
Abstract
Sepsis-associated encephalopathy (SAE) is a neurological complication of sepsis, characterized by brain dysfunction without any direct central nervous system infection. The diagnosis of SAE is currently a challenge. In fact, problems in making a diagnosis of SAE cause a great variability of incidence that can reach up to 70% of all septic patients. Even more, despite SAE is the most frequent type of encephalopathy occurring in critically ill patients, the molecular mechanisms that guide its progression have not been completely elucidated. On the other hand, miRNAs have proven to be excellent biomarkers for both diagnosis and prognosis, especially in brain pathologies because of their small size they can cross the blood–brain barrier easier than other biomolecules. The identification of new miRNAs as biomarkers may help to improve SAE diagnosis and prognosis and also to design new therapies for this clinical manifestation that produces diffuse cerebral dysfunction. This review is focused on SAE physiopathology and the need to have clear criteria for its diagnosis; thus, this work postulates some miRNA candidates to be used for SAE biomarkers because of their role in both, neurological damage and sepsis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
According to the NIH, encephalopathy is defined as any diffuse disease of the brain that alters brain function or structure [1]. Sepsis-associated encephalopathy (SAE) is a brain dysfunction due to sepsis, and it is linked to a systemic inflammatory response syndrome (SIRS) [2, 3]. In sepsis, a potentially fatal organ dysfunction through a dysregulated host response to infection occurs (Sepsis-3 guidelines 2019) [4]. This process can progress to septic shock, a subtype of sepsis in which circulatory, cellular, and metabolic abnormalities are associated with a dramatic increase in mortality [4]. Therefore, SAE is a multifactorial syndrome categorized as a diffuse brain dysfunction induced by the systemic response to an infection, in the absence of direct central nervous system (CNS) infection or other types of encephalopathy, and it can appear as the first organic dysfunction in many septic patients [5, 6]. Sepsis has a global incidence of around 48 million cases per year and causes 11 million deaths, being the leading cause of death around the world, even more than acute myocardial infarction [7,8,9]. SAE incidence is predicted between 9 and 71% of septic patients, depending on the inclusion and exclusion criteria used [3, 10,11,12,13]. In fact, SAE is the most common cause of encephalopathy in the intensive care unit (ICU) worldwide [14,15,16], but the disparity of data and incidence of SAE at ICUs suggests that new and more specific criteria for SAE diagnosis are needed.
Despite sepsis is associated with high mortality incidence, about two out of every three patients survive a sepsis episode. In addition, sepsis can compromise the quality of life in survivors as they have sequelae after sepsis, including immunosuppression, increased cognitive impairment, depression, increased cardiovascular risk, and post-traumatic stress disorder [17,18,19]. Furthermore, sepsis is associated with increased morbidity and mortality, as occurs in SAE, which is characterized by psychiatric disorders and long-term cognitive impairments in sepsis survivors [6, 20,21,22,23,24]. Moreover, despite SAE is described as an acute reversible syndrome in most cases, there is mounting evidence that there are many substantial risks for long-term cognitive impairments. It appears to be linked to direct cellular damage in the brain, mitochondrial and endothelial dysfunction, neurotransmission disturbances, and imbalances of calcium homeostasis in brain tissue, which may impair learning memory and cognitive function [25, 26]. Disturbances in mental processing speed, executive function, attention, and visual-spatial disabilities are other features associated with SAE [5]. Furthermore, it is noteworthy that patients with lower scores on the Glasgow Coma Scale (GCS) and higher Acute Physiology and Chronic Health Evaluation II (APACHE II) scores are more likely to suffer SAE [13].
Diagnosis of SAE represents a challenge due to lack of specific test or diagnostic criteria to define the SAE condition, as well as the scarce number of biomarkers available or these available biomarkers have low specificity and sensitivity [27,28,29,30]. However, although currently available tests do not allow a specific diagnosis of SAE, these tests actually help to exclude other pathologies [5, 12, 31]. In fact, nowadays, SAE is being diagnosed by ruling out direct infection or primary pathologies in the CNS, or consequence of deleterious effects produced by different drugs, toxins, and metabolic disorders [6, 12, 31]. A study performed by Zhang et al. [13] showed that patients admitted to ICU usually manifest pre-existing or chronic kidney or liver failure, blood glucose disturbances, electrolyte imbalances, or pre-existing CNS disease, among others. For all these reasons, SAE is managed as sepsis; so the approach is based on controlling properly metabolic disturbances and avoiding neurotoxic drugs [12, 27, 31].
Clinically, SAE is manifested in two different forms: an early predictable form and a late form, which is usually accompanied by complex metabolic encephalopathy that can lead to irreversible brain damage [11]. It is noteworthy that sometimes SAE is associated with high levels of aromatic amino acids (AAA) in plasma [32] as a consequence of energy deficit, which in turn causes a breakdown of the metabolic pathways related to gluconeogenesis and obtain energy in muscle [33, 34]. The muscles are able to degrade branched-chain amino acids but not AAA, causing their increase in plasma [33,34,35,36]. Higher levels of AAA have been observed in septic patients in comparison to healthy subjects [33, 34] and also in patients with SAE with no serious liver abnormalities [32]. Nevertheless, SAE can be induced by a hepatic failure, which is commonly present in septic patients and in patients suffering from chronic liver disease or cirrhosis [37,38,39]. Hepatic failure is highly related to encephalopathy (hepatic encephalopathy), where a high amount of amino acids released into the bloodstream are found, due to the impossibility of the liver to catabolize amino acids, especially the AAA [35, 40]. In addition, Basler et al. [32] demonstrated that as the disease progressed, the levels of the amino acid were unbalanced (the ratio of branched-chain to aromatic amino acids were decreased), while inflammation markers (such as procalcitonin (PTC) or IL-6) increased, sharpening as time passed. Furthermore, it established a relation between sepsis severity and the level of amino acids in plasma: those patients who did not survive sepsis had higher levels of aromatic and sulfur-containing amino acids (Met and Cys) in comparison to septic survivors [33, 34, 41]. Importantly, it was proposed that the outcome of septic patients might be positively affected using combined therapy with glucose, insulin, and branched-chain amino acids [33, 34, 42]. However, these results need further research.
SAE is characterized by decreased fluctuating attention and confusion in early stages that can progress to delirium, agitation, and coma in late stages [5, 12, 27, 31]. Notably, up to 70% of patients with advanced SAE show critical illness neuromyopathy [15].
It is noteworthy that according to the Sepsis-3 consensus, severe COVID-19 is related to sepsis [43]. Interestingly, COVID-19-associated acute brain dysfunction has been recently described including encephalitis, Guillain-Barré syndrome, ischemic and hemorrhagic stroke, and COVID-19 SAE [44]. These heterogeneous events occurring in some cases of severe COVID-19 patients may further compromise the clinical course and outcomes of severe COVID-19 patients [45]. Therefore, the identification of such events is very important for early empirical combination therapy to survive severe COVID-19 [44].
Physiopathology of SAE
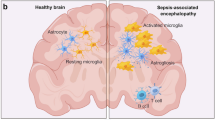
Several mechanisms are involved in SAE pathogenesis, such as disturbance of the blood–brain barrier (BBB), neuronal apoptosis, endothelial activation, hyperinflammation produced by inflammatory cytokines release, oxidative stress, neurotransmission disturbances by alteration in neurotransmitter level, altered brain signaling, altered microcirculation, and dysregulated metabolism [5, 46]. Recently, Kodali et al. demonstrated that cerebral endothelial cells (CECs) are the first activated cells during the earliest stages of acute neuroinflammation, defined as a spinal cord or brain inflammatory response, mediated by cytokine, chemokine, and oxidative stress production [47, 48]. Kodali et al. [47] suggested that CECs are the main source of proinflammatory mediators, which in turn can promote glial cell activation, such as microglial activation. Furthermore, Kodali et al. [47] observed that SAE continued by activating apoptotic signaling in CECs, which is known that causes a BBB disruption, allowing the entrance of peripheral cytokines into the CNS and thus causing an exacerbate gliosis. Finally, it causes a vicious neuroinflammatory cascade, which is commonly observed during SAE. This process is summarized in Fig. 1.

