease progression [111]. In ALS pre-symptomatic phase (4–6 weeks old mice), the A2AR-mediated excitation of the neuromuscular transmission had a higher magnitude than that found in age-matched controls, as demonstrated by increases in the mean amplitude and quantal content of EPPs, as well as in the frequency of MEPPs and appearance of giant MEPPs. Contrariwise, the CGS 21,680-induced A2AR-mediated facilitation was absent from symptomatic SOD1(G93A) mice (12–14 weeks old). In addition, the A2AR is overexpressed in lymphocytes from ALS patients, leading to intracellular cAMP accumulation in these cells [118]. This may indicate a putative role of A2AR in immunosuppression observed in ALS patients.
Regarding the controversies associated with the expression and function of A3R and A2BR at the NMJ, further investigations are mandatory to elucidate the role of these low-affinity ADO receptors on neurotransmitter release using animal models of skeletal muscle ischaemia/reperfusion, congenital and toxicological myopathies, and/or autoimmune and inflammatory insults where extracellular ADO (and inosine) levels may increase dramatically.
ADO Modulates Cholinergic and Peptidergic Signalling at the NMJ
Modulation of Cholinergic Autoreceptors Function
In addition to direct modulation of the neuromuscular transmission, evidence points towards crosstalk between co-localised P1 receptors, mAChRs and nAChRs at the NMJ [6, 73, 119, 120]. Motor nerve terminals are endowed with facilitatory M1 and inhibitory M2 muscarinic receptors (M1Rs and M2Rs, respectively), which coordinate their actions to adjust ACh release to neuronal activity [6, 119, 120]. The M1R positive feedback loop predominates during low-frequency neuronal firing, but it fades out upon increasing the neuronal firing rate. The opposite occurs concerning the M2R-mediated inhibition of ACh release (Fig. 1a and b) [6]. The muscarinic M1/M2 activation balance may also depend on the duration of neuronal activation [7]. Immunofluorescence staining of the mouse levator auris longus showed that M1Rs are absent from the motor endplate region but can be detected at presynaptic sites, possibly on the nerve terminal or PSCs [120].
Besides acting on mAChRs, ACh facilitates its release through the activation of fast desensitising nAChRs located on motor nerve terminals of both mice and rats [5, 121, 122]. Nicotinic facilitation of ACh release from motor nerve terminals of the rat phrenic nerve exhibits a pharmacological profile suggesting the involvement of α3β2 subunits-containing nAChRs (Fig. 1a) [4, 123]. The mechanism(s) underlying the facilitation of ACh release by presynaptic M1Rs and α3β2-containing nAChRs is still elusive. Experimental data suggest that release facilitation by M1Rs relies mainly on intracellular Ca2+ recruitment from inositol 1,4,5-triphosphate (IP3)-sensitive stores (Fig. 1a), being relatively insensitive to blockage of PKC activity [101, 124,125,126]. Most likely, α3β2-containing nAChRs may facilitate ACh release either through direct Ca2+ influx via the nicotinic pore or through indirect focal depolarisation of the nerve terminal membrane [4].
Increasing evidence suggests that cholinergic facilitation of ACh release is fine-tuning modulated by endogenous ADO generated at the neuromuscular synapse to adapt the neuromuscular transmission to the neuronal firing rate. Low endogenous levels of the nucleoside are generated when the neuronal firing pace is slow, which favours activation of inhibitory A1Rs, thus keeping the facilitation of neurotransmitter release by muscarinic M1 and α3β2 nAChRs fully operative to reduce failures of skeletal muscle contraction in response to nerve activity [4, 123]. Upon increasing the motoneuronal firing rate, the endogenous formation of ADO from released adenine nucleotides dramatically increases fostering the activation of facilitatory A2ARs [46]. This shift from A1R to A2AR signalling contributes to attenuated facilitation of ACh release by M1Rs and α3β2 nAChRs, thus favouring the inhibitory control of the neuromuscular transmission handled by muscarinic M2Rs [6, 46, 127]. Tonic activation of A2ARs suppresses α3β2 nAChR-mediated facilitation of ACh by a mechanism dependent on cAMP generation (Fig. 1b) [121, 123] and PKA activation [99, 101], which accelerates the desensitisation of nAChRs [128]. Moreover, the adaptive shift also triggers the recruitment of normally quiescent L-type (Cav1) VGCCs, which might compensate for the loss of fast desensitising P/Q-type (Cav2.1) currents to attenuate/prevent tetanic-induced synaptic depression during high-frequency neuronal activity (Fig. 1a and b; see above) [77, 86].
The mechanism by which M2Rs inhibits evoked ACh release remains largely unexplored but might involve a reduction in cAMP levels due to Gi/o protein coupling [125, 129] or to the modulation of a presynaptic effector mechanism downstream of Ca2+ entry, like that occurring with presynaptic A1Rs [127]. Neurochemical and electrophysiological data show that activation of A1 and M2 inhibitory receptors is mutually exclusive [6, 73, 127]. Besides preventing M2R-mediated inhibition of ACh release, full operation of the M1R also attenuates the A2AR-mediated facilitation through a mechanism involving PKC-induced phosphorylation of AC and/or its downstream intracellular signalling cascade (Fig. 1a) [101, 125].
Modulation of Peptidergic Neurotransmission
A1R and A2AR play a pivotal role in the facilitation of nerve-evoked ACh release by neuropeptides at neuromuscular synapses [14, 130,131,132]. Calcitonin gene-related peptide (CGRP) released from large dense-core vesicles of mammalian motor nerve terminals [133, 134] facilitates ACh release by a mechanism involving stimulation of the AC activity [133, 135, 136], providing that A2AR are synchronously activated by endogenously generated ADO [14]. The mechanism underlying the synergism between A2A and CGRP receptors to facilitate nerve-evoked ACh release is similar to that occurring between the purinergic receptor and forskolin, a direct AC activator [100], probably involving the coupling and/or recruitment of Gs proteins before activation of the AC [14, 137].
Cholinergic motor nerve terminals of the rat diaphragm also contain the vasoactive intestinal peptide (VIP) [138]. Neurochemical and electrophysiological data show that VIP acts presynaptically to facilitate ACh release at amphibian and mammalian NMJs [139]. Interestingly, VIP-induced facilitation of ACh release was only apparent when high extracellular levels of ADO accumulated at the neuromuscular synapse as a consequence of high-frequency neuronal firing episodes [46, 132]. Like that occurring with CGRP (see above), our findings show that synergism with A2ARs is also required to trigger VIP-induced facilitation of ACh release from motor nerve terminals [14, 132].
Moreover, A2ARs also play a pivotal role in the neuromuscular transmission facilitation caused by the neurotrophin brain-derived neurotrophic factor (BDNF). The source of BDNF at the skeletal motor endplate is still a matter of debate, even though it may be released from thrombin-activated skeletal muscle fibres [130]. Pre- and postsynaptic tropomyosin receptor kinase B (TrkB) receptors can mediate the actions of BDNF at the NMJ [130, 131, 140, 141]. BDNF-induced potentiation of synaptic transmission associated with increases in the release probability involves the functional coupling between A2AR-dependent PKA and neurotrophin-triggered PLCγ and mitogen-activated protein kinase pathway [130, 131]. Overall, these findings suggest a common pattern indicating that the neuromuscular influence of neuropeptides and/or neurotrophins critically depends on endogenous ADO generation to extracellular levels high enough to activate A2AR.
Perisynaptic Schwann Cells (PSCs): a Third-party Player in Neuromuscular Transmission Tuning
PSCs Regulate Neuromuscular Development and Homeostasis
PSCs play significant roles in NMJ synaptogenesis, development, and repair [2]. Since PSCs directly shield the NMJ, their importance for the development and maintenance of this specialised synapse has been extensively investigated over the years. At amphibian NMJs, PSCs appear shortly after the formation of the first few muscle connections and maintain close contact with the neuromuscular synapse during its development. Throughout ontogeny, PSCs extend sprouts that spread well beyond the borders of the NMJ, which lead to nerve terminal growth; these processes disappear at later stages to give rise to the mature NMJ architecture. As such, PSC sprouts likely to play an important role in synaptic growth and maturation at the developing NMJ [142]. In line with such evidence, selective ablation of PSCs from amphibian NMJs causes early (within a few days) retraction of nerve terminals, synaptic growth inhibition, and nerve-evoked muscle contraction impairment [143]. Regeneration of NMJs also implicates PSCs via mechanisms similar to those described for synaptogenesis. Following nerve injury, PSCs elaborate extensive sprouts to guide adequately the regenerating nerve terminals [144, 145].
Overall, PSCs account for important processes related to the formation, maintenance, and repair of the NMJ. By receiving inputs from both skeletal muscle fibres and motor neurons, PSCs may respond concurrently by expressing a plethora of molecular effectors. The relevance of PSCs in this process is highlighted given that their absence, or of its molecular effectors, significantly impairs the formation, maintenance and repair of NMJs, resulting in structural and functional abnormalities of motor endplates. Therefore, PSCs are situated to sense the functional state of motor endplates, which might be critical for the development and preservation of NMJ integrity [2, 146].
