
Abstract
Purines have physiologically important functions throughout the nervous system. In both the central (CNS) and peripheral nervous systems (PNS), purines in the form of adenosine triphosphate and adenosine can play a number of roles in neuronal activation and inhibition. In addition, purines are known to be important for glial cell signaling in both the CNS and PNS. In the PNS, the neuromuscular junction (NMJ) is an excellent model for studying simple synaptic interactions. It is well suited to investigations of neuron-glia interactions because synaptic properties are well defined and perisynaptic Schwann cells (PSCs), glial cells at the NMJ, dynamically interact with the pre- and postsynaptic elements. At the NMJ, purines are critical for presynaptic modulation but also for neuron-glia interactions. Purines signal to PSCs through metabotropic and ionotropic receptors and activation of these receptors can have both modulatory and activating functions. This review will discuss recent developments in our understanding of purinergic modulation of the NMJ with an emphasis on the involvement of purines in neuron-glia interactions at this synapse.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Adenosine triphosphate (ATP) was found to be coreleased with acetylcholine (ACh) at neuromuscular junctions (NMJs) more than 30 years ago [37]. It is interesting to note that purines can also be released from muscle fibers [36, 40] and Schwann cells [24]. Since then, the functions of purines at NMJs were examined in a number of different preparations, revealing diverse functions. A number of different receptors are present on all three elements of the NMJ: the presynaptic nerve terminal, postsynaptic muscle, and perisynaptic Schwann cells (PSCs). These receptors include P1 adenosine receptors on presynaptic terminals [3, 10, 38] and PSCs [31, 35] and P2 ATP receptors on PSCs [31, 35], nerve terminals [20, 27], and muscle fibers [7, 8, 36]. Purinergic function in the peripheral nervous system (PNS) and at the NMJ indicates roles during development [13] at mature NMJs [10, 30], in PNS myelination [42], and neuron-glia interactions [31, 35]. This review will discuss the role of purines in the modulation of NMJ function giving special focus on the involvement of purines in neuron-glia interactions. Some elements of this review were published as part of a Novartis Foundation symposium [45].
Purine receptors and presynaptic modulation
Purinergic modulation of NMJ function can occur in a bidirectional manner as either potentiation or depression of transmitter release [10]. The outcome of the purinergic modulation depends on a number of factors. For instance, the function of purinergic signaling changes with the developmental stage of the NMJ where during the development of the tadpole NMJ, purines seem to potentiate neurotransmitter release [13], while in the adult frog, the effect is opposite [17]. Dual effects of purines were also reported at adult NMJs. For instance, using a mammalian preparation, Correia-de-Sa et al. [10] demonstrated purine-mediated potentiation and depression of transmitter release depending on the frequency and pattern of stimulation. In this study, activation of P1 adenosine receptors of the A1 or A2A subtypes were responsible for inducing the depressive and potentiating effects, respectively. This model suggests that the A1 receptor effect dominates during low-frequency activity, while higher levels of synaptic adenosine are required for the activation of A2A receptors. These higher levels of adenosine are achieved during short-duration, high-frequency bursts of activity that cause the release of larger amounts of ATP, which is then converted into adenosine by ectonucleotidases [47]. However, it is suggested that during prolonged high-frequency activation, release of ATP leads to the inhibition of ectonucleotidases such that the production of adenosine is reduced [11]. Correia-de-Sa et al. [10, 29] suggested that during high-frequency bursts of nerve activity, the accumulation of synaptic adenosine is greater and the activation of adenosine receptors shifts such that transmitter release is potentiated through the activation of A2A receptors. These phenomena are thought to function to preserve transmitter during times of prolonged release and to potentiate release during times where run-down has already occurred to ensure muscle activation.
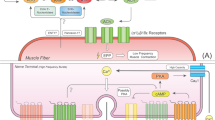
Other mechanisms of modulation can occur through direct activation of ATP receptors [18, 17, 20, 27]. These actions were also shown to be potentiating or depressing depending on the receptors involved. When metabotropic P2 receptors are activated, they have an inhibitory effect similar to that of adenosine [17]. However, Moores et al. [27] found evidence for presynaptic ionotropic P2X7 receptors that potentiate transmitter release due to an increase in nonselective cation conductance. Therefore, it appears that multiple types of purinergic receptors may have similar actions and can operate in parallel to modulate transmitter release at the NMJ (Fig. 1).