Schematic overview of defined SAE physiopathology. This figure shows a normal physiopathology in a healthy brain and makes a comparison with an altered brain physiopathology, which is observed during SAE. An activation of CECs and the apoptosis induced on them causes a BBB disruption, which loses its selectivity, and an entrance of proinflammatory cytokines is produced. These processes promote glial cell activation, producing reactive astrocytes and activated microglia, which finally causes gliosis. Furthermore, in an SAE brain, an overexpression of glutamate as a consequence of neuroinflammation is produced. Ischemic lesions cause an altered microcirculation in the SAE brain. Finally, a vicious neuroinflammatory cascade produced during SAE, causing brain atrophy. BBB, blood–brain barrier; NT, neurotransmission; ROS, reactive oxygen species
SAE patients present a hyperinflammatory response mainly in the hippocampus, which can be quantified by measuring the levels of the NLRP3 inflammasome, IL-1β, IL-6, and gliosis [2, 27, 31, 49]. The tumor necrosis factor alpha (TNF-α) is another cytokine released in the brain when SAE occurs [27, 31, 50]. Furthermore, the hyperinflammatory response was associated with the activation of the inflammasome in the microglia, being, the pyroptosis, an important nonapoptotic inflammatory cell death determinant of neurodegeneration [51]. In fact, Sui et al. [2] showed that the antioxidant resveratrol was able to inhibit the NLRP3 expression and IL-1β cleavage in a dose-dependent manner. They conclude that resveratrol treatment improved spatial memory and also decreased inflammation by inhibiting the NLRP3/IL-1β axis in the microglia in a mice model of SAE [2].
Moreover, it is now accepted that pyroptosis contributes to the development of many neurological diseases, including SAE [51, 52]. It is noteworthy that the CNS is able to recognize damage-associated molecular patterns (DAMPs) through pattern recognition receptors (PRR), which are present mainly on microglia and astrocytes. These receptors are localized on the membrane’s surface, for extracellular signal recognition, and in the cell cytoplasm for intracellular signal transmission [52]. There exist many studies that demonstrated the expression of NLRP1, NLRP2, and NLRP3 in some CNS-related diseases, especially under stress conditions [53,54,55,56,57]. Neurons, astrocytes, and microglia are the main cells able to suffer pyroptosis in the CNS and thus able to express the pyroptosis-related cytokines (IL-1α, IL-1β, and IL-18) and the receptors related to this process [58,59,60].
It has been demonstrated that inflammation is able to increase cytokine transcription of IL-1β TNF-α and IL-6. This is of special relevance because IL-1β can activate microglia, which has neurotoxic properties when it produces the release of nitric oxide (NO) and reactive oxygen species (ROS) [5, 27, 31, 61]. TNF-α causes neutrophil infiltration in brain tissue, neuronal apoptosis, and brain edema [31]. Furthermore, at the early stages of neuroinflammation, it is able to cause neurotoxicity [62]. Meanwhile, IL-6 can induce cyclooxygenase-2 (COX-2) expression, which in turn increases the levels of the vasodilatory prostaglandin I2 (PGI2) [31, 63], causing fever and behavioral disturbances [5]. Endothelial adhesion molecules, such as V-CAM and I-CAM, also increase their expression in cerebrovascular endothelial cells, increasing the permeability of the BBB [5, 31, 64] and allowing the transfer of toxic factors from the peripheral circulation to the brain [12, 27], causing the BBB loss of selectivity, leading to neuroinflammation and microglia activation [65].
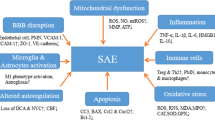
Neuroinflammation, which plays a central role in SAE onset and development, can be produced by a reduction in the proportion and in the total perfused brain vessel density, as well as the alteration of the microcirculation [6, 22, 66, 67]. Notably, the main contributors to brain dysfunction are uncontrolled neuroinflammation and ischemic injury, which can cause the liberation of cytokines and able to activate the microglia [68]. Systemic inflammatory mediators produced by sepsis can enter the brain due to the BBB disturbance, causing the activation of microglia and neuroinflammation by releasing more pro-inflammatory cytokines [69]. The overproduction of pro-inflammatory cytokines can induce cholinergic neuron apoptosis, thus being reduced the cholinergic activity and the acetylcholine (ACh) neurotransmitter levels [69]. The reduction in cholinergic activity can lead to delirium and cognitive decline, characteristics of SAE. Furthermore, this reduction causes a decreased cholinergic inhibition of activated microglia, thus accelerating the microglia activation and, in turn, increasing the cytokine levels [69]. In addition, microglial activation causes hypothalamic neuroinflammation triggering neurologic alterations such as neurotransmission disturbances, as can be the release increment of glucocorticoid hormone or a cell death increment [6, 63]. This cascade leads to the immunosuppressive response, characteristic of sepsis and SAE, and exacerbates the infection worsening the outcome [69]. In the CNS, the cytokine increase is associated with infections, trauma, and injuries, among others [70, 71]. There is also an increase of transforming growth factor beta (TGF-β) and monocyte chemoattractant protein-1 (MCP-1) [72]. These mediators modify the expression of N-methyl-D-aspartate receptors (NMDARs) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) in neurons contributing to brain dysfunction [73]. Importantly, NMDARs and AMPARs are glutamate receptors. As it is known, glutamate is the most important excitatory neurotransmitter in the human brain, and when its levels are above the physiological range, excitotoxic effects are produced. This deleterious action of glutamate is induced in different types of brain insults, including neuroinflammation (for a recent review, see Joy and Carmichael [74]). Some studies have linked increased IL-1β levels with altered modulation of NMDARs, which may derive from functional disturbances in cognition and behavior and contribute to cognitive decline and depression in sepsis survivors [75,76,77]. It is also known that tryptophan, an AAA, is metabolized to quinolinic acid which can be synthesized in activated macrophages, acting as an excitatory transmitter stimulating NMDAR. The activation of these receptors can activate the neuronal isoform of the nitric oxide synthase (nNOS) and other calcium-dependent enzymes, releasing free radicals which can damage the DNA and activate the nuclear enzyme poly-ADP-ribose-synthetase (PARS), which its rapid activation depletes the intracellular concentration of NAD + , slowing the rate of glycolysis and ATP formation, so resulting in energy depletion, cell dysfunction, and death [78, 79]. These metabolic and molecular pathways are described in Fig. 2.