PSCs Modulate the Neuromuscular Synaptic Transmission
In addition to their role in synaptogenesis, development, and repair, PSCs are crucial players in neurotransmission regulation both in amphibian [12, 13] and in mammalian [8,9,10] NMJs. This agrees with the tripartite arrangement concept for the neuromuscular synapse and complies with that found in several synapses of the CNS [147]. At the frog NMJ, injection of guanosine 5′-O-(3-thiotriphosphate) (GTPγS, a non-hydrolysable analogue of guanosine 5′-triphosphate) in PSCs reduces nerve-evoked transmitter release through pertussis-toxin-sensitive and insensitive G-proteins [13]. This suggests that nonspecific activation of G proteins in PSCs depresses neuromuscular transmission, with cholinergic and/or purinergic metabotropic receptors as strong candidates to fulfil this task. Contrariwise, Ca2+ mobilisation from IP3-sensitive stores in PSCs potentiates synaptic activity, while injection of the fast Ca2+ chelator, BAPTA, into these cells favours synaptic depression [12]. Overall, it has been proposed that mammalian PSCs may be important to decode, either facilitating or inhibiting synaptic activity based on intracellular Ca2+ oscillations and the dynamic interplay with purinoceptors [10]. Increasing evidence demonstrates a direct link between the activation of α7 nAChRs at the surface of PSCs and depression of the neuromuscular transmission [8, 9]. PSCs are also endowed with plasma membrane-bound purinoceptors, which may be critical for these cells to sense and adapt neuromuscular transmission on a moment-to-moment basis [9, 11]. Unravelling the complex network of interactions between different cells at the NMJ will certainly upgrade our understanding of the molecular mechanisms involved in the regulation of ACh release and the organisation of tripartite synapses, both in health and disease conditions.
PSCs Modulate Synaptic Activity via Purinergic Signalling
The presence of purinoceptors, side by side with cholinoceptors and receptors for other signalling molecules [9, 11], allows PSCs to sense and regulate the neuromuscular synaptic activity through a variety of different mechanisms [8,9,10, 36]. ATP and ADO have been identified as putative gliotransmitters released from PSCs in response to synaptic activity to either inhibit or facilitate ACh release through the activation of various purinoceptor subtypes [8, 10].
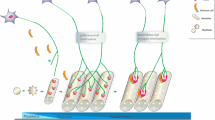
The presence of functional homo-pentameric nAChRs containing α7 subunits (α7 nAChRs) was demonstrated on the surface of PSCs from adult rodents by immunohistochemistry [8, 9]. These receptors were initially assumed to be located on presynaptic nerve terminals, most likely because of their ability to affect electrophysiological recordings using sharp intracellular microelectrodes impaling skeletal muscle fibres, which do not distinguish presynaptic from perisynaptic-originated signals [148]. α7 nAChRs were also identified in the sarcolemma of developing or denervated NMJs [9, 149], but they were absent from motor nerve terminals and skeletal muscle fibres of healthy adult animals [9, 150]. Unlike the muscle-type α1-containing nAChRs, the α7 nAChRs desensitise rapidly, exhibit higher Ca2 permeability, and are equally activated by ACh and its metabolite, choline [8, 151]. The perisynaptic localisation of these receptors on PSCs prompts a sensing activity that is predominant when ACh spills over from the motor endplate during intense neuronal firing or in the presence of cholinesterase inhibitors used to reverse surgical neuromuscular block and myasthenic crisis. α7 nAChRs present at the surface of PSCs operate a negative-feedback mechanism to downregulate nerve-evoked ACh release (Fig. 2) [9].

Sensing role PSCs to control ACh spillover from the neuromuscular synapse in the presence of cholinesterase inhibitors: on the role of α7 nAChRs-induced adenosine release and retrograde activation of A1 receptors. See text for details. Bold arrows indicate an increase in the strength of the proposed pathways; dashed arrows indicate a decrease in the strength of the proposed pathways. Predominant receptors operating in each condition appear as fully opaque, while non-predominant receptors appear as semi-transparent. The plus green symbol in panel (A) stands for enhancement of Ca2+ influx and acetylcholine release, while the minus red symbol in panel (B) accounts for a decrease in both Ca2+ influx and neurotransmitter release. Abbreviations: ACh, acetylcholine; AChE, acetylcholinesterase; ADK, adenosine kinase; ADO, ADO; AMP, adenosine 5′-monophosphate; ENT1, equilibrative nucleoside transporter 1; EPP, endplate potential; VGCC, voltage-gated Ca2+ channel
The fact that (1) α7 nAChRs are absent from motor nerve terminals and that (2) α7 nAChR-induced inhibition of transmitter exocytosis required inhibition of butyrylcholinesterase anchored to the surface of PSCs raised questions about (i) the physiological meaning of the need for surplus ACh accumulation at the neuromuscular synapse and (ii) the chemical nature of the gliotransmitter responsible for the communication between the PSC and the underlying motor nerve terminal (Fig. 2a). Using neurochemical and real-time fluorescence microscopy assays, our group showed that selective activation of α7 nAChRs increased intracellular Ca2+ inside PSCs while decreasing transmitter exocytosis elicited by high-frequency nerve stimulation bursts [8]. All these effects were abrogated by the glial cell metabolic uncoupler, sodium fluoroacetate [152, 153]. Data also showed that α7 nAChRs control ACh spillover from the neuromuscular synapse by promoting ADO outflow from PSCs via ENT1, with subsequent retrograde activation of presynaptic inhibitory A1Rs. These findings demonstrate that ADO is the gliotransmitter involved in this mechanism. This was concluded because (1) removal of endogenous ADO with ADA, (2) inhibition of ADO release via ENT1 transporter, and (3) blockage of presynaptic A1Rs, all prevented nerve-evoked ACh release inhibition caused by α7 nAChR activation on PSCs [8]. Moreover, the pharmacology of α7 nAChR-induced down-modulation of ACh release was remarkably similar to that observed by inhibiting ADK [154], a situation that is known to increase the intracellular accumulation of the nucleoside, thus forcing its translocation to the extracellular milieu via ENTs (Fig. 2b).
There is, however, a gap in our knowledge about the molecular mechanism linking α7 nAChR activation to ADO outflow from PSCs via ENT1. It is known that both human and mouse ENT1 are directly phosphorylated by PKA and PKC [155] and that PKC-mediated phosphorylation of ENT1 increases its transport efficacy and/or mobilisation to the plasma membrane, thus contributing to facilitating ADO outflow [156, 157]. Alternatively, PKC-mediated ADK inhibition may foster ADO outflow via ENT1 by increasing the intracellular concentration of the nucleoside [154]. Experimental data ruled out the involvement of PLC and IP3 receptor-mediated mechanisms [8], as well as the participation of atypical PKC isoforms [158] in the inhibitory role of α7 nAChR on neuromuscular transmission. This suggests that other effectors, like Ca2+/calmodulin-dependent protein kinase II and ryanodine receptors, may be involved instead [159].
Nerve-evoked PSC Ca2+ transients increase in magnitude and duration when high-frequency tetanic trains are delivered in the presence of the cholinesterase inhibitor, neostigmine [8]. These findings may be clinically relevant given that cholinesterase inhibitors are frequently used to improve neuromuscular transmission in patients with myasthenia gravis and to reverse the residual neuromuscular block caused by non-depolarising muscle relaxants. Activation of perisynaptic α7 nAChRs by ACh spilling over from the NMJ may also explain the paradoxical reduction of nerve-evoked neurotransmitter release (e.g. “train-of-four fade”, meaning the reduction of the fourth to the first twitch height in a stimulation train delivered at 2 Hz frequency) observed with widely used cholinesterase inhibitors, like neostigmine, leading to partial or longer recovery from the neuromuscular block even in the presence of atropine [160,161,162]. A similar mechanism may occur regarding the effects of muscle relaxants exhibiting significant cholinesterase activity, like cis-atracurium [163,164,165].
The nerve stimulation pattern also differentially affects synaptic activity at the mouse NMJ, and these nuances are intimately linked to the glial modulation of neurotransmitter release (Fig. 3). At the mouse soleus, prolonged continuous stimulation trains have been shown to favour post-tetanic potentiation and phasic Ca2+ responses in PSCs, while intermittent bursting paradigms favour post-tetanic depression and oscillatory Ca2+ responses in these cells [10, 36]. Chelation of Ca2+ transients inside glial cells abolishes these effects, strengthening the idea that PSCs interpret and effectively modulate synaptic activity using Ca2+ as a second messenger [12, 13]. By mimicking Ca2+ oscillations inside PSCs using flash photolysis of caged Ca2+, Todd and co-workers [10] showed that (i) synaptic plasticity events were prevented by inhibiting the extracellular ADO formation from the catabolism of adenine nucleotides, and that (ii) the post-tetanic depression and potentiation were dependent on the activation of A1R (Fig. 3a) and A2AR (Fig. 3b), respectively. These findings suggest that PSCs release ATP in response to Ca2+ transients secondary to motor neuron activation which after being sequentially metabolised into ADO activates A1R or A2AR depending on the stimulation paradigm [46].

