
Abstract
Sepsis is defined as the host's reaction to infection and characterised by a systemic inflammatory response with important clinical implications. Central nervous system dysfunction secondary to sepsis is associated with local generation of pro- and anti-inflammatory cytokines, impaired cerebral microcirculation, an imbalance of neurotransmitters, apoptosis and cognitive impairment. It's known that the IL-1β is one of the first cytokines to be altered. Thus, the objective of this study was to evaluate the role of IL-1β in cognitive parameters in brain tissue through the use of an IL-1β (IL-1ra) receptor antagonist up to 10 days and to assess blood–brain barrier permeability, cytokine levels, oxidative parameters and energetic metabolism up to 24 h, after sepsis induction. To this aim, we used sham-operated Wistar rats or submitted to the cecal ligation and perforation (CLP) procedure. Immediately after, the animals received one dose of 10 μg of IL-1ra. After 24 h, the rats were killed and were evaluated for biochemical parameters in the pre-frontal cortex, hippocampus and striatum. After 10 days, the animals were submitted to the habituation to the open field and step-down inhibitory avoidance task. We observed that the use of IL-1ra reverted the increase of blood–brain barrier permeability in the pre-frontal cortex, hippocampus and striatum; the increase of IL-1β, IL1-6 and TNF-α levels in the pre-frontal cortex and striatum; the decrease of complex I activity in the pre-frontal, hippocampus and striatum; the increase of oxidative parameters in pre-frontal cortex, hippocampus and striatum; and cognitive impairment. In conclusion, the results observed in this study reinforce the role of acute brain inflammatory response, in particular, the IL1β response, in the cognitive impairment associated with sepsis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Central nervous system (CNS) dysfunction secondary to sepsis can occur in 8–70 % of septic patients [1] and is associated with neuronal dysfunction and degeneration [2, 3] as well as long-term memory impairment [4, 5]. Indeed, the systemic inflammatory response has a major effect on CNS function [6, 7]. Pro-inflammatory cytokines, in particular, interleukin 1β (IL-1β) and tumor necrosis factor α (TNF-α), which are generated in the periphery, communicate with the brain mainly by primary autonomic afferents or by the circumventricular region, where the blood–brain barrier (BBB) is discontinuous or nonexistent [8]. In this context, sepsis is associated with increase in the levels of TNF- α and IL-1 in the cerebrospinal fluid (CSF) [9] and brain tissue such as hippocampus and cortex.
Thus, IL-1 can be considered the prototypic multifunctional and pleiotropic cytokine due to its widespread effects on immune signaling, CNS functions, and its prominence in many disease states [10, 11]. IL-1β is a potent pro-inflammatory cytokine that is crucial for host's defense responses to infection and injury [10] and it serves as soluble and principally extracellular activators of the IL-1 system, whereas IL-1 receptor antagonist (IL-1ra) is a competitive inhibitor that prevents IL-1α and IL-1β from interacting with the IL-1 receptor 1 (IL-1R1) [12]. Overall, much of what is known about IL-1 bioaction is derived from work using IL-1 and IL-1R1 knock out (KO) mice or administered IL-1ra.
The interaction between bacteria and/or their products and the innate immune system is the crucial moment in the pathophysiology of inflammatory diseases. This interaction defines the severity of the clinical manifestations and the survival or not of the patient and it is of most importance to study the mechanisms involved in this initial phase and in the search for new therapies that may reduce mortality and avoid any major changes in the CNS improving quality of life. Therefore, the purpose in this study is to continue our previous research studies focused on understanding the cognitive damage after sepsis. To this aim, we sought to evaluate the effects of the IL-1β receptor antagonist in brain structures such as the hippocampus, pre-frontal cortex and striatum of animals subjected to sepsis by CLP evaluating the action of this inhibitor in early changes as in cytokine levels, BBB permeability, oxidative damage, metabolism energy as well as the late changes such as cognitive impairment.
Material and Methods
Animals
Male Wistar rats (3–4 months old, 220–310 g) were obtained from our breeding colony. The animals were housed five to a cage with food and water available ad libitum, and maintained on a 12-h light/dark cycle (lights on at 7:00 a.m.). All experimental procedures involving animals were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and the Brazilian Society for Neuroscience and Behavior (SBNeC) recommendations for animal care. All procedures were approved by the Animal Care and Experimentation Committee of the UNESC, Brazil (protocol number 89|2011).