Metabolic alterations during SAE pathogenesis. The main alterations observed in SAE physiopathology cause a hyperinflammation, which derives from microglial activation and brain dysfunction. At the molecular level, the NLRP3 inflammasome and the COX-2 pathways are activated, being inflammation the most relevant mechanism during SAE pathophysiology. SAE, sepsis-associated encephalopathy; BBB, blood–brain barrier; NLRP3, NLR family pyrin domain 3; ACh, acetylcholine; TNF-α, tumor necrosis factor alpha; IL, interleukin; NO, nitric oxide; ROS, reactive oxygen species; COX-2, cyclooxygenase-2; PGI2, prostaglandin I2. Gray arrow indicates a cause; orange arrow indicates a release; green arrow indicates an activation; black line indicates an inhibition. Figure based on Chung et al. [46]
Role of miRNAs As a Potential Biomarker for SAE
microRNAs (miRNAs) are small noncoding RNAs which in recent years have been proposed as key biological regulators in many tissues and cell types, playing important roles in processes such as cell differentiation, growth, proliferation, apoptosis, metabolism, and cellular homeostasis [80,81,82,83]. miRNAs regulate the expression of many genes by linking their bases to complementary sequences of the 3′-untranslated region (3′-UTR), producing translation repression, or in 5′-UTR which stabilizes mRNA structure and facilitates its transcription [80, 84].
miRNAs have been postulated to play essential roles in normal brain functions and in many neuropathological conditions [85,86,87]. They are especially relevant in the brain because their small size miRNAs can cross the BBB easier than proteins or other biomolecules [88, 89]. When miRNAs are dysregulated, they can modify the expression levels of their mRNA targets, upregulating or downregulating gene expression and altering transcriptional programs, as demonstrated in many diseases, including sepsis [90, 91]. For that reason, the potential of miRNAs as biomarkers is obvious. Moreover, miRNAs have other features that make them optimal candidates as biomarkers, such as the fact they are present in biofluids, including blood, urine, and saliva, allowing relatively noninvasive sample collection [80, 92]. In addition to their accessibility, miRNAs are highly stable in biofluids and in different biospecimens making them relatively easy to work with and analyze using different methods (i.e., small RNA sequencing, arrays, qPCR, and ddPCR) [93,94,95,96].
Focusing on neurological inflammation, microglia have been postulated to play a central role in SAE since microglia mediates the immune response and the hyperinflammatory status in the brain [97, 98]. In this regard, microglia serves as brain-resident myeloid cells participating in cerebral development, ischemia, neurodegeneration, and neuro-viral infection [51, 99]. In this scenario, miRNAs are one of the most important regulators mediating microglial activation, polarization, and autophagy and subsequently affecting neuroinflammation and the outcome of CNS disease [100,101,102].
Although very few miRNAs have been described in SAE, it is expected that some miRNAs could show a similar expression pattern that those observed in sepsis, especially miRNAs related to hyperinflammation. Moreover, other miRNAs related to brain damage can also have dysregulated levels, although they could not be specific for SAE.
Many circulating miRNAs have been associated with sepsis diagnosis and prognosis [103]. Puskarich et al. [91] observed a correlation between sepsis and inflamma-miRs such as miR-146a, miR-223, and miR-150 in plasma. Interestingly, miR-146a is involved in the regulatory T cells (Treg) survival and suppressor function [68], thus regulating immune response and targeting the tumor necrosis factor receptor-associated factor 6 (TRAF6), a ubiquitin-conjugating enzyme that mediates NF-κB activation [104]. Moreover, miR-223 directly participates in inflammation by targeting NF-κB [105]. NF-κB is closely related to inflammasome activation and therefore in pyroptosis, and importantly, it is upregulated in SAE [51]. Low levels of miR-223 and miR-146a were found in patients with severe sepsis [106], so we can suggest that low levels of miR-223 may contribute to maintaining high transcription of NF-κB. Regarding miR-150, its expression was correlated with mortality [91]. Moreover, Vasilescu et al. [107] also found that miR-486 and miR-182 expression was higher in septic patients than in healthy subjects. Importantly, some miRNAs such as miR-146a or miR-155 demonstrated their role in neuroinflammation. This is because both miRNAs regulate the overexpression and activation of NF-κB and therefore induce neural pyroptosis through activation of the IL-1β signaling pathway [51, 108, 109].
Notably, miR-155, miR-27a, and miR-210 have been widely postulated as biomarkers in sepsis, and recent studies showed that they play a central role in microglia function [90, 110]. Moreover, some studies have shown that microglial cells are the front line target in the brain for lipopolysaccharide (LPS) action, reinforcing the idea of the key role of microglia in SAE [111]. Regarding miR-155, it was shown that its inhibition in microglia contributes to the development of endotoxin tolerance through an immunohomeostatic reaction [112]. This tolerance is caused by repeated exposure to LPS that maintains an altered response in immune cells, resulting in inhibition of the proinflammatory response and resolution of inflammation [112]. Since microglia is the first line of defense in the brain and the first cells which are the target of LPS in this tissue, it is expected that downregulation of miR-155 may protect microglia against LPS-induced inflammatory injury [111, 113, 114], which is frequent in septic infections. For that reason, it is expected to find high levels of miR-155 in SAE patients when bacterial LPS is found in septic patients. However, miR-155 is not specific, because it is also found at high levels in a wide variety of neurological diseases, for example, in Alzheimer’s disease [88].
miR-27a was also found to be expressed in LPS-activated microglia, so it is possible that levels of miR-27a were reduced in SAE, postulating this miRNA as an interesting factor to be investigated in SAE. Moreover, miR-27a was able to inhibit microglia-produced inflammatory cytokines, including IL-6, IL-1β, and TNF-α, and blocking the expression of TLR4 and IL-1 receptor-associated kinase 4 (IRAK4) [115]. The capacity of miR-27a to regulate the expression of some key inflammatory mediators in microglia makes it a good candidate biomarker for SAE.
In addition, miR-210 is upregulated under hypoxic conditions; therefore, it is considered an important regulator of hypoxia response through the control of many functions such as DNA repair, mitochondrial respiration, angiogenesis, and cell proliferation [116, 117]. Likewise, low miR-210 levels have shown neuroprotective effects on mice with hypoxic-ischemic encephalopathy, due to its capacity for activating microglia, so it is upregulated during the development of the pathology [118,119,120]. Interestingly, miR-210 was related to ROS generation and inflammation in the brain through the ischemia–reperfusion process [119, 121]. Interestingly, due to ischemia–reperfusion injury is usually associated with SAE, miR-210 represents a good biomarker candidate to diagnose SAE. Moreover, targeting miR-210 is a promising approach to develop a miRNA-based therapy since it has been demonstrated that higher levels of this miRNA play a neuroprotective role [119].
Focusing on specific biomarkers for SAE, miR-370 is the most characterized biomarker associated with SAE. Visitchanakun et al. [122] demonstrated that mice with SAE had high levels of miR-370 in brain tissue, and the results were corroborated in plasma samples of SAE patients. This result suggests that miR-370 is very specific for SAE since it shows undetectable levels in patients with sepsis and other inflammatory diseases. Despite the specific role of miR-370 in SAE is not fully elucidated, some authors have postulated that miR-370 induces cell cycle arrest by targeting β-catenin that has a physiological role in controlling cell–cell adhesion and regulating gene transcription [123] and is able to inhibit the proliferation of human glioma cells [122].
In addition, some miRNAs postulated as key regulators in microglia have been shown to mediate inflammation in CNS pathologies [124]; therefore, they may play relevant roles in SAE pathology. In fact, transcriptome analyses comparing microglia, myeloid, and other immune cells identified 239 genes and 8 microRNAs that were highly expressed and specific for microglia: miR-29a, miR-29b, miR-342-3p, miR-103, miR-99a, miR-322, miR-125b-5p, and miR-30a [125].
Interestingly, miR-181b has been postulated to have a protective role in the hippocampus of septic rats. In fact, Dong et al. showed that the expression of hippocampal miR-181b was significantly decreased in septic rats. In this way, the upregulation of miR-181b can inhibit the activation of the NF-κβ signaling pathway and the release of the inflammatory cytokine IL-1β and TNF-α, which are elevated in plasma patients with SAE, therefore alleviating the inflammatory reaction and hippocampus injury in septic rats [126].