PSCs detect patterns of synaptic activity and subsequentially provide feedback to motor neurons by releasing ATP. Depending on the paradigm of neuronal firing, ATP released by PSCs can either decrease (A) or increase (B) ACh release via its rapid hydrolysis into adenosine and activation of A1 or A2ARs, respectively. See text for details. Bold arrows indicate an increase in the strength of the proposed pathways. The minus red symbol in panel (A) stands for a decrease in Ca2+ influx and ACh release, while the plus green symbol in panel (B) accounts for an increase in both Ca2+ influx and neurotransmitter release. Abbreviations: ACh, acetylcholine; ADO, adenosine; ATP, adenosine 5′-triphosphate; EPP, endplate potential; VGCC, voltage-gated Ca2+ channel
Despite the aforementioned findings, it is still unclear which receptors are involved in the ability of PSCs to detect and modulate the activity of the neuromuscular synapse. The broad-spectrum nicotinic antagonist, d-tubocurarine, is commonly used to paralyse skeletal muscle fibres during electrophysiological recordings, which might eliminate any source of modulation undertaken by α7 nAChRs located in the plasma membrane of PSCs. It is likely that under such conditions PSCs detect and modulate nerve-evoked ACh release via muscarinic and/or purinergic receptors [11], resulting in ATP and/or ADO release to the synaptic cleft. The presence of mAChRs has been functionally demonstrated in PSCs of amphibian NMJs; this assumption was based on the fact that both electrical nerve stimulation and exogenous application of mAChR agonists were able to induce PSCs Ca2+ oscillations in the presence of d-tubocurarine [166, 167]. This conclusion is strengthened by the fact that both pertussis-toxin-sensitive and insensitive G-proteins and Ca2+ recruitment form PSC internal stores were able to influence neuromuscular transmission at the frog NMJ [12, 13]. In this context, it might be possible that the effects attributed to facilitatory M1Rs at motor endplates reflect, at least in part, the effect of a retrograde gliotransmitter released by PSCs [168]. Likewise, activation of P2X, P2Y, and A1Rs on PSCs causes intracellular Ca2+ oscillations in these cells [11, 34], strengthening the neuromuscular plasticity phenomena reported by Todd and co-workers [10]. However, there are still gaps in our knowledge concerning the putative role of perisynaptic muscarinic and purinergic receptors on neuromuscular transmission adaptations, which are worth pursuing in the future.
Considering that A1R may be present and operate Ca2+ rises inside PSCs at mammalian NMJs [11], one may hypothesise that these receptors act synergistically with the α7 nAChR-mediated sensing mechanism to control spillover of the neurotransmitter from the motor endplate [8, 9]. Thus, α7 nAChR-induced Ca2+ oscillations may be further strengthened by fostering ADO outflow from PSCs, which results in autocrine activation of A1Rs. While this mechanism may be functionally relevant to amplify the sensing role of the α7 nAChR on PSCs, it still does not explain the inhibitory repercussion of this receptor on neurotransmitter exocytosis, unless ADO simultaneously acts as a retrograde gliotransmitter via inhibitory A1Rs on motor nerve terminals [8, 46, 78]. Compelling experimental data indicate that the inhibitory tone operated via A1Rs on nerve-evoked ACh release is normally silent during high-frequency neuronal bursts [46] unless α7 nAChR on PSCs are activated to restrain ACh spillover from the neuromuscular synapse [8]. Thus, the integrated action of α7 nAChR and A1Rs on PSCs may act as a secondary self-sustained ADO-mediated break supplementing the muscarinic M2R auto-inhibition to avoid exhaustion of ACh reservoirs during prolonged high-frequency neuronal activity, which would endanger the integrity of motor nerve terminals and, thus, neuromuscular transmission efficacy.
Concluding Remarks
At mature NMJs, adenine nucleotides are intimately involved in the control of ACh release from stimulated motor nerve terminals, as well as in muscular contractile activity. ATP is released together with ACh from activated nerve terminals. The release of ATP from contracting skeletal myotubes has also been observed. While the latter may involve pannexin-1 hemichannels, ATP releases together with ACh implies exocytosis of synaptic vesicles. Once in the extracellular space, ATP may be rapidly broken down into ADP, AMP, and ADO [24, 60]. However, there are still gaps in our knowledge concerning the mechanisms implicated in the control of neuromuscular transmission by adenine nucleotides, though data suggest that they might differ between species and developing or adult motor endplates.
ADP-sensitive P2Y12 and P2Y13Rs play inhibitory roles in quantal ACh release from nerve terminals of amphibian and mammalian NMJs, respectively [41, 59]. Gq/11-coupled P2YRs were also involved in the inhibition of non-quantal ACh release [39, 40], yet their full molecular and pharmacological characterisation is still missing. Likewise, activation of P2YRs on PSCs triggers intracellular Ca2+ transients, but the physiological meaning of this effect remains to be elucidated [34]. Regarding ATP-sensitive P2XRs, evidence has been gathered indicating that they might be present on presynaptic motor nerve terminals as well as on PSCs. Activation of neuronal P2X7Rs is associated with increases in vesicular exocytosis, while activation of P2XRs on PSCs favours intracellular Ca2+ oscillations [34, 49]. Given the limited information regarding P2 receptor characterisation and function at the neuromuscular junction, further studies are encouraged before any firm conclusion can be drawn from their role in neuromuscular transmission and plasticity.
At most motor endplates, ADO can be released as such or originated from the extracellular catabolism of release ATP. The nucleoside plays a predominant inhibitory role in nerve-evoked ACh release under resting conditions via the activation of inhibitory A1Rs [37, 38, 46, 88]. This scenario dramatically changes during high-frequency stimuli or upon focal depolarisation of motor nerve terminals, where co-localised A2ARs contribute to facilitating neurotransmitter release [46, 86, 111], which may be critical to overcome the tetanic depression of the neuromuscular transmission (Fig. 1a and 1b) [77]. Low-affinity A2B and A3Rs have also been identified at the mouse NMJ; the presence of the A3R subtype raises the possibility that the ADO metabolite inosine can also inhibit nerve-evoked ACh release [104, 108].
Close association and putative functional interplay between the ADO-generating enzyme, ecto-5′-nucleotidase/CD73, and facilitatory A2ARs has been demonstrated. This explains why the extracellular breakdown of adenine nucleotides delivers the nucleoside directly to this receptor subtype, thus facilitating ACh release in many different synapses [24, 95,96,97,98]. Activation of A2ARs fosters Ca2+ influx into stimulated motor nerve terminals through the recruitment of normally “quiescent” L-type (Cav1) channels by an AC/cAMP/PKA-dependent pathway; this mechanism contributes to bypass P/Q-type (Cav2.1) channel desensitisation and sustains ACh release during high-frequency neuronal bursts [46, 75, 77, 86]. Failure of this mechanism has been associated with neuromuscular transmission deficits in myasthenic patients, leading us to hypothesise that targeting the endogenous ADO formation by ecto-5′-nucleotidase/CD73 and A2AR activation might restore neuromuscular competence while also suppressing activation of the immune system in myasthenia gravis patients [28, 69, 114].
ADO also plays a pivotal role in regulating the cholinergic and peptidergic modulation of ACh release at the NMJ. Activation of A1Rs suppresses cholinergic auto-inhibition by M2Rs, thus allowing the M1R-mediated facilitation to play a predominant role in resting conditions [6, 73]. As a shift from A1-inhibitory to A2A-facilitatory tonus occurs during high neuronal firing rates, the activity of facilitatory M1Rs and α3β2 nAChRs is suppressed, and that of M2Rs is unmasked [6, 121, 123]. Furthermore, activation of A2ARs by ADO also unmasks the facilitatory role of neuropeptides such as CGRP [14], VIP [132], and BDNF [130, 131] by a mechanism involving the AC/cAMP/PKA pathway.
As emphasised above, PSCs are endowed with A1Rs coupled to intracellular Ca2+ mobilisation [11, 34, 104]. Autocrine stimulation of A1Rs by ADO released from PSCs (via ENT1 transporters) in response to α7 nAChR activation may significantly enhance the sensing ability of the latter receptors to control ACh spillover from the motor endplate region and prevent exhaustion of the neurotransmitter during high-frequency neuronal bursts; this could otherwise endanger subsequent neuromuscular transmission performance and motor endplate integrity [8, 9]. Thus, A1Rs located on PSCs and motor nerve terminals may act in tandem to control neurotransmitter exocytosis once α7 nAChRs sense an excess of ACh emerging from the neuromuscular synapse and trigger the release of ADO, which most likely functions as a retrograde gliotransmitter at mammalian motor endplates. This newly evidenced mechanism may add an explanation to the paradoxical reductions of nerve-evoked neurotransmitter release often observed in medical interventions with cholinesterase inhibitors, like neostigmine [160,161,162], and with skeletal muscle relaxants exhibiting significant cholinesterase activity, like cis-atracurium [163,164,165].
In summary, purines play extremely important and dynamic roles in modulating neuromuscular transmission in health and disease conditions. Adenine nucleotides and nucleosides are also engaged in the mechanisms by which PSCs govern neuromuscular transmission, emphasising the role of these purines as gliotransmitters in tripartite synapses, like the NMJ. Using animal models of neuromuscular diseases, it became evident that fostering the A2AR-mediated reinforcement of the neuromuscular transmission may be a good therapeutic strategy to overcome the neuromuscular deficits associated with myasthenia gravis and ALS, providing the conclusion of ongoing preclinical studies and the development of novel and safer drug compounds entitled to be used in clinical trials.
Data Availability
Not applicable.