Experimental Procedure
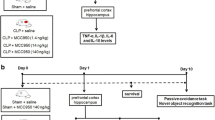
The animals were subjected to CLP as previously described [4]. Briefly, 26 rats (21 rats were subjected to CLP and 5 to Sham) were anesthetised with a mixture of ketamine (80 mg/kg) and xylazine (10 mg/kg) administered i.p. Then, a 3-cm midline laparotomy was performed to allow exposure of the cecum with the adjoining intestine. The cecum was tightly ligated with a 3.0-silk suture at its base, below the ileocecal valve, and perforated once with a 14-gauge needle. The cecum was then gently squeezed to extrude a small amount of feces from the perforation site and then returned into the peritoneal cavity, and the laparotomy was closed with 4.0-silk sutures. All animals were returned to their cages with free access to food and water. In the sham-operated group, the rats were subjected to all surgical procedures, but the cecum was neither ligated nor perforated. After the surgery, all the groups received ‘basic support’ (50 ml/kg saline immediately and 12 h after CLP).
Intervention
Immediately after sepsis induction, the rats received intracisternally a single dose of 10 μg of IL-1ra (Sigma Aldrich, Brazil) or 10 μg of ACSF (control) and were divided into three groups: Sham (n = 20), CLP (n = 20) and CLP + IL-1ra (n = 20). Twenty four hours after the treatment, ten animals of each group were killed by decapitation and had the hippocampus, pre-frontal cortex and striatum immediately dissected, isolated and stored at −80°C for biochemical analyses. After 10 days, ten rats underwent the behavior tests.
Biochemical Analyses
BBB Permeability
A 2 % solution of Evans blue in saline was injected into the tail vein 1 h before harvesting of brain tissues and allowed to circulate for 60 min. Subsequently, the chest was surgically opened under anesthesia and the intravascular dye was removed by saline perfusion (40–50 ml) through the left heart ventricle. The brain was then removed and quantitative evaluation of BBB disruption was achieved by measuring the Evans blue content in the brain regions. Briefly, the rat brain was homogenised in 50 % wt/vol trichloroacetic acid, after centrifugation the supernatant was diluted fourfold with ethanol, and fluorescence intensity (ng/ml) was measured on a microplate fluorescence reader. The total Evans blue content (ng) in each sample was derived from the concentrations of external standards [13].
Lipid Peroxidation
Lipid peroxidation was measured by formation of thiobarbituric acid (TBA) reactive substances (TBARS) after the method of Draper and Hadley [14]. Following the brain dissection, brain structures were washed with PBS, harvested and lysed. TBA 0.67 % was added to each tube and vortexed. The optical density of each solution was measured in a spectrophotometer at 535 nm. Data were expressed as nmol of TBARS equivalents per mg of protein.
Protein Carbonyl Formation
Protein carbonyl content was measured in brain homogenates using 2,4-dinitrophenylhydrazine (DNPH) in a spectrophotometric assay [15]. Absorbance was recorded in a spectrophotometerat 370 nm for both DNPH-treated and HCl-treated samples. Protein carbonyl levels were expressed as nmol of carbonyl per mg of protein.
Mitochondrial Respiratory Chain Enzymes Activities
Brain structures were homogenised (1:10, w/v) in SETH buffer (250 mM sucrose, 2 mM EDTA, 10 mM Trizma base, 50 IU/ml heparin, pH 7.4) for determination of mitochondrial respiratory chain enzyme activities (complexes I, II, II–III and IV). NADH dehydrogenase (complex I) was evaluated according to the method described by Cassina and Radi [16] by the rate of NADH-dependent ferricyanide reduction at 420 nm [16]. The activities of succinate: DCIP oxidoreductase (complex II) and succinate: cytochrome c oxidoreductase (complexes II–III) were determined according to the method of Fischer et al. [17]. Complex II activity was measured by following the decrease in absorbance due to the reduction of 2,6-DCIP) at 600 nm. Complexes II–III activity was measured by cytochrome c reduction from succinate. The activity of cytochrome c oxidase (complex IV) was assayed by following the decrease in absorbance due to the oxidation of previously reduced cytochrome c at 550 nm [18]. The activities of the mitochondrial respiratory chain complexes were expressed as nmol/min mg protein [18].