Schematic representation of the purinergic modulation of synaptic functions at the NMJ. Diagram depicting the presynaptic nerve terminal (green), PSC (blue), and postsynaptic muscle fiber (yellow). Sources of purines include presynaptic corelease with ACh, release from muscle fibers, and possibly from PSCs. A first site of purine modulation is the presynaptic terminal where they regulate transmitter release. Both A1 and P2Y receptors decrease transmitter release where P2Y receptors are thought to inhibit calcium channels through production of AA [20]. In the mouse A1, receptors were shown to cause inhibition of calcium channels through the interaction of \(G_{{\beta \mathord{\left/ {\vphantom {\beta Y}} \right. \kern-\nulldelimiterspace} Y}}\) with the SNARE complex protein, syntaxin, which also interacts with the calcium channels [39]. Transmitter release can also be upregulated through activation of P2X and A2A receptors. P2X, ionotropic receptors increase nonspecific cation conductance leading to Ca2+ entry while A2A receptors are involved in the activation of L-type calcium channels [29]. Further inhibitory action can be derived from the production of ROS such as H2O2. The location for production of ROS is not isolated to any specific compartments, however, muscle fibers are a likely source. Their production is linked to the activation of P2Y receptors [18]. Other short-term modulation of the NMJ comes from purine-mediated signaling with PSCs. Purines are at least partially involved in feedback modulation of transmitter release by PSCs [5, 32]. Purines are also involved in long-term regulation of the NMJ, in part, through P2Y1-mediated activation of transcription of nicotinic ACh receptor (nAChR) subunits and acetylcholinesterase (AChE) [6, 7]
Mechanisms of presynaptic modulation
The mechanisms of purinergic modulation vary depending on the receptors involved and, to some extent, on the species being investigated. Purinergic depression of transmitter release in amphibians seems to modulate the mechanisms of transmitter release without altering presynaptic calcium [22, 30, 34]. However, recently, Grishin et al. [20] demonstrated a decrease in presynaptic calcium currents at the frog NMJ mediated by P2Y activation. This effect was mimicked with the addition of arachidonic acid (AA), suggesting its involvement as a second messenger. It is unclear why the difference in results found in this new study occurred. However, Grishin investigated P2Y receptors while Robitaille et al. [34] investigated actions of adenosine as Huang et al. [22] and Redman and Silinsky [30] did. It is interesting to note that it was observed that presynaptic calcium currents might be altered by purinergic signaling in other NMJs. This type of modulation was well established at mammalian NMJs [21, 38] where modulation of calcium currents is thought to occur through hydrolysis of ATP to adenosine, which then activates adenosine receptors [21]. Once activated, adenosine A1 receptors seem to inhibit P/Q-type calcium channels (Fig. 1) that in mammals are responsible for calcium entry during transmitter release [38]. It is interesting to note that the inhibition of calcium currents is not the only site of purinergic modulation. Recently, Silinsky [39] demonstrated that disruption of the presynaptic release machinery, specifically the soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) complex, decreased the effect of adenosine signaling on calcium currents. This occurs through interactions with the synaptic protein syntaxin, which has binding domains for P/Q calcium channels and the \(G_{{{\text{ $\beta $ }} \mathord{\left/ {\vphantom {{\text{ $\beta $ }} {\text{ $\gamma $ }}}} \right. \kern-\nulldelimiterspace} {\text{ $\gamma $ }}}}\) subunit. The \(G_{{{\text{ $\beta $ }} \mathord{\left/ {\vphantom {{\text{ $\beta $ }} {\text{ $\gamma $ }}}} \right. \kern-\nulldelimiterspace} {\text{ $\gamma $ }}}}\) interaction with syntaxin decreases P/Q channel activity and ultimately inhibits transmitter release (Fig. 1) [39]. These results are a clear demonstration of G protein modulation of presynaptic release machinery. It would be interesting to know whether similar interactions between purinergic receptors and presynaptic machinery also exist in amphibian NMJs.