References
Fagerlund MJ, Eriksson LI (2009) Current concepts in neuromuscular transmission. Br J Anaesth 103:108–114. https://doi.org/10.1093/bja/aep150
Ko CP, Robitaille R (2015) Perisynaptic Schwann cells at the neuromuscular synapse: adaptable, multitasking glial cells. Cold Spring Harb Perspect Biol 7. https://doi.org/10.1101/cshperspect.a020503
Bittner EA, Martyn JAJ (2019) 21 - neuromuscular physiology and pharmacology. In: Hemmings HC, Egan TD (eds) Pharmacology and Physiology for Anesthesia, 2nd edn. Elsevier, Philadelphia, pp 412–427
Faria M, Oliveira L, Timóteo MA, Lobo MG, Correia-de-Sá P (2003) Blockade of neuronal facilitatory nicotinic receptors containing α3β2 subunits contribute to tetanic fade in the rat isolated diaphragm. Synapse 49:77–88. https://doi.org/10.1002/syn.10211
Wessler I, Scheuer B, Kilbinger H (1987) [3H]acetylcholine release from the phrenic nerve is increased or decreased by activation or desensitization of nicotine receptors. Eur J Pharmacol 135:85–87. https://doi.org/10.1016/0014-2999(87)90760-6
Oliveira L, Timóteo MA, Correia-de-Sá P (2002) Modulation by adenosine of both muscarinic M1-facilitation and M2-inhibition of [3H]-acetylcholine release from the rat motor nerve terminals. Eur J Neurosci 15:1728–1736. https://doi.org/10.1046/j.1460-9568.2002.02020.x
Wessler I, Karl M, Mai M, Diener A (1987) Muscarine receptors on the rat phrenic nerve, evidence for positive and negative muscarinic feedback mechanisms. Naunyn-Schmiedeb Arch Pharmacol 335:605–612. https://doi.org/10.1007/BF00166975
Noronha-Matos JB, Oliveira L, Peixoto AR, Almeida L, Castellão-Santana LM, Ambiel CR, Alves-do Prado W, Correia-de-Sá P (2020) Nicotinic α7 receptor-induced adenosine release from perisynaptic Schwann cells controls acetylcholine spillover from motor endplates. J Neurochem 154:263–283. https://doi.org/10.1111/jnc.14975
Petrov KA, Girard E, Nikitashina AD, Colasante C, Bernard V, Nurullin L, Leroy J, Samigullin D et al. (2014) Schwann cells sense and control acetylcholine spillover at the neuromuscular junction by alpha7 nicotinic receptors and butyrylcholinesterase. J Neurosci 34:11870–11883. https://doi.org/10.1523/JNEUROSCI.0329-14.2014
Todd KJ, Darabid H, Robitaille R (2010) Perisynaptic glia discriminate patterns of motor nerve activity and influence plasticity at the neuromuscular junction. J Neurosci 30:11870–11882. https://doi.org/10.1523/JNEUROSCI.3165-10.2010
Rochon D, Rousse I, Robitaille R (2001) Synapse-glia interactions at the mammalian neuromuscular junction. J Neurosci 21:3819–3829. https://doi.org/10.1523/JNEUROSCI.21-11-03819.2001
Castonguay A, Robitaille R (2001) Differential regulation of transmitter release by presynaptic and glial Ca2+ internal stores at the neuromuscular synapse. J Neurosci 21:1911–1922. https://doi.org/10.1523/jneurosci.21-06-01911.2001
Robitaille R (1998) Modulation of synaptic efficacy and synaptic depression by glial cells at the frog neuromuscular junction. Neuron 21:847–855. https://doi.org/10.1016/S0896-6273(00)80600-5
Correia-de-Sá P, Ribeiro JA (1994) Potentiation by tonic A2a-adenosine receptor activation of CGRP-facilitated [3H]-ACh release from rat motor nerve endings. Br J Pharmacol 111:582–588. https://doi.org/10.1111/j.1476-5381.1994.tb14777.x
Pinard A, Robitaille R (2008) Nitric oxide dependence of glutamate-mediated modulation at a vertebrate neuromuscular junction. Eur J Neurosci 28:577–587. https://doi.org/10.1111/j.1460-9568.2008.06355.x
Ribeiro JA, Cunha RA, Correia-de-Sá P, Sebastião AM (1996) Purinergic regulation of acetylcholine release. Prog Brain Res 109:231–241. https://doi.org/10.1016/S0079-6123(08)62107-X
Wood SJ, Slater CR (2001) Safety factor at the neuromuscular junction. Prog Neurobiol 64:393–429. https://doi.org/10.1016/S0301-0082(00)00055-1
Burnstock G (2007) Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev 87:659–797. https://doi.org/10.1152/physrev.00043.2006
Silinsky EM (1975) On the association between transmitter secretion and the release of adenine nucleotides from mammalian motor nerve terminals. J Physiol 247:145–162. https://doi.org/10.1113/jphysiol.1975.sp010925
Wagner JA, Carlson SS, Kelly RB (1978) Chemical and physical characterization of cholinergic synaptic vesicles. Biochem 17:1199–1206. https://doi.org/10.1021/bi00600a010
Volknandt W, Zimmermann H (1986) Acetylcholine, ATP, and proteoglycan are common to synaptic vesicles isolated from the electric organs of electric eel and electric catfish as well as from rat diaphragm. J Neurochem 47:1449–1462. https://doi.org/10.1111/j.1471-4159.1986.tb00778.x
Smith DO (1991) Sources of adenosine released during neuromuscular transmission in the rat. J Physiol 432:343–354. https://doi.org/10.1113/jphysiol.1991.sp018388
Verkhratsky A, Krishtal O (2009) Adenosine triphosphate (ATP) as a neurotransmitter. In: Squire LR (ed) Encyclopaedia of Neuroscience. Academic Press, Oxford, pp 115–123
Magalhães-Cardoso MT, Pereira MF, Oliveira L, Ribeiro JA, Cunha RA, Correia-de-Sá P (2003) Ecto-AMP deaminase blunts the ATP-derived adenosine A2A receptor facilitation of acetylcholine release at rat motor nerve endings. J Physiol 549:399–408. https://doi.org/10.1113/jphysiol.2003.040410
Cunha RA, Sebastião AM (1993) Adenosine and adenine nucleotides are independently released from both the nerve terminals and the muscle fibres upon electrical stimulation of the innervated skeletal muscle of the frog. Pflügers Arch 424:503–510. https://doi.org/10.1007/BF00374914
Vizi ES, Nitahara K, Sato K, Sperlágh B (2000) Stimulation-dependent release, breakdown, and action of endogenous ATP in mouse hemidiaphragm preparation: the possible role of ATP in neuromuscular transmission. J Auton Nerv Syst 81:278–284. https://doi.org/10.1016/S0165-1838(00)00129-6
Santos DA, Salgado AI, Cunha RA (2003) ATP is released from nerve terminals and from activated muscle fibres on stimulation of the rat phrenic nerve. Neurosci Lett 338:225–228. https://doi.org/10.1016/S0304-3940(02)01419-2
Noronha-Matos JB, Morais T, Trigo D, Timóteo MA, Magalhães-Cardoso MT, Oliveira L, Correia-de-Sá P (2011) Tetanic failure due to decreased endogenous adenosine A2A tonus operating neuronal Cav1 (L-type) influx in myasthenia gravis. J Neurochem 117:797–811. https://doi.org/10.1111/j.1471-4159.2011.07216.x
Buvinic S, Almarza G, Bustamante M, Casas M, López J, Riquelme M, Sáez JC, Huidobro-Toro JP et al (2009) ATP released by electrical stimuli elicits calcium transients and gene expression in skeletal muscle. J Biol Chem 284:34490–34505. https://doi.org/10.1074/jbc.M109.057315
Liu GJ, Werry EL, Bennett MR (2005) Secretion of ATP from Schwann cells in response to uridine triphosphate. Eur J Neurosci 21:151–160. https://doi.org/10.1111/j.1460-9568.2004.03831.x
Shin YH, Lee SJ, Jung J (2012) Secretion of ATP from Schwann cells through lysosomal exocytosis during Wallerian degeneration. Biochem Biophys Res Commun 429:163–167. https://doi.org/10.1016/j.bbrc.2012.10.121
Wei ZY, Qu HL, Dai YJ, Wang Q, Ling Z, Su WF, Zhao YY, Shen WX et al. (2021) Pannexin 1, a large-pore membrane channel, contributes to hypotonicity-induced ATP release in Schwann cells. Neural Regen Res 16:899–904. https://doi.org/10.4103/1673-5374.290911
Ralevic V, Burnstock G (1998) Receptors for purines and pyrimidines. Pharmacol Rev 50:413–492
Robitaille R (1995) Purinergic receptors and their activation by endogenous purines at perisynaptic glial cells of the frog neuromuscular junction. J Neurosci 15:7121–7131. https://doi.org/10.1523/JNEUROSCI.15-11-07121.1995
De Lorenzo S, Veggetti M, Muchnik S, Losavio A (2006) Presynaptic inhibition of spontaneous acetylcholine release mediated by P2Y receptors at the mouse neuromuscular junction. Neurosci 142:71–85. https://doi.org/10.1016/j.neuroscience.2006.05.062
Todd KJ, Robitaille R (2006) Purinergic modulation of synaptic signalling at the neuromuscular junction. Pflügers Arch 452:608–614. https://doi.org/10.1007/s00424-006-0068-3
Sokolova E, Grishin S, Shakirzyanova A, Talantova M, Giniatullin R (2003) Distinct receptors and different transduction mechanisms for ATP and adenosine at the frog motor nerve endings. Eur J Neurosci 18:1254–1264. https://doi.org/10.1046/j.1460-9568.2003.02835.x