ELISA Analyses
Brain structures were homogenised in extraction solution containing PBS. The concentration of cytokines (IL-1β, IL-6 and TNF-α) were determined by ELISA (R&D Systems, Minneapolis, MN). All samples were assayed in duplicate. Briefly, the capture antibody (13 ml, contains 0.1 % sodium azide) was diluted in phosphate-buffered saline (PBS), added to each well and left overnight at 4 °C. The plate was washed four times with PBS and 0.05 % Tween 20 (Sigma, St. Louis, MO, USA). The plate was blocked with 1 % bovine serum albumin and incubated for 1 h at room temperature before washing four times with PBS and 0.05 % Tween 20. The samples and standards were added and the plate was incubated overnight at 4 °C. After washing the plate, detection antibody (concentration provided by the manufacturer) diluted in PBS was added. The plate was incubated for 2 h at room temperature. After washing the plate, streptavidin (DuoSet R&D Systems, Minneapolis, MN, USA) was added and the plate was incubated for 30 min. At last, color reagent o-phenylenediamine (Sigma, St. Louis, MO, USA) was added to each well, and the reaction was allowed to develop in the dark for 15 min. The reaction was stopped with the addition of 1 M H2SO4 to each well. The absorbance was read on a plate reader at 492 nm wavelength (Emax, Molecular Devices, Minneapolis, MN, USA). Sham and CLP samples were placed in the plate for the ELISA analysis.
All the results were normalised by protein concentration measured by the Lowry assay [19].
Behavioral Tests
The animals separately underwent two behavioral tasks: habituation to an open field and step-down inhibitory avoidance task. Thus, using this design, we do not assess time-dependent memory, but assess memory over time (with new training at each test session). All behavioral procedures were conducted between 13:00 and 16:00 h in a sound-isolated room, and a single animal performed only one behavior test in only one time point after surgery. All behavioral tests were recorded by the same person blind to the animal group.
Habituation to an Open Field Task
This task evaluates motor performance in the training section and non-associative memory in the retention test session. Habituation to an open field was carried out in a 20 × 30 cm open field surrounded by 30-cm-high walls made of brown plywood with a frontal glass wall. The floor of the open field was divided into 12 equal rectangles by black lines. The animals were gently placed on the left rear quadrant and left to explore the arena for 5 min (training session). Immediately following this, the animals were taken back to their home cage and 24 h later submitted again to a similar open-field session (test session). Crossing of the black lines and rearing performed in both sessions were counted. The decrease in the number of crossings and rearings between the two sessions was taken as a measure of the retention of habituation [20].
Step-Down Inhibitory Avoidance Task
This task evaluates aversive memory. The apparatus and procedures have been described in previous reports. Briefly, the training apparatus was a 25 × 12 × 12 cm acrylic box whose floor consisted of parallel caliber stainless steel bars (1-mm diameter) spaced 1 cm apart. A 7-cm-wide, 2.5-cm-high platform was placed on the floor of the box against the left wall. In the training trial, animals were placed on the platform and their latency to step down on the grid with all four paws was measured with an automatic device. Immediately after stepping down on the grid, the animals received a 0.2 mA, 2.0 s foot shock and were returned to their home cage. A retention test trial was performed 24 h after training (long-term memory). The retention test trial was procedurally identical to training, except that no foot shock was presented. The retention test step-down latency (maximum 180 s) was used as a measure of inhibitory avoidance retention. Reactivity to the foot shock was evaluated in the same apparatus used for inhibitory avoidance, except that the platform was removed. Each animal was placed on the grid and allowed a 1-min habituation period prior to the start of a series of shocks (0.5 s), delivered at 10-s intervals. Shock intensities ranged from 0.1 to 0.5 mA in 0.1-mA increments. The adjustments in shock intensity were made in accordance to each animal's response. The intensity was raised by 1 unit when no response occurred and lowered by 1 unit when a response was made. A ‘flinch’ response was defined as withdrawal of one paw from the grid floor, and a ‘jump’ response was defined as rapid withdrawal of three or four paws. Two measurements of the ‘flinch’ threshold were made and then two measurements of the ‘jump’ threshold were made. For each animal, the mean of the two scores for the flinch and the jump thresholds was calculated [21].