Another miRNA postulated as a regulator of microglia differentiation and inflammation was miR-101. Reiko et al. demonstrated that miR-101, which is enriched in the brain, regulates microglial morphology and inflammation, usually altered in SAE patients through the downregulation of the expression of MAPK phosphatase-1 [127]. Other authors showed that through the MAPK pathway, miR-101 also regulates cellular autophagy in the brain [128].
Another interesting miRNA is miR-203, which can inhibit ischemia induced by the activation of microglia, by targeting directly MyD88, a protein that plays a central role in the responses of microglia to pathogen-associated molecular patterns (PAMPs) through Toll-like receptors (TLRs) [90, 129]. Moreover, the overexpression of miR-203 in the brain induces the repression of NF-κβ signaling and prevents subsequent microglial activation ameliorating neuronal injury induced by hyperinflammation [130].
An important mechanism that induces an inflammatory state in CNS is the one mediated by inflammasomes. Some authors showed that the NLRP3-inflammasome complex plays an important role in sepsis through inflammation. NLRP3 inflammasome is considered one of the most important mechanisms that mediate the pro-inflammatory status in the early phases of sepsis [131, 132]. Importantly, it has been shown that NLRP3/caspase-1 pathway-induced pyroptosis mediates cognitive deficits in a mouse model of SAE [133]. Moreover, the inhibition of the NLRP3/IL-1β axis in the microglia improves spatial memory in mice with SAE [2]. Regarding miRNAs, Zhou et al. showed that miR-7 was able to inhibit the activation of microglial NLRP3 inflammasome in vitro. Importantly, stereotactic injection of miR-7 mimics into the mouse striatum ameliorated microglial activation, concomitant with attenuation of dopaminergic neuron degeneration in a mouse model [134].
Similarly, Kumar and Nerurkar demonstrated that mice infected with West Nile virus showed differential expression of miR-196a, miR-202-3p, miR-449c, and miR-125a-3p in brain tissue, leading to neuroinflammation and neuronal death [135]. Furthermore, the upregulation of miR-32 was correlated with the levels of neuroinflammatory molecules [136]. In this regard, it has been demonstrated that the inhibition of miR-32 ameliorated inflammatory cytokine production in LPS-treated microglia, such as IL-1α, IL-1β, and NF-κβ pathway [88].
Finally, in a study performed by El-Assaad et al. [137], specific expression profiles of miRNA in the brain tissue after bacterial infection were found. They found that let-7i, miR-27a, miR-150, miR-126, miR-210, and miR-155 were differentially expressed in the brain. These findings suggest the possibility of measuring specific patterns of miRNA expression in the brain under bacterial infection, which may help to identify brain-related deleterious effects in sepsis. However, since miRNA expression profiles should not be specific for SAE, the use in combination with other biomarkers associated with sepsis may be an effective method to diagnose SAE. Potential biomarkers for SAE based on miRNAs are summarized in Table 1.
Despite there are no optimal inclusion and exclusion criteria for SAE, it is difficult to apply a specific treatment. Some researchers are looking for good therapeutic strategies and drugs for SAE treatment; nevertheless, to perform a successful clinical trial, it is important to properly define inclusion and exclusion criteria [138]. Beyond the necessity of biomarkers to identify appropriate therapies, there is also a need to identify biomarkers to clearly define SAE. Some molecules that are being postulated for SAE treatment are showing promising results in animal models. Rocha Catalão et al. [49] showed that Simvastatin prevents long-term cognitive deficits in sepsis survivor rats, which is one of the main problems in sepsis and SAE survivor patients, by reducing neuroinflammation and neurodegeneration. They observed, in the hippocampus, a reduction of gliosis, nitrate, IL1-β, and IL-6 and overexpression of Bcl-2 protein levels, which was correlated with a decrease of apoptosis. Likewise, animals exposed to Simvastatin presented a better performance in tasks involving habituation, aversive and discriminative memory, and a reduction of neurodegeneration [49]. In another study, Zhang et al. demonstrated the ability of amitriptyline to reduce sepsis-induced brain damage. This compound works through the tropomyosin receptor kinase A (TrkA) signaling pathway, implicated in neuron survival and differentiation [139]. It has been demonstrated that amitriptyline is able to reduce cerebral inflammation by increasing the levels of IL-10, a potent anti-inflammatory cytokine, and reducing pro-inflammatory cytokine generation. Therefore, it is able to control gliosis and ROS production during the SAE physiopathology [139,140,141]. Another postulated therapy is pentamidine; Huang et al. [142] demonstrated the role of this antiprotozoal drug for inhibiting the S100B/RAGE/NF-κB signaling pathway, thus reducing neuroinflammation in the mouse hippocampus, attenuating ROS generation and gliosis.
Conclusions
SAE is a multifactorial syndrome categorized as a diffuse brain dysfunction with irreversible brain damage, which is associated with sepsis. SAE is the first organic dysfunction in many septic patients, and up to 70% of them can suffer from this syndrome. However, depending on the inclusion criteria, this percentage varies because there is no clear diagnostic method or biomarker to identify those patients. So the diagnosis of SAE represents one of the main challenges in SAE. Nowadays, molecular mechanisms underlying the SAE pathophysiology are not completely understood, but it seems that changes in brain metabolism, hyperinflammatory phenotypes, and alteration of the immune response play a central role in the SAE development and progression. Likewise, the microglia appear as a key player due to their function on the cerebral immune defense and their role in inflammation. miRNAs are involved in the brain immune response by regulating microglia, representing an effective form to assist clinicians to diagnose SAE and prognosticate SAE outcome, and helping to decrease high associated mortality. However, a single biomarker could be not enough to diagnose this heterogeneous syndrome, and a combination of biomarkers may improve the performance of the diagnostic methods. In this regard, some miRNAs have been postulated as biomarkers for SAE, such as inflamma-miRs and microglial-specific miRNAs. Specifically, circulating miR-370 was postulated as a feasible candidate for SAE diagnosis, alone or in combination with other miRNAs. Nevertheless, despite the advances in understanding SAE physiopathology, there is a clinically unmet need for the appropriate diagnosis of SAE. In this sense, identifying (bio)markers, single or in combination with other (diagnostic tools), with the potential to achieve a specific diagnostic of SAE, will improve patient care and reduce future morbidities in these patients.
Data availability
Not applicable.