Another effect of purinergic signaling on calcium channels was recently described by Oliveira et al. [29] where they investigated a potentiating mechanism mediated by A2A receptors. In this model, during high-frequency continuous nerve activity, the inhibitory A1 effects were dominant. However, during high-frequency bursts of activity, levels of ACh release are increased through the activation of A2A receptors, which leads to the recruitment of L-type calcium channels (Fig. 1). They suggested that this modulation might be an endogenous mechanism for ensuring release during repetitive burst activity.
The purinergic modulation described above implies a direct regulation mediated by presynaptic autoreceptors for ATP and adenosine. However, there is evidence that ATP may act also via a diffusible retrograde messenger. Indeed, Giniatullin and colleagues demonstrated the involvement of reactive oxygen species (ROS) in purine-mediated inhibition. The production of ROS, such as H2O2, seemed to be dependent on ATP signaling but not on adenosine (Fig. 1). Fluorescent staining for ROS with carboxy-2′,7′-dichlorodihydrofluorescein revealed ATP-mediated production of ROS in muscle fibers; however, general staining was observed in other synaptic components. The authors suggested that PSCs could be involved in the production of ROS although it was not tested directly. Regardless of the source, the production of ROS results in feedback modulation of the NMJ such that ACh release is decreased.
Purinergic signaling can alter calcium currents, calcium-independent processes, and recruit other signaling cascades. Overall, this leads to a fine balance of NMJ signaling such that sufficient presynaptic release can be maintained at all times. The exact mechanisms for many of these processes have yet to be resolved; however, it appears that redundant mechanisms exist for both potentiation and inhibition of release.
Purinergic signaling to Schwann cells
Purines appear to be important factors for signaling to glia in both the central nervous system (CNS) and PNS. We will only discuss purine signaling to PNS glia (Schwann cells) in this review. In the PNS, there are three types of Schwann cells: the myelinating and nonmyelinating Schwann cells associated with axons and the PSCs located at NMJs. PSCs perform similar functions to CNS astrocytes where they participate in synapse–glia signaling, modulate synaptic function, and are in all sense of the term, active synaptic partners [2]. Purinergic signaling to Schwann cells was investigated in culture and in situ revealing roles for purine signaling in both axonal Schwann cells and PSCs.
Myelinating Schwann cells were shown to express both ionotropic [9, 19] and metabotropic [1, 25] ATP receptors. Expression of these receptors appears to be dependent on neuronal interaction [26], demonstrating the synergistic relationship between neurons and glia in roles such as structural support, maintenance, and stability differentiation and development. Purinergic signaling between neurons and Schwann cells was also shown to be important for regulating the developmental progression of myelination [42, 43]. These data demonstrated that signaling through P2 receptors could arrest developing Schwann cells in a premyelinating state through the activation of mitogen-activated protein kinase (MAPK) [42]. After this, it was shown that ATP and adenosine could have different effects on Schwann cell development [43]. Activation of A2A receptors resulted in the inhibition of Schwann cell proliferation through the activation of the extracellular signal-regulated kinase/MAPK pathway. It appears that this cascade is regulated separately from the P2 initiated cascade, which leads to inhibition of differentiation rather than proliferation [43]. These results demonstrate the complex nature of purinergic signaling where different outcomes can be mediated through similar cascades. In addition, it indicates a role of purinergic signaling in long-term regulation of Schwann cells. It is interesting to note that Schwann cells were also shown to release ATP through mechanisms requiring activation of P2Y2 receptors [24]. Release of ATP from Schwann cells is suggested to provide excitatory feedback to axons, however, this has yet to be confirmed in situ. These studies indicate the importance of purinergic signaling for Schwann cell function and proper myelination during development.