Giniatullin RA, Sokolova EM (1998) ATP and adenosine inhibit transmitter release at the frog neuromuscular junction through distinct presynaptic receptors. Br J Pharmacol 124:839–844. https://doi.org/10.1038/sj.bjp.0701881
Malomouzh AI, Nikolsky EE, Vyskočil F (2011) Purine P2Y receptors in ATP-mediated regulation of non-quantal acetylcholine release from motor nerve endings of rat diaphragm. Neurosci Res 71:219–225. https://doi.org/10.1016/j.neures.2011.07.1829
Galkin AV, Giniatullin RA, Mukhtarov MR, Švandová I, Grishin SN, Vyskočil F (2001) ATP but not adenosine inhibits nonquantal acetylcholine release at the mouse neuromuscular junction. Eur J Neurosci 13:2047–2053. https://doi.org/10.1046/j.0953-816x.2001.01582.x
Giniatullin A, Petrov A, Giniatullin R (2015) The involvement of P2Y12 receptors, NADPH oxidase, and lipid rafts in the action of extracellular ATP on synaptic transmission at the frog neuromuscular junction. Neurosci 285:324–332. https://doi.org/10.1016/j.neuroscience.2014.11.039
Giniatullin AR, Grishin SN, Sharifullina ER, Petrov AM, Zefirov AL, Giniatullin RA (2005) Reactive oxygen species contribute to the presynaptic action of extracellular ATP at the frog neuromuscular junction. J Physiol 565:229–242. https://doi.org/10.1113/jphysiol.2005.084186
Giniatullin A, Petrov A, Giniatullin R (2019) Action of hydrogen peroxide on synaptic transmission at the mouse neuromuscular junction. Neurosci 399:135–145. https://doi.org/10.1016/j.neuroscience.2018.12.027
Ziganshin AU, Khairullin AE, Hoyle CHV, Grishin SN (2020) Modulatory roles of ATP and adenosine in cholinergic neuromuscular transmission. Int J Mol Sci 21. https://doi.org/10.3390/ijms21176423
Arkhipova OV, Grishin SN, Sitdikova GF, Zefirov AL (2006) The presynaptic effects of arachidonic acid and prostaglandin E2 at the frog neuromuscular junction. Neurosci Behav Physiol 36:307–312. https://doi.org/10.1007/s11055-006-0017-9
Correia-de-Sá P, Timóteo MA, Ribeiro JA (1996) Presynaptic A1 inhibitory/A2A facilitatory adenosine receptor activation balance depends on motor nerve stimulation paradigm at the rat hemidiaphragm. J Neurophysiol 76:3910–3919. https://doi.org/10.1152/jn.1996.76.6.3910
Cunha RA, Ribeiro JA (2000) ATP as a presynaptic modulator. Life Sci 68:119–137. https://doi.org/10.1016/S0024-3205(00)00923-1
Deuchars SA, Atkinson L, Brooke RE, Musa H, Milligan CJ, Batten TFC, Buckley NJ, Parson SH, Deuchars J (2001) Neuronal P2X7 receptors are targeted to presynaptic terminals in the central and peripheral nervous systems. J Neurosci 21:7143–7152. https://doi.org/10.1523/jneurosci.21-18-07143.2001
Moores TS, Hasdemir B, Vega-Riveroll L, Deuchars J, Parson SH (2005) Properties of presynaptic P2X7-like receptors at the neuromuscular junction. Brain Res 1034:40–50. https://doi.org/10.1016/j.brainres.2004.12.001
Salgado AI, Cunha RA, Ribeiro JA (2000) Facilitation by P2 receptor activation of acetylcholine release from rat motor nerve terminals: interaction with presynaptic nicotinic receptors. Brain Res 877:245–250. https://doi.org/10.1016/S0006-8993(00)02679-2
Oliveira L, Costa AC, Noronha-Matos JB, Silva I, Cavalcante WLG, Timóteo MA, Corrado AP, Dal Belo CA, Ambiel CR, Alves-do-Prado W, Correia-de-Sá P (2015) Amplification of neuromuscular transmission by methylprednisolone involves activation of presynaptic facilitatory adenosine A2A receptors and redistribution of synaptic vesicles. Neuropharmacol 89:64–76. https://doi.org/10.1016/j.neuropharm.2014.09.004
Ziganshin AU, Khairullin AE, Teplov AY, Gabdrakhmanov AI, Ziganshina LE, Hoyle CHV, Ziganshin BA, Grishin SN (2019) The effects of ATP on the contractions of rat and mouse fast skeletal muscle. Muscle Nerve 59:509–516. https://doi.org/10.1002/mus.26423
Khairullin AE, Teplov AY, Grishin SN, Farkhutdinov AM, Ziganshin AU (2019) The thermal sensitivity of purinergic modulation of contractile activity of locomotor and respiratory muscles in mice. Biophysics 64:812–817. https://doi.org/10.1134/S0006350919050075
Ziganshin AU, Khairullin AE, Zobov VV, Ziganshina LE, Gabdrakhmanov AI, Ziganshin BA, Grishin SN (2017) Effects of ATP and adenosine on contraction amplitude of rat soleus muscle at different temperatures. Muscle Nerve 55:417–423. https://doi.org/10.1002/mus.25263
Miteva AS, Gaydukov AE, Shestopalov VI, Balezina OP (2018) Mechanism of P2X7 receptor-dependent enhancement of neuromuscular transmission in pannexin 1 knockout mice. Purinergic Signal 14:459–469. https://doi.org/10.1007/s11302-018-9630-7
Noronha-Matos JB, Coimbra J, Sá-e-Sousa A, Rocha R, Marinhas J, Freitas R, Guerra-Gomes S, Ferreirinha F et al. (2014) P2X7-induced zeiosis promotes osteogenic differentiation and mineralization of postmenopausal bone marrow-derived mesenchymal stem cells. FASEB J 28:5208–5222. https://doi.org/10.1096/fj.14-257923
Volonte C, Apolloni S, Skaper SD, Burnstock G (2012) P2X7 receptors: channels, pores and more. CNS Neurol Disord Drug Targets 11:705–721. https://doi.org/10.2174/187152712803581137
Miteva AS, Gaydukov AE, Shestopalov VI, Balezina OP (2017) The role of pannexin 1 in the purinergic regulation of synaptic transmission in mouse motor synapses. Biochem (Mosc) Suppl A: Membr Cell Biol 11:311–320. https://doi.org/10.1134/S1990747817040067
Guarracino JF, Cinalli AR, Fernández V, Roquel LI, Losavio AS (2016) P2Y13 receptors mediate presynaptic inhibition of acetylcholine release induced by adenine nucleotides at the mouse neuromuscular junction. Neurosci 326:31–44. https://doi.org/10.1016/j.neuroscience.2016.03.066
Cunha RA, Sebastião AM (1991) Extracellular metabolism of adenine nucleotides and adenosine in the innervated skeletal muscle of the frog. Eur J Pharmacol 197:83–92. https://doi.org/10.1016/0014-2999(91)90368-Z
Braun N, Sévigny J, Robson SC, Hammer K, Hanani M, Zimmermann H (2004) Association of the ecto-ATPase NTPDase2 with glial cells of the peripheral nervous system. Glia 45:124–132. https://doi.org/10.1002/glia.10309
Meghji P, Pearson JD, Slakey LL (1992) Regulation of extracellular adenosine production by ectonucleotidases of adult rat ventricular myocytes. Am J Physiol Heart Circ Physiol 263:H40–H47. https://doi.org/10.1152/ajpheart.1992.263.1.H40
Sebastião AM, Cristóvão-Ferreira S, Ribeiro JA (2013) Downstream pathways of adenosine. In: Masino S, Boison D (eds) Adenosine: A Key Link between Metabolism and Brain Activity. Springer, New York, New York, NY, pp 131–156
Cunha RA (2001) Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: different roles, different sources and different receptors. Neurochem Int 38:107–125. https://doi.org/10.1016/S0197-0186(00)00034-6
Arch JRS, Newsholme EA (1978) Activities and some properties of 5′-nucleotidase, adenosine kinase and adenosine deaminase in tissues from vertebrates and invertebrates in relation to the control of the concentration and the physiological role of adenosine. Biochem J 174:965–977. https://doi.org/10.1042/bj1740965
Boison D (2013) Adenosine kinase: exploitation for therapeutic gain. Pharmacol Rev 65:906–943. https://doi.org/10.1124/pr.112.006361
Matzner H, Parnas H, Parnas I (1988) Presynaptic effects of d-tubocurarine on neurotransmitter release at the neuromuscular junction of the frog. J Physiol 398:109–121. https://doi.org/10.1113/jphysiol.1988.sp017032
Cruz LJ, Gray WR, Olivera BM, Zeikus RD, Kerr L, Yoshikami D, Moczydlowski E (1985) Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J Biol Chem 260:9280–9288
Oliveira L, Correia A, Cristina Costa A, Guerra-Gomes S, Ferreirinha F, Magalhães-Cardoso MT, Vilanova M, Correia-de-Sá P (2015) Deficits in endogenous adenosine formation by ecto-5′-nucleotidase/CD73 impair neuromuscular transmission and immune competence in experimental autoimmune myasthenia gravis. Mediators Inflamm 2015. https://doi.org/10.1155/2015/460610
Ko S-F, Huang C-C, Hsieh M-J, Ng S-H, Lee C-C, Lee C-C, Lin T-K, Chen M-C, Lee L (2008) 31P MR spectroscopic assessment of muscle in patients with myasthenia gravis before and after thymectomy: initial experience. Radiol 247:162–169. https://doi.org/10.1148/radiol.2471070591
Thomas S, Robitaille R (2001) Differential frequency-dependent regulation of transmitter release by endogenous nitric oxide at the amphibian neuromuscular synapse. J Neurosci 21:1087–1095. https://doi.org/10.1523/JNEUROSCI.21-04-01087.2001