Statistical Analysis
Data from the habituation to an open field and biochemical analyses are reported as means ± S.E.M and were analysed by the ANOVA Tukey post hoc. Data from the inhibitory avoidance is reported as median and interquartile ranges and comparisons among groups were performed using Mann–Whitney U tests. Wilcoxon tests was used within individual groups. p < 0.05 was considered statistically significant.
Results
Fig. 1 shows the BBB permeability 24 h after induction. Sepsis caused an increase of BBB permeability in pre-frontal cortex, hippocampus and striatum and the use of IL-1ra decreased the BBB permeability in all structures studied. In Fig. 2, the cytokine levels were demonstrated 24 h after induction. There was increase of IL-1β (Fig. 2A), IL-6 (Fig. 2A) and TNF-α (Fig. 2C) levels in pre-frontal cortex and hippocampus in the sepsis groups. The use of IL1ra reversed the increase of IL-1β, IL-6 and TNF-α in both structures. There weren't alterations in the striatum in all groups. The energetic metabolism is shown in Fig. 3. Sepsis caused an increase of complex I activity (Fig. 3A) 24 h after induction in pre-frontal cortex, hippocampus and striatum and the treatment with IL-1ra reversed these alterations in all structures. There weren't alterations in the activity of complexes II (b), III (Fig. 3C) and IV (Fig. 3D).

BBB permeability 24 h after induction in pre-frontal cortex, hippocampus and striatum. Data are presented as mean ± SEM, n = 7 rats per group.*p < 0.05 versus Sham and #p < 0.05 versus CLP

Cytokines levels 24 h after induction in pre-frontal cortex, hippocampus and striatum. Data from the IL1-β (A), IL-6 (B) and TNF-α (C) are presented as mean ± SEM, n = 7 rats per group. *p < 0.05 versus Sham and #p < 0.05 versus CLP

Energetic metabolism 24 h after induction in pre-frontal cortex, hippocampus and striatum. Data from the Complex I (A), Complex II (B), Complex III (C) and Complex IV (D) activity are presented as mean ± SEM, n = 7 rats per group. *p < 0.05 versus Sham and #p < 0.05 versus CLP
Fig. 4 shows oxidative damage 24 h after sepsis induction. There was an increase of lipid peroxidation (Fig. 4A) indicated by TBARS levels in the pre-frontal cortex, hippocampus and striatum and an increase of protein peroxidation (Fig. 4B) indicated by carbonyl levels in the pre-frontal cortex and hippocampus in the sepsis group. The treatment with IL-1ra decreased these levels in all structures evaluated. Finally, Fig. 5 demonstrates the cognition evaluation up to 10 days of the induction. In Fig. 5A, there weren't statistical differences in the number of crossings and rearings between training and test in the sepsis group, demonstrating that sepsis caused habituation memory impairment. There weren't statistical differences in the number of crossings and rearings in the training session, demonstrating that there wasn't locomotor impairment. In Fig. 5B, there weren't statistical differences in the latency time between training and test in the sepsis group, demonstrating that sepsis caused aversive memory impairment. There weren't statistical differences in the latency time in the training session, demonstrating that there wasn't locomotor impairment.

Oxidative damage 24 h after induction in pre-frontal cortex, hippocampus and striatum. Data from the lipid peroxidation (A) and protein peroxidation (B) are presented as mean ± SEM, n = 7 rats per group.*p < 0.05 versus Sham and #p < 0.05 versus CLP

Behavior tests. The animals underwent two behavioral tasks: habituation to an open field (A) and step-down inhibitory avoidance (B). Data from the habituation to an open field are presented as mean ± SEM and data from step-down inhibitory avoidance are presented as median ± interquartile ranges, n = 10 rats per group. *p < 0.05 versus training
Since sepsis could affect sensory processing during training, such as the rats' reactivity to the foot shock rather than memory, we evaluated the effects of sepsis on foot shock sensitivity in all analysed time points, and there were no significant differences between groups in the flinch or the jump nociceptive thresholds, showing that sepsis did not affect the animal's reactivity to the foot shock (data not shown).
Discussion
In short, we observed that the treatment with IL-1ra (1) decreased the BBB permeability; (2) decreased the IL-1β, IL-6 and TNFα levels; (3) decreased the complex I activity; (4) decreased the oxidative damage; and (5) reversed the cognitive impairment.