Purinergic signaling for PSCs has also revealed multiple receptor pathways and signaling cascades [31, 35]. Purinergic signaling to PSCs was first described in 1992 and was observed in acutely isolated muscles using calcium imaging [23]. Since the initial description of ATP signaling to PSCs, information was provided regarding similar signaling pathways in amphibian [31] and mouse PSCs [35]. It is important to note that it was shown that multiple receptors are expressed at amphibian and mammalian NMJs, leading to the possibility of complex regulation of PSC function and neuron-glia interactions.
At the frog NMJ, PSCs express both ATP and adenosine receptors. The expression of ATP receptors appears to be heterogeneous with the expression of both P2X and P2Y receptors [31]. The presence of both ionotropic and metabotropic receptors likely allows for differential signaling through calcium entry. Calcium responses to purines are efficacious with almost 100% of PSCs responding to exogenous local application. This is also true for the mammalian NMJ [35]. In the mouse, calcium rises can also be elicited after the application of ATP or adenosine. It also appears that both P2X and P2Y receptors are present. The exact subtype of these receptors have not been clearly identified although the pharmacology suggests that they might be of the P2X1 or P2X5 group while the P2Y receptors are probably of group 1. Activation of P2X receptors trigger the entry of Ca2+ from the external milieu while P2Y receptors elicit intracellular rises in calcium, most likely through inositol triphosphate (IP3)-mediated stores. This is supported by recent work in our laboratory at the mouse NMJ. This was shown through the inhibition of ATP signaling in the presence of the phospholipase C inhibitor U73122, a step upstream of IP3 production (Todd and Robitaille, unpublished results). Regarding the adenosine receptors, they appear to be of the A1 type while there is no evidence for the presence of A2A receptors [31]. Although these results confirm the presence and action of these receptors, they do not address the involvement of purines in PSC signaling during evoked activity.
At the frog NMJ, Robitaille [31] determined the contribution of purines to PSC calcium responses. This was done through nerve stimulation in the presence and absence of the general P2 receptor antagonist suramin. In the presence of suramin, PSC calcium responses were reduced by about 50%. This suggests that at this synapse, purines provide about half of the stimulus for the observed response. The other part of the response is due to the activation of muscarinic receptors that are also expressed on the PSCs [33]. This suggests parallel pathways for PSC activation that may have different roles in short- or long-term signaling. In his study, Robitaille did not find any evidence for a direct activation of PSCs by adenosine during sustained synaptic activity. The contribution of purines to PSC responses observed during nerve activity at the mouse NMJ is not very clear. Also, it seems that the purinergic activation may not be driven by P2 receptors but rather by adenosine P1 receptors [35]. At the mouse NMJ, however, muscarinic signaling to PSCs also appears to be important for the activation of PSCs.
The presence of purinergic and cholinergic signaling systems on PSCs suggests the ability to differentially modulate PSC function. Although the contribution of each receptor family to PSC responses during nerve activity was not investigated in the mouse, it is thought to be similar to the amphibian PSCs. It is interesting to note that purinergic and cholinergic signaling systems were shown to interact at the mammalian NMJ nerve terminal [28] where adenosine, acting through A1 and A2A receptors, can decrease presynaptic M1 and M2 receptor activity. This was addressed in both the frog and mouse NMJs, but no evidence for interaction between purinergic and muscarinic systems was observed in PSCs [35]. Although these two pathways do not seem to interact, other mechanisms for modulation of PSC signaling do exits.
Modulation of PSC signaling
Because purinergic signaling is clearly the most efficacious calcium-dependent pathway on PSCs, it stands to reason that purinergic modulatory pathways would also exist. In particular, modulation by peptides and diffusible transmitters appears to be important [4, 44].
Exogenous application of substance P (SP) at the frog NMJ induced calcium rises in glial cells [4]. Induction of PSC calcium responses by SP occurred through the activation of NK-1 receptors. It is interesting to note that this signaling appears to be involved in modulation of PSC responses during nerve activity. During repetitive evoked activity, PSC responses are gradually reduced. However, when Bourque and Robitaille [4] blocked NK-1 activation, the rundown of PSC responses was prevented. This reveals a peptidergic modulation of PSC signaling. This endogenous modulation was, in part, due to SP effects on the purinergic signaling as suggested by the observation that PSC responses to exogenously applied ATP were reduced by half in the presence of bath-applied SP. This is one pathway regulating purinergic signaling in PSCs and ultimately of neuron-glia communication.