References
Lin L-C, Chen Y-Y, Lee W-T et al (2010) Heat shock pretreatment attenuates sepsis-associated encephalopathy in LPS-induced septic rats. Brain Dev 32:371–377. https://doi.org/10.1016/j.braindev.2009.06.002
Sui D, Xie Q, Yi W et al (2016) Resveratrol protects against sepsis-associated encephalopathy and inhibits the NLRP3/IL-1β axis in microglia. Mediators Inflamm 2016:1045657. https://doi.org/10.1155/2016/1045657
Young GB, Bolton CF, Austin TW et al (1990) The encephalopathy associated with septic illness. Clin Invest Med 13:297–304
Singer M, Deutschman CS, Seymour C et al (2016) The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA - J Am Med Assoc 315:801–810. https://doi.org/10.1001/jama.2016.0287
Chaudhry N, Duggal AK (2014) Sepsis associated encephalopathy Adv Med 2014:1–16. https://doi.org/10.1155/2014/762320
Gofton TE, Young GB (2012) Sepsis-associated encephalopathy. Nat Rev Neurol 8:557–566. https://doi.org/10.1038/nrneurol.2012.183
Reinhart K, Daniels R, Kissoon N et al (2017) Recognizing sepsis as a global health priority — a WHO resolution. N Engl J Med 377:414–417. https://doi.org/10.1056/NEJMp1707170
Rudd KE, Johnson SC, Agesa KM et al (2020) Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet (London, England) 395:200–211. https://doi.org/10.1016/S0140-6736(19)32989-7
Torio CM, Andrews RM (2013) STATISTICAL BRIEF # 160 National Inpatient Hospital Costs : The Most. Healthc Cost Util Proj Stat Br #160 Natl Inpatient Hosp Costs Most Expens Cond by Payer, 2011 31:1–12. https://doi.org/10.1056/NEJMoa1500896
Sprung CL, Peduzzi PN, Shatney CH et al (1990) Impact of encephalopathy on mortality in the sepsis syndrome. Crit Care Med 18:801–806. https://doi.org/10.1097/00003246-199008000-00001
Molnar L, Fülesdi B, Németh N, Molnár C (2018) Sepsis-associated encephalopathy: a review of literature. Neurol India 66:352. https://doi.org/10.4103/0028-3886.227299
Iacobone E, Bailly-Salin J, Polito A et al (2009) Sepsis-associated encephalopathy and its differential diagnosis. Crit Care Med 37:331–336. https://doi.org/10.1097/CCM.0b013e3181b6ed58
Zhang L, Wang X, Ai Y et al (2012) Epidemiological features and risk factors of sepsis-associated encephalopathy in intensive care unit patients: 2008–2011. Chin Med J (Engl) 125:828–831
Bleck T, Smith M, Pierre-Louis S et al (1993) Neurologic complications of critical medical illnesses. Crit Care Med 21:98–103. https://doi.org/10.1097/00003246-199301000-00019
Eidelman LA, Putterman D, Putterman C, Sprung CL (996) The spectrum of septic encephalopathy. Definitions, etiologies, and mortalities. JAMA - J Am Med Assoc 275:470‐473
Ely EW (2004) Delirium as a predictor of mortality in mechanically ventilated patients in the intensive care unit. JAMA 291:1753. https://doi.org/10.1001/jama.291.14.1753
Shih CJ, Chao PW, Ou SM, Chen YT (2017) Long-term risk of cardiovascular events in patients with chronic kidney disease who have survived sepsis: a nationwide cohort study. J Am Heart Assoc 6:233-245.e004613
Prescott HC, Osterholzer JJ, Langa KM et al (2016) Late mortality after sepsis: propensity matched cohort study. BMJ 353:1–8. https://doi.org/10.1136/bmj.i2375
Dantzer R, O’Connor JC, Freund GG et al (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56. https://doi.org/10.1038/nrn2297
Carson WF, Cavassani KA, Dou Y, Kunkel SL (2011) Epigenetic regulation of immune cell functions during post-septic immunosuppression. Epigenetics 6:273–283. https://doi.org/10.4161/epi.6.3.14017
Prescott HC, Angus DC (2018) Enhancing recovery from sepsis: a review. JAMA - J Am Med Assoc 319:62–75. https://doi.org/10.1001/jama.2017.17687
Ji M-H, Qiu L-L, Tang H et al (2015) Sepsis-induced selective parvalbumin interneuron phenotype loss and cognitive impairments may be mediated by NADPH oxidase 2 activation in mice. J Neuroinflammation 12:182. https://doi.org/10.1186/s12974-015-0401-x
Iwashyna TJ, Ely EW, Smith DM, Langa KM (2010) Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 304:1787. https://doi.org/10.1001/jama.2010.1553
Mina F, Comim CM, Dominguini D et al (2014) Il1-β involvement in cognitive impairment after sepsis. Mol Neurobiol 49:1069–1076. https://doi.org/10.1007/s12035-013-8581-9
Zhan RZ, Fujiwara N, Shimoji K (1996) Regionally different elevation of intracellular free calcium in hippocampus of septic rat brain. Shock 6:293–297. https://doi.org/10.1097/00024382-199610000-00012
Zampieri FG, Park M, Machado FS, Azevedo LCP (2011) Sepsis-associated encephalopathy: not just delirium. Clinics 66:1825–1831. https://doi.org/10.1590/S1807-59322011001000024
Sonneville R, Verdonk F, Rauturier C et al (2013) Understanding brain dysfunction in sepsis. Ann Intensive Care 3:15. https://doi.org/10.1186/2110-5820-3-15
Hosokawa K, Gaspard N, Su F et al (2014) Clinical neurophysiological assessment of sepsis-associated brain dysfunction: a systematic review. Crit Care 18:674. https://doi.org/10.1186/s13054-014-0674-y
Gunther ML, Morandi A, Ely EW (2008) Pathophysiology of delirium in the intensive care unit. Crit Care Clin 24:45–65. https://doi.org/10.1016/j.ccc.2007.10.002
Pandharipande PP, Morandi A, Adams JR et al (2009) Plasma tryptophan and tyrosine levels are independent risk factors for delirium in critically ill patients. Intensive Care Med 35:1886–1892. https://doi.org/10.1007/s00134-009-1573-6
Pampín-Huerta FR, Lozano-Requelme ML, Galeiras-Vázquez RM, Moreira-Gómez D (2016) Encefalopatía asociada a la sepsis como presentación de una infección urinaria bacteriémica por Proteus mirabilis. Infectio 20:169–171. https://doi.org/10.1016/j.infect.2015.09.001
Basler T, Meier-Hellmann A, Bredle D, Reinhart K (2002) Amino acid imbalance early in septic encephalopathy. Intensive Care Med 28:293–298. https://doi.org/10.1007/s00134-002-1217-6
Freund H, Atamian S, Holroyde J, FISCHER JE, (1979) Plasma amino acids as predictors of the severity and outcome of sepsis. Ann Surg 190:571–576. https://doi.org/10.1097/00000658-197911000-00003
Freund HR, Ryan JA, Fischer JE (1978) Amino acid derangements in patients with sepsis. Ann Surg 188:423. https://doi.org/10.1097/00000658-197809000-00017
Munro H, Fernstrom J, Wurtman R (1975) Insulin, plasma aminoacid imbalance, and hepatic coma. Lancet 305:722–724. https://doi.org/10.1016/S0140-6736(75)91632-3
Druml W, Heinzel G, Kleinberger G (2001) Amino acid kinetics in patients with sepsis. Am J Clin Nutr 73:908–913. https://doi.org/10.1093/ajcn/73.5.908
Fischer JE, Rosen HM, Ebeid AM et al (1976) The effect of normalization of plasma amino acids on hepatic encephalopathy in man. Surgery 80(77–91)
Sax HC (1986) Clinical use of branched-chain amino acids in liver disease, sepsis, trauma, and burns. Arch Surg 121:358. https://doi.org/10.1001/archsurg.1986.01400030120019