It is known that the IL-1 pathway regulates inflammation, angiogenesis, hematopoiesis and cognition [22, 23] and that a sustained systemic inflammation may contribute to prolong or aggravate brain dysfunction [24, 25]. Sepsis induces activation of cerebral endothelial cells, which result in BBB dysfunction and release of various mediators in the brain [26]. In this study, we showed that the treatment with IL-1ra decreased the BBB permeability in pre-frontal cortex, hippocampus and striatum and lowered the levels of IL-1β, IL-6 and TNF-α in pre-frontal cortex and hippocampus after 24 h of induction, showing that IL-1β may be involved in brain dysfunction. Several studies have suggested that IL-1β, IL-6 and TNF-α are key inflammatory mediators during the progression of brain damage. Allan and collaborators [27] demonstrated that a synergistic interaction between IL-1β and other cytokines, such as TNFα and IL-6, enhances this cognitive dysfunction [27].
In this context, learning and memory processes largely rely on the hippocampus, and this brain region expresses the highest density of IL-1 receptors, making it vulnerable to the adverse consequences of neuroinflammation [28, 29]. Although IL-1β is required for normal learning and memory processes, exogenous administration or excessive endogenous levels produce detrimental cognitive behavioral effects [22, 30, 31]. Recently, our group demonstrated higher CSF levels of IL-1β and TNF-α in septic animals at 24 h that had a worse performance in the inhibitory aversive task, and this was associated with lower BDNF levels in hippocampus [32]. In the same line, high levels of inflammatory cytokines have been associated with a decrease in the levels of BDNF and with changes in hippocampal neural function, even in humans [33]. Imamura and collaborators [7] showed that pre-incubation with IL-1R1 antagonist for 30 min before recording of field excitatory post-synaptic potentials in the hippocampus canceled long-term memory deficiency after CLP. Terrando and collaborators [34] suggest that by blocking IL-1 signaling, the inflammatory cascade to lipopolysaccharide (LPS) is attenuated, thereby reducing microglial activation and preventing the behavioral abnormality.
We had previously demonstrated that oxidative damage and inflammation occur early in the brain after sepsis [35, 36] are resolved, in part, when long-term cognitive impairment occurs [37]. In the immune system, once activated, inflammatory cells produce reactive oxygen species (ROS) that are primarily directed to kill microorganisms. However, excessive amounts of ROS can attack cellular components and lead to cell damage [38]. ROS are also involved in several intracellular pathways that ultimately lead to the activation of the innate immune system [39]. In addition, oxidised proteins and lipids could stimulate cytokine release from macrophages through the activation of membrane receptors [40]. We observed that the use of IL-1ra reversed the effect of sepsis on the BBB permeability and cytokines levels, as described above. This inhibitor also decreased the oxidative damage and enhanced the energetic metabolism, especially complex I activity, in brain tissue after 24 h of induction. The main sites of ROS productions are the complexes I and III of the Electron Transporter Chain (ETC.) [41, 42]. In conclusion, this study showed that the use of a single dose of IL-1βra immediately after sepsis can modulate cognitive impairment up to 10 days after induction by reversing the alterations associated with sepsis pathophysiology as high levels of pro-inflammatory cytokines, oxidative stress and alterations in the energetic metabolism. Thus, we believe that IL-1β has a pivotal role in sustaining the neuroinflammatory response and closely interacts with memory processing and long-term potentiation.