Another pathway involved in modulating purinergic signaling to PSCs is the production of nitric oxide (NO). Synthesis of NO was observed in both PSCs and muscle fibers at the frog NMJ [12]. This was determined through localization of NO synthase to these compartments. Production of NO occurs both tonically, providing a basal level of NO, and in response to activity where it is involved in mediating synaptic depression at the NMJ [44]. It is interesting to note that during high levels of nerve activity NO may signal to PSCs, resulting in a reduction of purinergic calcium responses as suggested by the observation that exogenous application of NO donors resulted in a reduction of ATP-induced calcium responses [12].
Another important effect of NO at the NMJ is its ability to modulate adenosine-mediated depression of synaptic release because NO chelation before adenosine-mediated depression attenuated the adenosine effect [44]. The role of NO in the modulation of NMJ activity could occur either directly or indirectly. Indeed, it is possible that NO modulates presynaptic activity through interactions with presynaptic adenosine receptors. However, it is also possible that these effects are occurring indirectly through modulation of PSC signaling that, in turn, differentially signals to the presynaptic terminal.
Changes in purinergic signaling could also be altered during development or denervation [33, 41]. It is possible that purinergic signaling could alter the expression of a number of genes in PSCs as seen in myelinating Schwann cells [42]. In addition, similar long-term roles for purines were described in muscle fibers where purinergic signaling is involved in regulating acetylcholinesterase and ACh receptor expression (Fig. 1) [7, 46]. However, purines were found to have no effect on the inhibition of the gene expression and protein synthesis of glial fibrillary acidic protein (GFAP) in PSCs [15]. Indeed, this inhibition is due to cholinergic signaling to PSCs via muscarinic receptors [16]. The expression of cytoskeletal components such as GFAP is required for the elaboration of new PSC processes that are formed during synaptic weakening or denervation. Therefore, decrease in cholinergic signaling to PSCs should facilitate this process.
Furthermore, the close interaction between the three NMJ elements is important for signaling the overall health and state of the NMJ. In the event of perturbation such as denervation, PSC receptor expression changes, indicating the sensitivity of PSCs to NMJ health [33]. It is interesting to note that changes in receptor expression at PSCs of the frog NMJ were observed in the cholinergic pathway [33], but there is no evidence for changes in purinergic signaling to PSCs (R. Robitaille, unpublished observation). This could provide some mechanism by which signaling at NMJs can be maintained to facilitate reinnervation. Indeed, the maintenance of purinergic signaling on PSCs makes intuitive sense. ATP released from muscle fibers and PSCs [36, 40] may allow for the maintenance of communication through purinergic signaling between PSCs and muscles of the NMJ during denervation. This may contribute to the proper environment for reinnervation by presynaptic terminals.
Purines have multiple pathways on PSCs to induce diverse signals. To date, it is known that these pathways are modulated by both NO and SP. Although only short-term roles in NMJ function were described for purines, it remains possible that they mediate changes in gene expression that have not yet been discovered. In addition, it is hypothesized that purines are crucial for the maintenance of signaling between denervated muscle and PSCs that may in part facilitate reinnervation.
Purine-mediated synapse–glia interactions
Up to this point, only the mechanisms for purinergic signaling and modulation at the NMJ were discussed. An additional aspect of purinergic signaling, however, is their involvement in synaptic function, in particular, their role in PSC-mediated synaptic modulation. This information is derived from experiments employing specific perturbation of PSC function.