Sprung CL (1990) The role of amino acid changes in septic encephalopathy. pp 60–68
James JH, Jeppsson B, Ziparo V, Fischer J (1979) Hyperammonæmia, plasma aminoacid imbalance, and blood-brain aminoacid transport: a unified theory of portal-systemic encephalopathy. Lancet 314:772–775. https://doi.org/10.1016/S0140-6736(79)92119-6
Fischer JE, Funovics JM, Aguirre A et al (1975) The role of plasma amino acids in hepatic encephalopathy. Surgery 78:276–90
Puskarich MA, McHugh C, Flott TL et al (2020) Serum levels of branched chain amino acids predict duration of cardiovascular organ failure in septic shock. Shock Publish Ah. https://doi.org/10.1097/SHK.0000000000001687
Beltrán-García J, Osca-Verdegal R, Pallardó FV et al (2020) Sepsis and coronavirus disease 2019. Crit Care Med Publish Ah. https://doi.org/10.1097/CCM.0000000000004625
Tong DM, Zhou YT, Wang YW (2021) COVID-19-associated acute brain dysfunction related to sepsis. J Clin Med Res 13:82–91. https://doi.org/10.14740/jocmr4437
Meppiel E, Peiffer-Smadja N, Maury A et al (2021) Neurologic manifestations associated with COVID-19: a multicentre registry. Clin Microbiol Infect 27:458–466. https://doi.org/10.1016/j.cmi.2020.11.005
Chung H-Y, Wickel J, Brunkhorst FM, Geis C (2020) Sepsis-associated encephalopathy: from delirium to dementia? J Clin Med 9(3):703. https://doi.org/10.3390/jcm9030703
Kodali MC, Chen H, Liao F-F (2020) Temporal unsnarling of brain’s acute neuroinflammatory transcriptional profiles reveals panendothelitis as the earliest event preceding microgliosis. Mol Psychiatry. https://doi.org/10.1038/s41380-020-00955-5
DiSabato DJ, Quan N, Godbout JP (2016) Neuroinflammation: the devil is in the details. J Neurochem 139:136–153. https://doi.org/10.1111/jnc.13607
Catalão CHR, Santos-Junior NN, da Costa LHA et al (2020) Simvastatin prevents long-term cognitive deficits in sepsis survivor rats by reducing neuroinflammation and neurodegeneration. Neurotox Res 38:871–886. https://doi.org/10.1007/s12640-020-00222-z
Alexander JJ, Jacob A, Cunningham P et al (2008) TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1. Neurochem Int 52:447–456. https://doi.org/10.1016/j.neuint.2007.08.006
McKenzie BA, Dixit VM, Power C (2020) Fiery cell death: pyroptosis in the central nervous system. Trends Neurosci 43:55–73. https://doi.org/10.1016/j.tins.2019.11.005
Zhao G, Xie Z (2014) Pyroptosis and neurological diseases. Neuroimmunol Neuroinflammation 1:60. https://doi.org/10.4103/2347-8659.139716
Yin Y, Yan Y, Jiang X et al (2009) Inflammasomes are differentially expressed in cardiovascular and other tissues. Int J Immunopathol Pharmacol 22:311–322. https://doi.org/10.1177/039463200902200208
Zhang W-H, Wang X, Narayanan M et al (2003) Fundamental role of the Rip2/caspase-1 pathway in hypoxia and ischemia-induced neuronal cell death. Proc Natl Acad Sci 100:16012–16017. https://doi.org/10.1073/pnas.2534856100
Pelegrin P, Surprenant A (2006) Pannexin-1 mediates large pore formation and interleukin-1β release by the ATP-gated P2X7 receptor. EMBO J 25:5071–5082. https://doi.org/10.1038/sj.emboj.7601378
Compan V, Baroja-Mazo A, López-Castejón G et al (2012) Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 37:487–500. https://doi.org/10.1016/j.immuni.2012.06.013
Kummer JA, Broekhuizen R, Everett H et al (2007) Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem 55:443–452. https://doi.org/10.1369/jhc.6A7101.2006
John GR, Lee SC, Song X et al (2005) IL-1-regulated responses in astrocytes: relevance to injury and recovery. Glia 49:161–176. https://doi.org/10.1002/glia.20109
Alboni S, Cervia D, Sugama S, Conti B (2010) Interleukin 18 in the CNS. J Neuroinflammation 7:9. https://doi.org/10.1186/1742-2094-7-9
Allan SM, Tyrrell PJ, Rothwell NJ (2005) Interleukin-1 and neuronal injury. Nat Rev Immunol 5:629–640. https://doi.org/10.1038/nri1664
Semmler A, Okulla T, Sastre M et al (2005) Systemic inflammation induces apoptosis with variable vulnerability of different brain regions. J Chem Neuroanat 30:144–157. https://doi.org/10.1016/j.jchemneu.2005.07.003
O’Callaghan JP, Sriram K, Miller DB (2008) Defining “neuroinflammation.” Ann N Y Acad Sci 1139:318–330. https://doi.org/10.1196/annals.1432.032
Santos-Junior NN, Catalão CHR, Costa LHA et al (2018) Experimental sepsis induces sustained inflammation and acetylcholinesterase activity impairment in the hypothalamus. J Neuroimmunol 324:143–148. https://doi.org/10.1016/j.jneuroim.2018.08.013
Flierl MA, Rittirsch D, Huber-Lang MS, Stahel PF (2010) Pathophysiology of septic encephalopathy—an unsolved puzzle. Crit Care 14:165–173. https://doi.org/10.1186/cc9035
Hawkins BT, Davis TP (2005) The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev 57:173–185. https://doi.org/10.1124/pr.57.2.4
Taccone FS, Su F, Pierrakos C et al (2010) Cerebral microcirculation is impaired during sepsis: an experimental study. Crit Care 14:R140. https://doi.org/10.1186/cc9205
Michels M, Vieira AS, Vuolo F et al (2015) The role of microglia activation in the development of sepsis-induced long-term cognitive impairment. Brain Behav Immun 43:54–59. https://doi.org/10.1016/j.bbi.2014.07.002
Ren C, Yao R, Zhang H et al (2020) Sepsis-associated encephalopathy: a vicious cycle of immunosuppression. J Neuroinflammation 17:14. https://doi.org/10.1186/s12974-020-1701-3
Zhang Q-H, Sheng Z-Y, Yao Y-M (2014) Septic encephalopathy: when cytokines interact with acetylcholine in the brain. Mil Med Res 1:20. https://doi.org/10.1186/2054-9369-1-20
Michels M, Danielski L, Dal-Pizzol F, Petronilho F (2014) Neuroinflammation: microglial activation during sepsis. Curr Neurovasc Res 11:262–270. https://doi.org/10.2174/1567202611666140520122744
Shatz CJ (2009) MHC class I: an unexpected role in neuronal plasticity. Neuron 64:40–45. https://doi.org/10.1016/j.neuron.2009.09.044
Semmler A, Hermann S, Mormann F et al (2008) Sepsis causes neuroinflammation and concomitant decrease of cerebral metabolism. J Neuroinflammation 5:38–43. https://doi.org/10.1186/1742-2094-5-38
Stellwagen D, Malenka RC (2006) Synaptic scaling mediated by glial TNF-alpha. Nature 440:1054–1059. https://doi.org/10.1038/nature04671
Joy MT, Carmichael ST (2021) Encouraging an excitable brain state: mechanisms of brain repair in stroke. Nat Rev Neurosci 22:38–53. https://doi.org/10.1038/s41583-020-00396-7
Terrando N, Monaco C, Ma D et al (2010) Tumor necrosis factor-alpha triggers a cytokine cascade yielding postoperative cognitive decline. Proc Natl Acad Sci U S A 107:20518–20522. https://doi.org/10.1073/pnas.1014557107
Terrando N, Rei Fidalgo A, Vizcaychipi M et al (2010) The impact of IL-1 modulation on the development of lipopolysaccharide-induced cognitive dysfunction. Crit Care 14:R88. https://doi.org/10.1186/cc9019