Barroso A, Oliveira L, Campesatto-Mella E, Silva C, Timóteo MA, Magalhães-Cardoso MT, Alves-do-Prado W, Correia-de-Sá P (2007) L-citrulline inhibits [3H]acetylcholine release from rat motor nerve terminals by increasing adenosine outflow and activation of A1 receptors. Br J Pharmacol 151:541–550. https://doi.org/10.1038/sj.bjp.0707242
Oliveira L, Timóteo MA, Correia-de-Sá P (2009) Negative crosstalk between M1 and M2 muscarinic autoreceptors involves endogenous adenosine activating A1 receptors at the rat motor endplate. Neurosci Lett 459:127–131. https://doi.org/10.1016/j.neulet.2009.05.001
Ribeiro JA, Sebastião AM (1987) On the role, inactivation and origin of endogenous adenosine at the frog neuromuscular junction. J Physiol 384:571–585. https://doi.org/10.1113/jphysiol.1987.sp016470
Correia-de-Sá P, Ribeiro JA (1996) Adenosine uptake and deamination regulate tonic A2a receptor facilitation of evoked [3H]acetylcholine release from the rat motor nerve terminals. Neurosci 73:85–92. https://doi.org/10.1016/0306-4522(96)00028-0
Timóteo MA, Oliveira L, Campesatto-Mella E, Barroso A, Silva C, Magalhães-Cardoso MT, Alves-do-Prado W, Correia-de-Sá P (2008) Tuning adenosine A1 and A2A receptors activation mediates l-citrulline-induced inhibition of [3H]-acetylcholine release depending on nerve stimulation pattern. Neurochem Int 52:834–845. https://doi.org/10.1016/j.neuint.2007.09.016
Oliveira L, Timóteo MA, Correia-de-Sá P (2004) Tetanic depression is overcome by tonic adenosine A2A receptor facilitation of L-type Ca2+ influx into rat motor nerve terminals. J Physiol 560:157–168. https://doi.org/10.1113/jphysiol.2004.067595
Correia-de-Sá P, Sebastião AM, Ribeiro JA (1991) Inhibitory and excitatory effects of adenosine receptor agonists on evoked transmitter release from phrenic nerve ending of the rat. Br J Pharmacol 103:1614–1620. https://doi.org/10.1111/j.1476-5381.1991.tb09836.x
Fredholm BB, Ijzerman AP, Jacobson KA, Linden J, Müller CE (2011) International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors–an update. Pharmacol Rev 63:1–34. https://doi.org/10.1124/pr.110.003285
Jacobson KA, Gao ZG (2006) Adenosine receptors as therapeutic targets. Nat Rev Drug Discov 5:247–264. https://doi.org/10.1038/nrd1983
Ribeiro JA, Walker J (1975) The effects of adenosine triphosphate and adenosine diphosphate on transmission at the rat and frog neuromuscular junctions. Br J Pharmacol 54:213–218. https://doi.org/10.1111/j.1476-5381.1975.tb06931.x
Silinsky EM (1984) On the mechanism by which adenosine receptor activation inhibits the release of acetylcholine from motor nerve endings. J Physiol 346:243–256. https://doi.org/10.1113/jphysiol.1984.sp015019
Silinsky EM, Solsona CS (1992) Calcium currents at motor nerve endings: absence of effects of adenosine receptor agonists in the frog. J Physiol 457:315–328. https://doi.org/10.1113/jphysiol.1992.sp019380
Robitaille R, Thomas S, Charlton MP (1999) Effects of adenosine on Ca2+ entry in the nerve terminal of the frog neuromuscular junction. Can J Physiol Pharmacol 77:707–714. https://doi.org/10.1139/cjpp-77-9-707
Schwartz AD, Whitacre CL, Lin Y, Wilson DF (2003) Adenosine inhibits N-type calcium channels at the rat neuromuscular junction. Clin Exp Pharmacol Physiol 30:174–177. https://doi.org/10.1046/j.1440-1681.2003.03806.x
Correia-de-Sá P, Timóteo MA, Ribeiro JA (2000) A(2A) adenosine receptor facilitation of neuromuscular transmission: influence of stimulus paradigm on calcium mobilization. J Neurochem 74:2462–2469. https://doi.org/10.1046/j.1471-4159.2000.0742462.x
Giovannini F, Sher E, Webster R, Boot J, Lang B (2002) Calcium channel subtypes contributing to acetylcholine release from normal, 4-aminopyridine-treated and myasthenic syndrome auto-antibodies-affected neuromuscular junctions. Br J Pharmacol 136:1135–1145. https://doi.org/10.1038/sj.bjp.0704818
Silinsky EM (2004) Adenosine decreases both presynaptic calcium currents and neurotransmitter release at the mouse neuromuscular junction. J Physiol 558:389–401. https://doi.org/10.1113/jphysiol.2004.061457
Hong SJ, Chang CC (1995) Inhibition of acetylcholine release from mouse motor nerve by a P-type calcium channel blocker, omega-agatoxin IVA. J Physiol 482:283–290. https://doi.org/10.1113/jphysiol.1995.sp020517
Hirsh JK, Searl TJ, Silinsky EM (2002) Regulation by Rab3A of an endogenous modulator of neurotransmitter release at mouse motor nerve endings. J Physiol 545:337–343. https://doi.org/10.1113/jphysiol.2002.032516
Silinsky EM (2008) Selective disruption of the mammalian secretory apparatus enhances or eliminates calcium current modulation in nerve endings. Proc Natl Acad Sci USA 105:6427–6432. https://doi.org/10.1073/pnas.0708814105
Farah C, Michel LYM, Balligand JL (2018) Nitric oxide signalling in cardiovascular health and disease. Nat Rev Cardiol 15:292–316. https://doi.org/10.1038/nrcardio.2017.224
Cinelli MA, Do HT, Miley GP, Silverman RB (2020) Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med Res Rev 40:158–189. https://doi.org/10.1002/med.21599
Cunha RA, Correia-de-Sá P, Sebastião AM, Ribeiro JA (1996) Preferential activation of excitatory adenosine receptors at rat hippocampal and neuromuscular synapses by adenosine formed from released adenine nucleotides. Br J Pharmacol 119:253–260. https://doi.org/10.1111/j.1476-5381.1996.tb15979.x
Barros-Barbosa AR, Ferreirinha F, Oliveira Â, Mendes M, Lobo MG, Santos A, Rangel R, Pelletier J et al (2016) Adenosine A2A receptor and ecto-5’-nucleotidase/CD73 are upregulated in hippocampal astrocytes of human patients with mesial temporal lobe epilepsy (MTLE). Purinergic Signal 12:719–734. https://doi.org/10.1007/s11302-016-9535-2
Vieira C, Magalhães-Cardoso MT, Ferreirinha F, Silva I, Dias AS, Pelletier J, Sévigny J, Correia-de-Sá P (2014) Feed-forward inhibition of CD73 and upregulation of adenosine deaminase contribute to the loss of adenosine neuromodulation in postinflammatory ileitis. Mediat Inflamm 2014:254640. https://doi.org/10.1155/2014/254640
Augusto E, Matos M, Sévigny J, El-Tayeb A, Bynoe MS, Müller CE, Cunha RA, Chen J-F (2013) Ecto-5’-nucleotidase (CD73)-mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions. J Neurosci 33:11390–11399. https://doi.org/10.1523/JNEUROSCI.5817-12.2013
Duarte-Araújo M, Nascimento C, Timóteo MA, Magalhães-Cardoso MT, Correia-de-Sá P (2004) Dual effects of adenosine on acetylcholine release from myenteric motoneurons are mediated by junctional facilitatory A2A and extrajunctional inhibitory A1 receptors. Br J Pharmacol 141:925–934. https://doi.org/10.1038/sj.bjp.0705697
Correia-de-Sá P, Ribeiro JA (1994) Evidence that the presynaptic A2a-adenosine receptor of the rat motor nerve endings is positively coupled to adenylate cyclase. Naunyn-Schmiedeberg’s Arch Pharmacol 350:514–522. https://doi.org/10.1007/BF00173021
Correia-de-Sá P, Ribeiro JA (1993) Facilitation of [3H]-ACh release by forskolin depends on A2-adenosine receptor activation. Neurosci Lett 151:21–24. https://doi.org/10.1016/0304-3940(93)90035-J
Oliveira L, Correia-de-Sá P (2005) Protein kinase A and Cav1 (L-type) channels are common targets to facilitatory adenosine A2A and muscarinic M1 receptors on rat motoneurons. Neurosignals 14:262–272. https://doi.org/10.1159/000088642
Tarasova EO, Miteva AS, Gaidukov AE, Balezina OP (2015) The role of adenosine receptors and L-type calcium channels in the regulation of the mediator secretion in mouse motor synapses. Biochem (Mosc) Suppl Ser A Membr Cell Biol 9:318–328. https://doi.org/10.1134/S1990747815050141
Orenbuch A, Shalev L, Marra V, Sinai I, Lavy Y, Kahn J, Burden JJ, Staras K et al (2012) Synapsin selectively controls the mobility of resting pool vesicles at hippocampal terminals. J Neurosci 32:3969–3980. https://doi.org/10.1523/JNEUROSCI.5058-11.2012
Garcia N, Priego M, Hurtado E, Obis T, Santafe MM, Tomàs M, Lanuza MA, Tomàs J (2014) Adenosine A2B and A3 receptor location at the mouse neuromuscular junction. J Anat 225:109–117. https://doi.org/10.1111/joa.12188
Martinez-Pena Y, Valenzuela I, Pires-Oliveira M, Akaaboune M (2013) PKC and PKA regulate AChR dynamics at the neuromuscular junction of living mice. PLoS One 8:e81311. https://doi.org/10.1371/journal.pone.0081311