The involvement of purine receptors was investigated through the manipulation of the various G proteins recruited by cholinergic and purinergic signaling [31, 33]. These data indicate that muscarinic receptors on PSCs signal through G proteins that are insensitive to pertussis toxin (PTX), likely of the Gαi/o type [33]. On the other hand, purinergic receptors on PSCs are probably linked to Gαq type G proteins, which are sensitive to PTX [31]. The downstream signaling cascades recruited would involve purine-mediated production of IP3, while muscarinic signaling probably regulates levels of cyclic adenosine monophosphate. The involvement of these two pathways was investigated in terms of PSC-mediated synaptic depression. Robitaille [32] found that both PTX-sensitive and insensitive G proteins were involved in PSC-mediated modulation of synaptic transmission. Further work on this topic has revealed a specific role for IP3 production in PSCs [5]. These results revealed that injection of IP3 into PSCs potentiated synaptic transmission. These observations suggest the involvement of PSC purinergic signaling based on the inferred pathway recruited by these receptors.
It is important to note that the combined results where PSCs were specifically perturbed, provide evidence for bidirectional modulation of synaptic transmission by PSCs. As shown by Robitaille [32], PSCs can decrease high-frequency synaptic transmission. This seems to occur partially through muscarinic signaling because a PTX-insensitive pathway is involved. The follow-up study indicates PSC-mediated potentiation of transmitter release through the IP3 pathway [5]. PSCs, therefore, can dynamically respond to the ongoing changes in NMJ activity and appropriately respond with reciprocal signals that alter transmitter release. The existence of different signaling cascades recruited to mediate facilitation and depression is consistent with the synaptic properties of the NMJ because synaptic depression and potentiation both occur at this synapse and can be recruited at different times. Furthermore, this probably allows PSCs to adapt to the synaptic environment by favoring either depression or potentiation according to the state and health of the synapse.
Conclusion
Purinergic signaling at the NMJ has definite roles in synaptic modulation. In developmental models, ATP appears to potentiate synaptic transmission [13] and at mature synapses both potentiation [10] and depression [17] were described, the decisive factor being the type of receptor activated. Furthermore, the modulatory effects of purines can alter both quantal and nonquantal release [14]. Not only does the release of purines provide autofeedback to the presynaptic terminal, but purines also maintain communication with PSCs by inducing Ca2+ elevation in these cells [23, 31]. Purines also signal to muscle fibers through P2 receptors to induce short-term modulation [18] and long-term changes [6, 46]. Activation of purine receptors can have bidirectional effects on synaptic release and are quite important for neuron-glia communication and synaptic modulation. Finally, these factors could play a pivotal role during development and after injury to maintain signaling between muscle and glial components.
References
Ansselin AD, Davey DF, Allen DG (1997) Extracellular ATP increases intracellular calcium in cultured adult Schwann cells. Neuroscience 76:947–955
Auld DS, Robitaille R (2003) Perisynaptic Schwann cells at the neuromuscular junction: nerve- and activity-dependent contributions to synaptic efficacy, plasticity, and reinnervation. Neuroscientist 9:144–157
Baxter RL, Vega-Riveroll LJ, Deuchars J, Parson SH (2005) A2A adenosine receptors are located on presynaptic motor nerve terminals in the mouse. Synapse 57:229–234
Bourque MJ, Robitaille R (1998) Endogenous peptidergic modulation of perisynaptic Schwann cells at the frog neuromuscular junction. J Physiol 512:197–209
Castonguay A, Robitaille R (2001) Differential regulation of transmitter release by presynaptic and glial Ca2+ internal stores at the neuromuscular synapse. J Neurosci 21:1911–1922