References
Sprung CL, Peduzzi PN, Shatney CH, Schein RM, Wilson MF, Sheagren JN, Hinshaw LB (1990) Impact of encephalopathy on mortality in the sepsis syndrome. The Veterans Administration Systemic Sepsis Cooperative Study Group. Crit Care Med 18:801–806
Streck EL, Comim CM, Barichello T, Quevedo J (2008) The septic brain. Neurochem Res 33:2171–2177
Comim CM, Constantino LC, Barichello T, Streck EL, Quevedo J, Dal-Pizzol F (2009) Cognitive impairment in the septic brain. Curr Neurovasc Res 6:194–203
Tuon L, Comim CM, Petronilho F, Barichello T, Izquierdo I, Quevedo J, Dal-Pizzol F (2008) Time-dependent behavioral recovery after sepsis in rats. Intensive Care Med 34:1724–1731
Iwashyna TJ, Ely EW, Smith DM, Langa KM (2010) Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 304:1787–1794
Perry VH (2004) The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun 18(5):407–413
Imamura Y, Wang H, Matsumoto N, Muroya T, Shimazaki J, Ogura H, Shimazu T (2011) Interleukin-1β causes long-term potentiation deficiency in a mouse model of septic encephalopathy. Neuroscience 28(187):63–69
Quan N, Banks WA (2007) Brain-immune communication pathways. Brain Behav Immun 21(6):727–735
Waage A, Halstensen A, Shalaby R, Brandtzaeg P, Kierulf P, Espevik T (1989) Local production of tumor necrosis factor alpha, interleukin 1, and interleukin 6 in meningococcal meningitis. Relation to the inflammatory response. J Exp Med 170(6):1859–1867
Dinarello CA (1996) Biologic basis for interleukin-1 in disease. Blood 87(6):2095–2147
Murray CA, Lynch MA (1998) Evidence that increased hippocampal expression of the cytokine interleukin-1 beta is a common trigger for age- and stress-induced impairments in long-term potentiation. J Neurosci 18(8):2974–2981
Korherr C, Hofmeister R, Wesche H, Falk W (1997) A critical role for interleukin-1 receptor accessory protein in interleukin-1 signaling. Eur J Immunol 27:262–267
Liu W, Hendren J, Qin XJ, Shen J, Liu KJ (2009) Normobaric hyperoxia attenuates early blood–brain barrier disruption by inhibiting MMP-9-mediated occludin degradation in focal cerebral ischemia. J Neurochem 108:811–820
Draper HH, Hadley M (1990) Malondialdehyde determination as index of lipid peroxidation. Methods Enzymol 186:421–431
Levine RL, Williams JA, Stadtman ER, Shacter E (1994) Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol 233:346–357
Cassina A, Radi R (1996) Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys 328:309–316
Fischer JC, Ruitenbeek W, Berden JA, Trijbels JM, Veerkamp JH, Stadhouders AM, Sengers RC, Janssen AJ (1985) Differential investigation of the capacity of succinate oxidation in human skeletal muscle. Clin Chim Acta 153:23–36
Miro O, Cardellach F, Barrientos A, Casademont J, Rotig A, Rustin P (1998) Cytochrome c oxidase assay in minute amounts of human skeletal muscle using single wavelength spectrophotometers. J Neurosci Methods 80:107–111
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–267
Vianna MR, Alonso M, Viola H, Izquierdo I (2000) Role of hippocampal signaling pathways in long-term memory formation of a nonassociative learning task in the rat. Learn Mem 7:333–340
Quevedo J, Vianna MR, Roesler R, Izquierdo I (1999) Two time Windows of anisomycin-induced amnesia for inhibitory avoidance training in rats: protection from amnesia by pretraining but not pre-exposure to the task apparatus. Learn Mem 6:600–607
Rachal Pugh C, Fleshner M, Watkins LR, Maier SF, Rudy JW (2001) The immune system and memory consolidation: a role for the cytokine IL-1beta. Neurosci Biobehav Rev 25:29–41
Shieh JH, Peterson RH, Moore MA (1991) IL-1 modulation of cytokine receptors on bone marrow cells. In vitro and in vivo studies. J Immunol 147(4):1273–1278
McGrane S, Girard TD, Thompson JL, Shintani AK, Woodworth A, Ely EW (2011) Pandharipande PP: procalcitonin and C-reactive protein levels at admission as predictors of duration of acute brain dysfunction in critically ill patients. Crit Care 15:R78