Chavan SS, Huerta PT, Robbiati S et al (2012) HMGB1 mediates cognitive impairment in sepsis survivors. Mol Med 18:930–937. https://doi.org/10.2119/molmed.2012.00195
Eggers V, Schilling A, Kox WJ, Spies C (2003) Septic encephalopathy. Diagnosis und therapy. Anaesthesist 52:294–303. https://doi.org/10.1007/s00101-003-0496-9
Szabó C (1998) Role of poly(ADP-ribose)synthetase in inflammation. Eur J Pharmacol 350:1–19. https://doi.org/10.1016/S0014-2999(98)00249-0
Huang W (2017) microRNAs: biomarkers, diagnostics, and therapeutics. Methods Mol Biol 1617:57–67. https://doi.org/10.1007/978-1-4939-7046-9_4
Wang J, Chen J, Sen S (2016) microRNA as biomarkers and diagnostics. J Cell Physiol 231:25–30. https://doi.org/10.1002/jcp.25056
Bartel DP (2004) microRNAs. Cell 116:281–297. https://doi.org/10.1016/S0092-8674(04)00045-5
Chen C-Z (2004) microRNAs modulate hematopoietic lineage differentiation. Science (80- ) 303:83–86. https://doi.org/10.1126/science.1091903
Gu W, Xu Y, Xie X et al (2014) The role of RNA structure at 5′ untranslated region in microRNA-mediated gene regulation. RNA 20:1369–1375. https://doi.org/10.1261/rna.044792.114
Brettschneider J, Del TK, Lee VM-Y, Trojanowski JQ (2015) Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci 16:109–120. https://doi.org/10.1038/nrn3887
Pan Y-B, Sun Z-L, Feng D-F (2017) The role of microRNA in traumatic brain injury. Neuroscience 367:189–199. https://doi.org/10.1016/j.neuroscience.2017.10.046
Boudreau RL, Jiang P, Gilmore BL et al (2014) Transcriptome-wide discovery of microRNA binding sites in human brain. Neuron 81:294–305. https://doi.org/10.1016/j.neuron.2013.10.062
Guo Y, Hong W, Wang X et al (2019) microRNAs in microglia: how do microRNAs affect activation, inflammation, polarization of microglia and mediate the interaction between microglia and glioma? Front Mol Neurosci 12. https://doi.org/10.3389/fnmol.2019.00125
Di Pietro V, Yakoub KM, Scarpa U et al (2018) microRNA signature of traumatic brain injury: from the biomarker discovery to the point-of-care. Front Neurol 9. https://doi.org/10.3389/fneur.2018.00429
Szilágyi B, Fejes Z, Pócsi M et al (2019) Role of sepsis modulated circulating microRNAs. EJIFCC 30:128–145
Puskarich MA, Nandi U, Shapiro NI et al (2015) Detection of microRNAs in patients with sepsis. J Acute Dis 4:101–106. https://doi.org/10.1016/S2221-6189(15)30017-2
García-Giménez JL, Seco-Cervera M, Tollefsbol TO et al (2017) Epigenetic biomarkers: current strategies and future challenges for their use in the clinical laboratory. Crit Rev Clin Lab Sci 54:529–550. https://doi.org/10.1080/10408363.2017.1410520
García-Giménez JL (2016) Epigenetic biomarkers and diagnostic, 1st edn. Mica Haley, London, UK
Enuka Y, Lauriola M, Feldman ME et al (2016) Circular RNAs are long-lived and display only minimal early alterations in response to a growth factor. Nucleic Acids Res 44:1370–1383. https://doi.org/10.1093/nar/gkv1367
Morillon A (2018) Definition and families of long non-coding RNA. In: Long non-coding RNA. Elsevier, pp 25–53
Beltrán-García J, Osca-verdegal R, Romá-Mateo C et al (2020) Epigenetic biomarkers for human sepsis and septic shock : insights from immunosuppression. Epigenomics 1:1–21
Ginhoux F, Lim S, Hoeffel G et al (2013) Origin and differentiation of microglia. Front Cell Neurosci 7:45. https://doi.org/10.3389/fncel.2013.00045
Wang X-H, Wang T-L (2018) microRNAs of microglia: wrestling with central nervous system disease. Neural Regen Res 13:2067. https://doi.org/10.4103/1673-5374.241444
Freilich RW, Woodbury ME, Ikezu T (2013) Integrated expression profiles of mRNA and miRNA in polarized primary murine microglia. PLoS ONE 8:e79416. https://doi.org/10.1371/journal.pone.0079416
Slota B (2019) microRNAs in neuroinflammation: implications in disease pathogenesis, biomarker discovery and therapeutic applications. Non-Coding RNA 5:35. https://doi.org/10.3390/ncrna5020035
Ponomarev ED, Veremeyko T, Weiner HL (2013) microRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia 61:91–103. https://doi.org/10.1002/glia.22363
Christoforidou E, Joilin G, Hafezparast M (2020) Potential of activated microglia as a source of dysregulated extracellular microRNAs contributing to neurodegeneration in amyotrophic lateral sclerosis. J Neuroinflammation 17:135. https://doi.org/10.1186/s12974-020-01822-4
Tacke F, Roderburg C, Benz F et al (2014) Levels of circulating mir-133a are elevated in sepsis and predict mortality in critically ill patients. Crit Care Med 42:1096–1104. https://doi.org/10.1097/CCM.0000000000000131
Zeiser R (2019) Micro-RNA and kinase regulatory mechanisms and pathways in GVHD. In: Immune biology of allogeneic hematopoietic stem cell transplantation. Elsevier, pp 155–165
Ye D, Zhang T, Lou G, Liu Y (2018) Role of miR-223 in the pathophysiology of liver diseases. Exp Mol Med 50:128. https://doi.org/10.1038/s12276-018-0153-7
Wang J, Yu M, Yu G et al (2010) Serum miR-146a and miR-223 as potential new biomarkers for sepsis. Biochem Biophys Res Commun 394:184–188. https://doi.org/10.1016/j.bbrc.2010.02.145
Vasilescu C, Rossi S, Shimizu M et al (2009) microRNA fingerprints identify miR-150 as a plasma prognostic marker in patients with sepsis. PLoS ONE 4:1–19. https://doi.org/10.1371/journal.pone.0007405
Ceppi M, Pereira PM, Dunand-Sauthier I et al (2009) microRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc Natl Acad Sci 106:2735–2740. https://doi.org/10.1073/pnas.0811073106
Nahid MA, Satoh M, Chan EKL (2015) Interleukin 1β-responsive microRNA-146a is critical for the cytokine-induced tolerance and cross-tolerance to toll-like receptor ligands. J Innate Immun 7:428–440. https://doi.org/10.1159/000371517
Huang J, Sun Z, Yan W et al (2014) Identification of microRNA as sepsis biomarker based on miRNAs regulatory network analysis. Biomed Res Int 2014:1–12. https://doi.org/10.1155/2014/594350
Buttini M, Limonta S, Boddeke H (1996) Peripheral administration of lipopolysaccharide induces activation of microglial cells in rat brain. Neurochem Int 29:25–35. https://doi.org/10.1016/0197-0186(95)00141-7
Sun X, Sun J, Shao X et al (2018) Inhibition of microRNA-155 modulates endotoxin tolerance by upregulating suppressor of cytokine signaling 1 in microglia. Exp Ther Med. https://doi.org/10.3892/etm.2018.6032
Nimmerjahn A (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318. https://doi.org/10.1126/science.1110647
Yin H, Song S, Pan X (2017) Knockdown of miR-155 protects microglia against LPS-induced inflammatory injury via targeting RACK1: a novel research for intracranial infection. J Inflamm 14:17. https://doi.org/10.1186/s12950-017-0162-7