Choi RC, Man ML, Ling KK, Ip NY, Simon J, Barnard EA, Tsim KW (2001) Expression of the P2Y1 nucleotide receptor in chick muscle: its functional role in the regulation of acetylcholinesterase and acetylcholine receptor. J Neurosci 21:9224–9234
Choi RC, Siow NL, Cheng AW, Ling KK, Tung EK, Simon J, Barnard EA, Tsim KW (2003) ATP acts via P2Y1 receptors to stimulate acetylcholinesterase and acetylcholine receptor expression: transduction and transcription control. J Neurosci 23:4445–4456
Collet C, Strube C, Csernoch L, Mallouk N, Ojeda C, Allard B, Jacquemond V (2002) Effects of extracellular ATP on freshly isolated mouse skeletal muscle cells during pre-natal and post-natal development. Pflugers Arch 443:771–778
Colomar A, Amedee T (2001) ATP stimulation of P2X(7) receptors activates three different ionic conductances on cultured mouse Schwann cells. Eur J Neurosci 14:927–936
Correia-de-Sa P, Timoteo MA, Ribeiro JA (1996) Presynaptic A1 inhibitory/A2A facilitatory adenosine receptor activation balance depends on motor nerve stimulation paradigm at the rat hemidiaphragm. J Neurophysiol 76:3910–3919
Cunha RA (2001) Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: different roles, different sources and different receptors. Neurochem Int 38:107–125
Descarries LM, Cai S, Robitaille R, Josephson EM, Morest DK (1998) Localization and characterization of nitric oxide synthase at the frog neuromuscular junction. J Neurocytol 27:829–840
Fu WM, Poo MM (1991) ATP potentiates spontaneous transmitter release at developing neuromuscular synapses. Neuron 6:837–843
Galkin AV, Giniatullin RA, Mukhtarov MR, Svandova I, Grishin SN, Vyskocil F (2001) ATP but not adenosine inhibits nonquantal acetylcholine release at the mouse neuromuscular junction. Eur J Neurosci 13:2047–2053
Georgiou J, Robitaille R, Trimble WS, Charlton MP (1994) Synaptic regulation of glial protein expression in vivo. Neuron 12:443–455
Georgiou J, Robitaille R, Charlton MP (1999) Muscarinic control of cytoskeleton in perisynaptic glia. J Neurosci 19:3836–3846
Giniatullin RA, Sokolova EM (1998) ATP and adenosine inhibit transmitter release at the frog neuromuscular junction through distinct presynaptic receptors. Br J Pharmacol 124:839–844
Giniatullin AR, Grishin SN, Sharifullina ER, Petrov AM, Zefirov AL, Giniatullin RA (2005) Reactive oxygen species contribute to the presynaptic action of extracellular ATP at the frog neuromuscular junction. J Physiol 565:229–242
Grafe P, Mayer C, Takigawa T, Kamleiter M, Sanchez-Brandelik R (1999) Confocal calcium imaging reveals an ionotropic P2 nucleotide receptor in the paranodal membrane of rat Schwann cells. J Physiol 515(Pt 2):377–383
Grishin S, Shakirzyanova A, Giniatullin A, Afzalov R, Giniatullin R (2005) Mechanisms of ATP action on motor nerve terminals at the frog neuromuscular junction. Eur J Neurosci 21:1271–1279
Hamilton BR, Smith DO (1991) Autoreceptor-mediated purinergic and cholinergic inhibition of motor nerve terminal calcium currents in the rat. J Physiol 432:327–341
Huang SM, Kitamura A, Akita T, Narita K, Kuba K (2002) Adenosine depresses a Ca(2+)-independent step in transmitter exocytosis at frog motor nerve terminals. Eur J Neurosci 15:1291–1298
Jahromi BS, Robitaille R, Charlton MP (1992) Transmitter release increases intracellular calcium in perisynaptic Schwann cells in situ. Neuron 8:1069–1077
Liu GJ, Werry EL, Bennett MR (2005) Secretion of ATP from Schwann cells in response to uridine triphosphate. Eur J Neurosci 21:151–160
Lyons SA, Morell P, McCarthy KD (1994) Schwann cells exhibit P2Y purinergic receptors that regulate intracellular calcium and are up-regulated by cyclic AMP analogues. J Neurochem 63:552–560
Lyons SA, Morell P, McCarthy KD (1995) Schwann cell ATP-mediated calcium increases in vitro and in situ are dependent on contact with neurons. Glia 13:27–38
Moores TS, Hasdemir B, Vega-Riveroll L, Deuchars J, Parson SH (2005) Properties of presynaptic P2X7-like receptors at the neuromuscular junction. Brain Res 1034:40–50