van den Boogaard M, Kox M, Quinn KL, van Achterberg T, van der Hoeven JG, Schoonhoven L, Pickkers P (2011) Biomarkers associated with delirium in critically ill patients and their relation with long-term subjective cognitive dysfunction; indications for different pathways governing delirium in inflamed and noninflammed patients. Crit Care 15:R297
Handa O, Stephen J, Cepinskas G (2008) Role of endothelial nitric oxide synthase-derived nitric oxide in activation and dysfunction of cerebrovascular endothelial cells during early onsets of sepsis. Am J Physiol Heart Circ Physiol 295:H1712–H1719
Allan SM, Tyrrell PJ, Rothwell NJ (2005) Interleukin-1 and neuronal injury. Nat Rev Immunol 5:629–640
Parnet P, Amindari S, Wu C, Brunke-Reese D, Goujon E, Weyhenmeyer JA, Dantzer R, Kelley KW (1994) Expression of type I and type II interleukin-1 receptors in mouse brain. Brain Res Mol Brain Res 27:63–70
Gemma C, Fister M, Hudson C, Bickford PC (2005) Improvement of memory for context by inhibition of caspase-1 in aged rats. Eur J Neurosci 22:1751–1756
Katsuki H, Nakai S, Hirai Y, Akaji K, Kiso Y, Satoh M (1990) Interleukin-1 beta inhibits long-term potentiation in the CA3 region of mouse hippocampal slices. Eur J Pharmacol 181(3):323–326
Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW (2008) Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun 22:301–311
Biff D, Petronilho F, Constantino L, Vuolo F, Zamora-Berridi GJ, Dall'igna DM, Comim CM, Quevedo J, Kapczinski F, Dal-Pizzol F (2013) Correlation of acute phase inflammatory and oxidative markers with long-term cognitive impairment in sepsis survivors rats. Shock 40(1):45–48
Goldstein BI, Collinger KA, Lotrich F, Marsland AL, Gill MK, Axelson DA, Birmaher B (2011) Preliminary findings regarding proinflammatory markers and brain-derived neurotrophic factor among adolescents with bipolar spectrum disorders. J Child Adolesc Psychopharmacol 21(5):479–484
Terrando N, Rei Fidalgo A, Vizcaychipi M, Cibelli M, Ma D, Monaco C, Feldmann M, Maze M (2010) The impact of IL-1 modulation on the development of lipopolysaccharide-induced cognitive dysfunction. Crit Care 14(3):R88
Barichello T, Fortunato JJ, Vitali AM, Feier G, Reinke A, Moreira JC, Quevedo J, Dal-Pizzol F (2006) Oxidative variables in the rat brain after sepsis induced by cecal ligation and perforation. Crit Care Med 34(3):886–889
Comim CM, Vilela MC, Constantino LS, Petronilho F, Vuolo F, Lacerda-Queiroz N, Rodrigues DH, da Rocha JL, Teixeira AL, Quevedo J, Dal-Pizzol F (2011) Traffic of leukocytes and cytokine up-regulation in the central nervous system in sepsis. Intensive Care Med 37(4):711–718
Comim CM, Cassol-Jr OJ, Constantino LS, Felisberto F, Petronilho F, Rezin GT, Scaini G, Daufenbach JF, Streck EL, Quevedo J, Dal-Pizzol F (2011) Alterations in inflammatory mediators, oxidative stress parameters and energetic metabolism in the brain of sepsis survivor rats. Neurochem Res 36(2):304–311
Zhang H, Slutsky AS, Vincent JL (2000) Oxygen free radicals in ARDS, septic shock and organ dysfunction. Intensive Care Med 26:474–476
Victor VM, De La Fuente M (2003) Immune cells redox state from mice with endotoxin-induced oxidative stress. Involvement of NF-kappaB. Free Radic Res 37:19–27
de Souza LF, Ritter C, Pens Gelain D et al (2007) Mitochondrial superoxide production is related to the control of cytokine release from peritoneal macrophage after antioxidant treatment in septic rats. J Surg Res 141:252–256
Turrens JF, Alexandre A, Lehninger AL (1985) Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys 237:408–414
Sugioka K, Nakano M, Totsune-Nakano H et al (1988) Mechanism of O2- generation in reduction and oxidation cycle of ubiquinones in a model of mitochondrial electron transport systems. Biochim Biophys Acta 936:377–385
Acknowledgements
This research was supported by grants from CNPq (ELS, JQ and FD-P), FAPESC (ELS, JQ and FD-P) and UNESC (ELS, JQ and FD-P). ELS, FDP and JQ are CNPq research fellows.
Conflict of interest
None
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mina, F., Comim, C.M., Dominguini, D. et al. Il1-β Involvement in Cognitive Impairment after Sepsis. Mol Neurobiol 49, 1069–1076 (2014). https://doi.org/10.1007/s12035-013-8581-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-013-8581-9