Lv Y, Ou-yang A, Fu L (2017) microRNA-27a negatively modulates the inflammatory response in lipopolysaccharide-stimulated microglia by targeting TLR4 and IRAK4. Cell Mol Neurobiol 37:195–210. https://doi.org/10.1007/s10571-016-0361-4
Chen Z, Li Y, Zhang H et al (2010) Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 29:4362–4368. https://doi.org/10.1038/onc.2010.193
Huang X, Le Q-T, Giaccia AJ (2010) MiR-210 – micromanager of the hypoxia pathway. Trends Mol Med 16:230–237. https://doi.org/10.1016/j.molmed.2010.03.004
Li B, Dasgupta C, Huang L et al (2019) MiRNA-210 induces microglial activation and regulates microglia-mediated neuroinflammation in neonatal hypoxic-ischemic encephalopathy. Cell Mol Immunol. https://doi.org/10.1038/s41423-019-0257-6
Qiu J, Zhou X, Zhou X et al (2013) Neuroprotective effects of microRNA-210 on hypoxic-ischemic encephalopathy. Biomed Res Int 2013:1–5. https://doi.org/10.1155/2013/350419
Ma Q, Dasgupta C, Li Y et al (2016) Inhibition of microRNA-210 provides neuroprotection in hypoxic–ischemic brain injury in neonatal rats. Neurobiol Dis 89:202–212. https://doi.org/10.1016/j.nbd.2016.02.011
Rogobete AF, Sandesc D, Bedreag OH et al (2018) microRNA expression is associated with sepsis disorders in critically ill polytrauma patients. Cells 7:271. https://doi.org/10.3390/cells7120271
Visitchanakun P, Tangtanatakul P, Trithiphen O et al (2019) Plasma miR-370-3p as a biomarker of sepsis-associated encephalopathy, the transcriptomic profiling analysis of microRNA-arrays from mouse brains. Shock 1:1–9. https://doi.org/10.1097/SHK.0000000000001473
Valenta T, Hausmann G, Basler K (2012) The many faces and functions of β-catenin. EMBO J 31:2714–2736. https://doi.org/10.1038/emboj.2012.150
Karthikeyan A, Patnala R, Jadhav PS et al (2016) microRNAs: key players in microglia and astrocyte mediated inflammation in CNS pathologies. Curr Med Chem 23:3528–3546. https://doi.org/10.2174/0929867323666160814001040
Butovsky O, Jedrychowski MP, Moore CS et al (2014) Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci 17:131–143. https://doi.org/10.1038/nn.3599
Dong R, Hu D, Li Q et al (2019) Protective effects of microRNA-181b on aged rats with sepsis-induced hippocampus injury in vivo. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 31:857–861. https://doi.org/10.3760/cma.j.issn.2095-4352.2019.07.012
Saika R, Sakuma H, Noto D et al (2017) microRNA-101a regulates microglial morphology and inflammation. J Neuroinflammation 14:109. https://doi.org/10.1186/s12974-017-0884-8
Li Q, Wang Y, Peng W et al (2019) microRNA-101a regulates autophagy phenomenon via the MAPK pathway to modulate Alzheimer’s-associated pathogenesis. Cell Transplant 28:1076–1084. https://doi.org/10.1177/0963689719857085
Esen N, Kielian T (2006) Central role for MyD88 in the responses of microglia to pathogen-associated molecular patterns. J Immunol 176:6802–6811. https://doi.org/10.4049/jimmunol.176.11.6802
Yang Z, Zhong L, Zhong S et al (2015) MiR-203 protects microglia mediated brain injury by regulating inflammatory responses via feedback to MyD88 in ischemia. Mol Immunol 65:293–301. https://doi.org/10.1016/j.molimm.2015.01.019
Kumar V (2018) Inflammasomes: Pandora’s box for sepsis. J Inflamm Res 11:477–502. https://doi.org/10.2147/JIR.S178084
Danielski LG, Della GA, Bonfante S et al (2020) The NLRP3 inflammasome and its role in sepsis development. Inflammation 43:24–31. https://doi.org/10.1007/s10753-019-01124-9
Fu Q, Wu J, Zhou X-Y et al (2019) NLRP3/caspase-1 pathway-induced pyroptosis mediated cognitive deficits in a mouse model of sepsis-associated encephalopathy. Inflammation 42:306–318. https://doi.org/10.1007/s10753-018-0894-4
Zhou Y, Lu M, Du R-H et al (2016) microRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol Neurodegener 11:28. https://doi.org/10.1186/s13024-016-0094-3
Kumar M, Nerurkar VR (2014) Integrated analysis of microRNAs and their disease related targets in the brain of mice infected with West Nile virus. Virology 4:143–151. https://doi.org/10.1016/j.virol.2014.01.004
Zhang Y, Wang J, An W et al (2019) MiR-32 inhibits proliferation and metastasis by targeting EZH2 in glioma. Technol Cancer Res Treat 18:153303381985413. https://doi.org/10.1177/1533033819854132
El-Assaad F, Hempel C, Combes V et al (2011) Differential microRNA expression in experimental cerebral and noncerebral malaria. Infect Immun 79:2379–2384. https://doi.org/10.1128/IAI.01136-10
Lopes G, Parker JL, Willan A et al (2015) The role of biomarkers in improving clinical trial success: a study of 1,079 oncology drugs. J Clin Oncol 33:e17804–e17804. https://doi.org/10.1200/jco.2015.33.15_suppl.e17804
Zhang L, Peng X, Ai Y et al (2020) Amitriptyline reduces sepsis-induced brain damage through TrkA signaling pathway. J Mol Neurosci 70:2049–2057. https://doi.org/10.1007/s12031-020-01611-x
Kubera M, Holan V, Mathison R, Maes M (2000) The effect of repeated amitriptyline and desipramine administration on cytokine release in C57BL/6 mice. Psychoneuroendocrinology 25:785–797. https://doi.org/10.1016/S0306-4530(00)00026-3
Kandil EA, Abdelkader NF, El-Sayeh BM, Saleh S (2016) Imipramine and amitriptyline ameliorate the rotenone model of Parkinson’s disease in rats. Neuroscience 332:26–37. https://doi.org/10.1016/j.neuroscience.2016.06.040
Huang L, Zhang L, Liu Z et al (2019) Pentamidine protects mice from cecal ligation and puncture-induced brain damage via inhibiting S100B/RAGE/NF-κB. Biochem Biophys Res Commun 517:221–226. https://doi.org/10.1016/j.bbrc.2019.07.045
Funding
This paper was supported by AES2019 (ISCIII), grant number PI19/00994, co-financed by the European Regional Development Funds (ERDF), CaixaImpulse (LCF/TR/CI18/50030003) grant by Fundació la Caixa, and Research Projects grants by Fundación Mutua Madrileña (AP174352020). J.L.G-G. and F.V.P. thank the Spanish Ministry of Economy and Competitiveness, ISCIII through CIBERer (Biomedical Network Research Center for Rare Diseases and INGENIO2010). J.B-G. is supported by Grant Contracts i-PFIS grant (IFI18/00015) and co-financed by the European Social Fund. R.O.V was supported by Fundación Mutua Madrileña during the manuscript preparation. Currently, she is supported by the Grant Contract PFIS grant (FI20/00202).
Author information
Authors and Affiliations
Contributions
R. O-V and J. B-G.: manuscript drafting. J.L. G-G. and F.V. P: critical revision. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Osca-Verdegal, R., Beltrán-García, J., Pallardó, F.V. et al. Role of microRNAs As Biomarkers in Sepsis-Associated Encephalopathy. Mol Neurobiol 58, 4682–4693 (2021). https://doi.org/10.1007/s12035-021-02445-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02445-3