Oliveira L, Timoteo MA, Correia-de-Sa P (2002) Modulation by adenosine of both muscarinic M1-facilitation and M2-inhibition of [3H]-acetylcholine release from the rat motor nerve terminals. Eur J Neurosci 15:1728–1736
Oliveira L, Timoteo MA, Correia-de-Sa P (2004) Tetanic depression is overcome by tonic adenosine A(2A) receptor facilitation of L-type Ca(2+) influx into rat motor nerve terminals. J Physiol 560:157–168
Redman RS, Silinsky EM (1994) ATP released together with acetylcholine as the mediator of neuromuscular depression at frog motor nerve endings. J Physiol 477:117–127
Robitaille R (1995) Purinergic receptors and their activation by endogenous purines at perisynaptic glial cells of the frog neuromuscular junction. J Neurosci 15:7121–7131
Robitaille R (1998) Modulation of synaptic efficacy and synaptic depression by glial cells at the frog neuromuscular junction. Neuron 21:847–855
Robitaille R, Jahromi BS, Charlton MP (1997) Muscarinic Ca2+ responses resistant to muscarinic antagonists at perisynaptic Schwann cells of the frog neuromuscular junction. J Physiol 504:337–347
Robitaille R, Thomas S, Charlton MP (1999) Effects of adenosine on Ca2+ entry in the nerve terminal of the frog neuromuscular junction. Can J Physiol Pharmacol 77:707–714
Rochon D, Rousse I, Robitaille R (2001) Synapse–glia interactions at the mammalian neuromuscular junction. J Neurosci 21:3819–3829
Santos DA, Salgado AI, Cunha RA (2003) ATP is released from nerve terminals and from activated muscle fibres on stimulation of the rat phrenic nerve. Neurosci Lett 338:225–228
Silinsky EM (1975) On the association between transmitter secretion and the release of adenine nucleotides from mammalian motor nerve terminals. J Physiol 247:145–162
Silinsky EM (2004) Adenosine decreases both presynaptic calcium currents and neurotransmitter release at the mouse neuromuscular junction. J Physiol 558:389–401
Silinsky EM (2005) Modulation of calcium currents is eliminated after cleavage of a strategic component of the mammalian secretory apparatus. J Physiol 566:681–678
Smith DO (1991) Sources of adenosine released during neuromuscular transmission in the rat. J Physiol 432:343–354
Son YJ, Thompson WJ (1995) Schwann cell processes guide regeneration of peripheral axons. Neuron 14:125–132
Stevens B, Fields RD (2000) Response of Schwann cells to action potentials in development. Science 287:2267–2271
Stevens B, Ishibashi T, Chen JF, Fields RD (2004) Adenosine: an activity-dependent axonal signal regulating MAP kinase and proliferation in developing Schwann cells. Neuron Glia Biol 1:23–34
Thomas S, Robitaille R (2001) Differential frequency-dependent regulation of transmitter release by endogenous nitric oxide at the amphibian neuromuscular synapse. J Neurosci 21:1087–1095
Todd KJ, Robitaille R (2006) Neuron–glia interactions at the neuromuscular synapse. In: Chadwick DJ, Goode J (eds) Symposium on purinergic signalling in neuron–glia interactions, Novartis Foundation, London, 7–9 June 2006, no. 276
Tung EK, Choi RC, Siow NL, Jiang JX, Ling KK, Simon J, Barnard EA, Tsim KW (2004) P2Y2 receptor activation regulates the expression of acetylcholinesterase and acetylcholine receptor genes at vertebrate neuromuscular junctions. Mol Pharmacol 66:794–806
Zimmermann H, Braun N, Kegel B, Heine P (1998) New insights into molecular structure and function of ectonucleotidases in the nervous system. Neurochem Int 32:421–425
Acknowledgements
The authors thank Claude Gauthier for help with the figure preparation. This work was supported by grants to RR from the Canadian Institutes for Health Research, from the National Science and Engineering Research Council (NSERC), and by a group grant from Fonds de la recherché en Santé du Québec. KJT was supported by a NSERC studentship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Todd, K.J., Robitaille, R. Purinergic modulation of synaptic signalling at the neuromuscular junction. Pflugers Arch - Eur J Physiol 452, 608–614 (2006). https://doi.org/10.1007/s00424-006-0068-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-006-0068-3