Bernareggi A, Ren E, Giniatullin A, Luin E, Sciancalepore M, Giniatullin R, Lorenzon P (2018) Adenosine promotes endplate nAChR channel activity in adult mouse skeletal muscle fibers via low affinity P1 Receptors. Neurosci 383:1–11. https://doi.org/10.1016/j.neuroscience.2018.04.044
Ferrer-Montiel AV, Montal MS, Díaz-Muñoz M, Montal M (1991) Agonist-independent activation of acetylcholine receptor channels by protein kinase A phosphorylation. Proc Natl Acad Sci 88:10213–10217. https://doi.org/10.1073/pnas.88.22.10213
Cinalli AR, Guarracino JF, Fernandez V, Roquel LI, Losavio AS (2013) Inosine induces presynaptic inhibition of acetylcholine release by activation of A3 adenosine receptors at the mouse neuromuscular junction. Br J Pharmacol 169:1810–1823. https://doi.org/10.1111/bph.12262
Herman-de-Sousa C, Pinheiro AR, Paramos-de-Carvalho D, Costa MA, Ferreirinha F, Magalhães-Cardoso MT, Ribeiro S, Pelletier J et al. (2020) Opposing effects of adenosine and inosine in human subcutaneous fibroblasts may be regulated by third party ADA cell providers. Cells 9:651. https://doi.org/10.3390/cells9030651
Pousinha PA, Correia AM, Sebastião AM, Ribeiro JA (2012) Neuromuscular transmission modulation by adenosine upon aging. Neurobiol Aging 33:2869–2880. https://doi.org/10.1016/j.neurobiolaging.2012.01.008
Nascimento F, Pousinha PA, Correia AM, Gomes R, Sebastião AM, Ribeiro JA (2014) Adenosine A2A receptors activation facilitates neuromuscular transmission in the pre-symptomatic phase of the SOD1(G93A) ALS mice, but not in the symptomatic phase. PLoS One 9. https://doi.org/10.1371/journal.pone.0104081
Gilhus NE, Tzartos S, Evoli A, Palace J, Burns TM, Verschuuren JJGM (2019) Myasthenia gravis. Nat Rev Dis Primers 5. https://doi.org/10.1038/s41572-019-0079-y
El-Tayeb A, Iqbal J, Behrenswerth A, Romio M, Schneider M, Zimmermann H, Schrader J, Müller CE (2009) Nucleoside-5′-monophosphates as prodrugs of adenosine A2A receptor agonists activated by ecto-5′-nucleotidase. J Med Chem 52:7669–7677. https://doi.org/10.1021/jm900538v
Li N, Mu L, Wang J, Zhang J, Xie X, Kong Q, Tang W, Yao X et al. (2012) Activation of the adenosine A2A receptor attenuates experimental autoimmune myasthenia gravis severity. Eur J Immunol 42:1140–1151. https://doi.org/10.1002/eji.201142088
Menegon A, Bonanomi D, Albertinazzi C, Lotti F, Ferrari G, Kao H-T, Benfenati F, Baldelli P et al. (2006) Protein kinase A-mediated synapsin I phosphorylation is a central modulator of Ca2+-dependent synaptic activity. J Neurosci 26:11670–11681. https://doi.org/10.1523/JNEUROSCI.3321-06.2006
Sebastião AM, Rei N, Ribeiro JA (2018) Amyotrophic lateral sclerosis (ALS) and adenosine receptors. Front Pharmacol 9:267. https://doi.org/10.3389/fphar.2018.00267
Rojas P, Ramírez AI, Fernández-Albarral JA, López-Cuenca I, Salobrar-García E, Cadena M, Elvira-Hurtado L, Salazar JJ et al. (2020) Amyotrophic lateral sclerosis: a neurodegenerative motor neuron disease with ocular involvement. Frontiers in Neuroscience 14. https://doi.org/10.3389/fnins.2020.566858
Vincenzi F, Corciulo C, Targa M, Casetta I, Gentile M, Granieri E, Borea PA, Popoli P et al. (2013) A2A adenosine receptors are up-regulated in lymphocytes from amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener 14:406–413. https://doi.org/10.3109/21678421.2013.793358
Slutsky I, Parnas H, Parnas I (1999) Presynaptic effects of muscarine on ACh release at the frog neuromuscular junction. J Physiol 514:769–782. https://doi.org/10.1111/j.1469-7793.1999.769ad.x
Minic J, Molgó J, Karlsson E, Krejci E (2002) Regulation of acetylcholine release by muscarinic receptors at the mouse neuromuscular junction depends on the activity of acetylcholinesterase. Eur J Neurosci 15:439–448. https://doi.org/10.1046/j.0953-816x.2001.01875.x
Correia-de-Sá P, Ribeiro JA (1994) Tonic adenosine A2A receptor activation modulates nicotinic autoreceptor function at the rat neuromuscular junction. Eur J Pharmacol 271:349–355. https://doi.org/10.1016/0014-2999(94)90793-5
Vizi ES, Somogyi GT (1989) Prejunctional modulation of acetylcholine release from the skeletal neuromuscular junction: link between positive (nicotinic)- and negative (muscarinic)-feedback modulation. Br J Pharmacol 97:65–70. https://doi.org/10.1111/j.1476-5381.1989.tb11924.x
Timóteo MA, Faria M, Correia-de-Sá P (2003) Endogenous adenosine prevents post-tetanic release facilitation mediated by α3β2 nicotinic autoreceptors. Eur J Pharmacol 464:115–125. https://doi.org/10.1016/S0014-2999(03)01374-8
Castellão-Santana LM, Yumi Abiko P, Ambiel CR, Peixoto AR, Noronha-Matos JB, Correia-de-Sá P, Alves-Do-Prado W (2019) Tetanic facilitation of neuromuscular transmission by adenosine A2A and muscarinic M1 receptors is dependent on the uptake of choline via high-affinity transporters. Pharmacol 103:38–49. https://doi.org/10.1159/000494058
Oliveira L, Correia-de-Sá P (2006) Dissociation between M1-facilitation of acetylcholine release and crosstalk with A2A- and M2-receptors on rat motoneurons. Signal Transduct 6:19–31. https://doi.org/10.1002/sita.200500057
Arenson MS, Evans SC (2001) Activation of protein kinase C increases acetylcholine release from frog motor nerves by a direct action on L-type Ca2+ channels and apparently not by depolarisation of the terminal. Neurosci 104:1157–1164. https://doi.org/10.1016/S0306-4522(01)00114-2
Shakirzyanova AV, Bukharaeva EA, Nikolsky EE, Giniatullin RA (2006) Negative cross-talk between presynaptic adenosine and acetylcholine receptors. Eur J Neurosci 24:105–115. https://doi.org/10.1111/j.1460-9568.2006.04884.x
Nishizaki T, Sumikawa K (1998) Effects of PKC and PKA phosphorylation on desensitization of nicotinic acetylcholine receptors. Brain Res 812:242–245. https://doi.org/10.1016/S0006-8993(98)00836-1
Caulfield MP, Birdsall NJ (1998) International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev 50:279–290
Gaydukov A, Bogacheva P, Tarasova E, Molchanova A, Miteva A, Pravdivceva E, Balezina O (2019) Regulation of acetylcholine quantal release by coupled thrombin/BDNF signaling in mouse motor synapses. Cells 8:762. https://doi.org/10.3390/cells8070762
Pousinha PA, Diógenes MJ, Ribeiro JA, Sebastião AM (2006) Triggering of BDNF facilitatory action on neuromuscular transmission by adenosine A2A receptors. Neurosci Lett 404:143–147. https://doi.org/10.1016/j.neulet.2006.05.036
Correia-de-Sá P, Timóteo MA, Ribeiro JA (2001) Synergism between A2A-adenosine receptor activation and vasoactive intestinal peptide to facilitate [3H]-acetylcholine release from the rat motor nerve terminals. Neurosci Lett 309:101–104. https://doi.org/10.1016/S0304-3940(01)02030-4
Takami K, Kawai Y, Uchida S, Tohyama M, Shiotani Y, Yoshida H, Emson PC, Girgis S et al. (1985) Effect of calcitonin gene-related peptide on contraction of striated muscle in the mouse. Neurosci Lett 60:227–230. https://doi.org/10.1016/0304-3940(85)90248-4
Csillik B, Knyihár-Csillik E, Kreutzberg GW, Tajti L, Kereszturi A, Kovács T (1992) Calcitonin gene-related peptide is released from cholinergic synapses. Ann N Y Acad Sci 657:466–468. https://doi.org/10.1111/j.1749-6632.1992.tb22802.x
Mulholland MW, Jaffer S (1990) Stimulation of acetylcholine release in myenteric plexus by calcitonin gene-related peptide. Am J Physiol Gastrointest Liver Physiol 259:G934–G939. https://doi.org/10.1152/ajpgi.1990.259.6.G934
Takami K, Hashimoto K, Uchida S, Tohyama M, Yoshida H (1986) Effect of calcitonin gene-related peptide on the cyclic AMP level of isolated mouse diaphragm. Jpn J Pharmacol 42:345–350. https://doi.org/10.1254/jjp.42.345
Battaglia G, Norman AB, Hess EJ, Creese I (1986) Forskolin potentiates the stimulation of rat striatal adenylate cyclase mediated by D-1 dopamine receptors, guanine nucleotides, and sodium fluoride. J Neurochem 46:1180–1185. https://doi.org/10.1111/j.1471-4159.1986.tb00635.x
Azanza MJ, Garin P (1986) The autonomic innervation of the rat diaphragm. Gen Pharmacol 17:109–112. https://doi.org/10.1016/0306-3623(86)90021-2
Gold MR (1982) The effects of vasoactive intestinal peptide on neuromuscular transmission in the frog. J Physiol 327:325–335. https://doi.org/10.1113/jphysiol.1982.sp014234
Santafé MM, Garcia N, Tomàs M, Obis T, Lanuza MA, Besalduch N, Tomàs J (2014) The interaction between tropomyosin-related kinase B receptors and serine kinases modulates acetylcholine release in adult neuromuscular junctions. Neurosci Lett 561:171–175. https://doi.org/10.1016/j.neulet.2013.12.073
Garcia N, Tomàs M, Santafé MM, Besalduch N, Lanuza MA, Tomàs J (2010) The interaction between tropomyosin-related kinase B receptors and presynaptic muscarinic receptors modulates transmitter release in adult rodent motor nerve terminals. J Neurosci 30:16514–16522. https://doi.org/10.1523/JNEUROSCI.2676-10.2010
Herrera AA, Qiang H, Ko CP (2000) The role of perisynaptic Schwann cells in development of neuromuscular junctions in the frog (Xenopus laevis). J Neurobiol 45:237–254. https://doi.org/10.1002/1097-4695(200012)45:4%3c237::aid-neu5%3e3.0.co;2-j
Reddy LV, Koirala S, Sugiura Y, Herrera AA, Ko CP (2003) Glial cells maintain synaptic structure and function and promote development of the neuromuscular junction in vivo. Neuron 40:563–580. https://doi.org/10.1016/s0896-6273(03)00682-2
Koirala S, Qiang H, Ko CP (2000) Reciprocal interactions between perisynaptic Schwann cells and regenerating nerve terminals at the frog neuromuscular junction. J Neurobiol 44:343–360. https://doi.org/10.1002/1097-4695(20000905)44:3%3c343::Aid-neu5%3e3.0.Co;2-o
Son YJ, Thompson WJ (1995) Nerve sprouting in muscle is induced and guided by processes extended by schwann cells. Neuron 14:133–141. https://doi.org/10.1016/0896-6273(95)90247-3
Feng Z, Ko CP (2007) Neuronal glia interactions at the vertebrate neuromuscular junction. Curr Opin Pharm 7:316–324. https://doi.org/10.1016/j.coph.2006.12.003
Araque A, Parpura V, Sanzgiri RP, Haydon PG (1999) Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22:208–215. https://doi.org/10.1016/S0166-2236(98)01349-6
Gaydukov AE, Bogacheva PO, Tarasova EO, Balezina OP (2014) The mechanism of choline-mediated inhibition of acetylcholine release in mouse motor synapses. Acta Naturae 6:110–115
Fischer U, Reinhardt S, Albuquerque EX, Maelicke A (1999) Expression of functional α7 nicotinic acetylcholine receptor during mammalian muscle development and denervation. Eur J Neurosci 11:2856–2864. https://doi.org/10.1046/j.1460-9568.1999.00703.x
Jones SW, Salpeter MM (1983) Absence of [125I] alpha-bungarotoxin binding to motor nerve terminals of frog, lizard and mouse muscle. J Neurosci 3:326–331. https://doi.org/10.1523/JNEUROSCI.03-02-00326.1983
Albuquerque EX, Pereira EFR, Alkondon M, Rogers SW (2009) Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89:73–120. https://doi.org/10.1152/physrev.00015.2008
Vieira C, Ferreirinha F, Magalhães-Cardoso MT, Silva I, Marques P, Correia-de-Sá P (2017) Post-inflammatory ileitis induces non-neuronal purinergic signaling adjustments of cholinergic neurotransmission in the myenteric plexus. Front Pharmacol 8:811. https://doi.org/10.3389/fphar.2017.00811
MacEachern SJ, Patel BA, Keenan CM, Dicay M, Chapman K, McCafferty D-M, Savidge TC, Beck PL et al. (2015) Inhibiting inducible nitric oxide synthase in enteric glia restores electrogenic ion transport in mice with colitis. Gastroenterol 149:445-455.e443. https://doi.org/10.1053/j.gastro.2015.04.007
Sinclair CJD, Shepel PN, Geiger JD, Parkinson FE (2000) Stimulation of nucleoside efflux and inhibition of adenosine kinase by A1 adenosine receptor activation. Biochem Pharmacol 59:477–483. https://doi.org/10.1016/S0006-2952(99)00350-0
Reyes G, Nivillac NMI, Karim MZ, Desouza L, Siu KWM, Coe IR (2011) The equilibrative nucleoside transporter (ENT1) can be phosphorylated at multiple sites by PKC and PKA. Mol Membr Biol 28:412–426. https://doi.org/10.3109/09687688.2011.604861
Fernández-Calotti P, Galmarini CM, Cañones C, Gamberale R, Saénz D, Avalos JS, Chianelli M, Rosenstein R et al (2008) Modulation of the human equilibrative nucleoside transporter1 (hENT1) activity by IL-4 and PMA in B cells from chronic lymphocytic leukemia. Biochem Pharmacol 75:857–865. https://doi.org/10.1016/j.bcp.2007.10.017
Coe I, Zhang Y, McKenzie T, Naydenova Z (2002) PKC regulation of the human equilibrative nucleoside transporter, hENT1. FEBS Lett 517:201–205. https://doi.org/10.1016/S0014-5793(02)02622-4
Hughes SJ, Cravetchi X, Vilas G, Hammond JR (2015) Adenosine A1 receptor activation modulates human equilibrative nucleoside transporter 1 (hENT1) activity via PKC-mediated phosphorylation of serine-281. Cell Signal 27:1008–1018. https://doi.org/10.1016/j.cellsig.2015.02.023
Gaydukov AE, Balezina OP (2017) CaMKII is involved in the choline-induced downregulation of acetylcholine release in mouse motor synapses. Acta Naturae 9:110–113
Kent NB, Liang SS, Phillips S, Smith NA, Khandkar C, Eikermann M, Stewart PA (2018) Therapeutic doses of neostigmine, depolarising neuromuscular blockade and muscle weakness in awake volunteers: a double-blind, placebo-controlled, randomised volunteer study. Anaesthesia 73:1079–1089. https://doi.org/10.1111/anae.14386
de Paula RE, Antonio MB, Ambiel CR, Correia-de-Sá P, Alves-Do-Prado W (2014) Paradoxical neostigmine-induced TOFfade: on the role of presynaptic cholinergic and adenosine receptors. Eur J Pharmacol 723:389–396. https://doi.org/10.1016/j.ejphar.2013.11.001
Alves-do-Prado W, Corrado AP, Prado WA (1989) Train-of-four as an index of neuromuscular block in cats: changes induced by atropine. Braz J Med Biol Res 22:749–755
Pereira MW, Bornia ECS, Correia-de-Sá P, Alves-Do-Prado W (2011) Presynaptic muscarinic and adenosine receptors are involved in 2 Hz-induced train-of-four fade caused by antinicotinic neuromuscular relaxants in the rat. Clin Exp Pharmacol Physiol 38:764–770. https://doi.org/10.1111/j.1440-1681.2011.05588.x
Bornia ECS, Correia-de-Sá P, Alves-Do-Prado W (2011) Presynaptic facilitatory adenosine A2A receptors mediate fade induced by neuromuscular relaxants that exhibit anticholinesterase activity. Clin Exp Pharmacol Physiol 38:164–169. https://doi.org/10.1111/j.1440-1681.2011.05476.x
Bornia ECS, Bando É, Machinski M, Pereira MW, Alves-Do-Prado W (2009) Presynaptic M1, M2, and A1 receptors play roles in tetanic fade induced by pancuronium or cisatracurium. J Anesth 23:513. https://doi.org/10.1007/s00540-009-0790-z
Reist NE, Smith SJ (1992) Neurally evoked calcium transients in terminal Schwann cells at the neuromuscular junction. Proc Natl Acad Sci USA 89:7625–7629. https://doi.org/10.1073/pnas.89.16.7625
Robitaille R, Jahromi BS, Charlton MP (1997) Muscarinic Ca2+ responses resistant to muscarinic antagonists at perisynaptic Schwann cells of the frog neuromuscular junction. J Physiol 504:337–347. https://doi.org/10.1111/j.1469-7793.1997.337be.x
Wright MC, Potluri S, Wang X, Dentcheva E, Gautam D, Tessler A, Wess J, Rich MM et al. (2009) Distinct muscarinic acetylcholine receptor subtypes contribute to stability and growth, but not compensatory plasticity, of neuromuscular synapses. J Neurosci 29:14942–14955. https://doi.org/10.1523/jneurosci.2276-09.2009
Guarracino JF, Cinalli AR, Veggetti MI, Losavio AS (2018) Endogenous purines modulate K(+) -evoked ACh secretion at the mouse neuromuscular junction. J Neurosci Res 96:1066–1079. https://doi.org/10.1002/jnr.24223
Palma AG, Muchnik S, Losavio AS (2011) Excitatory effect of the A2A adenosine receptor agonist CGS-21680 on spontaneous and K+-evoked acetylcholine release at the mouse neuromuscular junction. Neurosci 172:164–176. https://doi.org/10.1016/j.neuroscience.2010.10.015
Funding
Open access funding provided by FCT|FCCN (b-on). The work was partially supported by the Fundação para a Ciência e a Tecnologia (FCT, FEDER funding, projects UIDB/04308/2020 and UIDP/04308/2020).
Author information
Authors and Affiliations
Contributions
Carlos Sousa-Soares: conceptualisation, writing—original draft, and writing—review and editing. José Bernardo Noronha-Matos: conceptualisation, writing—original draft, writing—review and editing, and supervision. Paulo Correia-de-Sá: conceptualisation, writing—original draft, writing—review and editing, supervision, and fundraising. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sousa-Soares, C., Noronha-Matos, J.B. & Correia-de-Sá, P. Purinergic Tuning of the Tripartite Neuromuscular Synapse. Mol Neurobiol 60, 4084–4104 (2023). https://doi.org/10.1007/s12035-023-03317-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03317-8