
Abstract
Sepsis is an organ dysfunction caused by an uncontrolled inflammatory response from the host to an infection. Sepsis is the main cause of morbidity and mortality in intensive care units (ICU) worldwide. One of the first organs to suffer from injuries resulting from sepsis is the brain. The central nervous system (CNS) is particularly vulnerable to damage, mediated by inflammatory and oxidative processes, which can cause the sepsis-associated encephalopathy (SAE), being reported in up to 70% of septic patients. This review aims to bring a summary of the main pathophysiological changes and dysfunctions in SAE, and the main focuses of current experimental studies for new treatments and therapies. The pathophysiology of SAE is complex and multifactorial, combining intertwined processes, and is promoted by countless alterations and dysfunctions resulting from sepsis, such as inflammation, neuroinflammation, oxidative stress, reduced brain metabolism, and injuries to the integrity of the blood-brain barrier (BBB). The treatment is limited once its cause is not completely understood. The patient’s sedation is far to provide an adequate treatment to this complex condition. Studies and experimental advances are important for a better understanding of its pathophysiology and for the development of new treatments, medicines, and therapies for the treatment of SAE and to reduce its effects during and after sepsis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sepsis is an organic dysfunction caused by the hosts uncontrolled inflammatory response to an infection [1, 2], represented by an imbalance in the pro-inflammatory and anti-inflammatory factors, leading to injury and multiple organ dysfunction and failure [3,4,5,6]. Sepsis remains as the main cause of ICU morbidity and mortality worldwide [7, 8], being responsible for approximately six million deaths, affecting predominantly low and middle-income countries [9]. The acute systemic inflammation resulting from sepsis causes a cascade of physiological changes that affects the central nervous system (CNS) [10, 11]. The CNS is particularly vulnerable to damage, mediated by inflammatory and oxidative processes, which can cause sepsis-associated encephalopathy (SAE) [11,12,13].
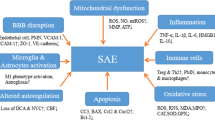
Although the exact mechanism of cerebral dysfunction is still not well understood, it is known that the pathophysiology of SAE is multifactorial [14,15,16,17]. Studies report that up to 70% of patients with sepsis develop SAE [18, 19]. One of the main processes involved in the brain changes and injuries during sepsis is the increased expression of pro-inflammatory cytokines [20,21,22], and the activation of microglia and astroglial cells causes changes in cerebral homeostasis [20, 23,24,25]. Neuroinflammation increases metabolic and bioenergetic demands, resulting in oxidative stress and mitochondrial dysfunction, with production of reactive oxygen species, stimulating a pro-apoptotic scenario that affects glial cells, neurons, and the blood-brain barrier (BBB) structure [26,27,28]. Alterations of the BBB integrity compromise the healthy brain function, considering its role as a highly selective barrier between the brain and the periphery [29, 30]. Metabolic and hemodynamic changes also precede cognitive impairment and structural changes in the brain, such as atrophy of white and gray matter [31,32,33]. Changes in brain metabolism may represent a key component to trigger encephalopathy during the pathological process of sepsis [16, 17, 34]. Once SAE’s cause remains unknown, its treatment is limited, and one of the therapeutic strategies, such as patient sedation, is clearly unsatisfactory as treatment of such complex condition [19].
This review provides a summary of the main pathophysiological changes and dysfunctions in SAE and describes the progress obtained from experimental studies, in the task force of better understanding the mechanisms related to this condition. Many efforts have been done to develop new treatments and therapies not only to treat SAE but also to improve the quality of life of the septic patient, during the hospitalization period and, mainly, after sepsis.
SAE Clinical Presentation and Dysfunctions
Encephalopathy associated with sepsis can be defined as a cognitive dysfunction associated with sepsis, in the absence of infection in the central nervous system or structural brain injury, after excluding other metabolic causes. Clinically, this syndrome is manifested by mental confusion, anxiety, irritability, depression, anhedonia, decreased social communication and environmental interest, and cognitive changes, including decreased concentration, learning capacity, and memory [35]. The researches about the mechanisms underlying this syndrome began in the 1950s [24, 36].
SAE is classified as acute when is manifested only during the course of sepsis, with patient improvement after its control [37, 38]. When symptoms last from weeks to months, SAE can be considered subacute, and if the symptoms persist for a year, it can be categorized as chronic. Subacute and chronic deficits require close monitoring and attention, since affected individuals may need rehabilitation or home care [36].
During sepsis, neurological dysfunctions can vary from mild mental confusion to cognitive impairment and coma [16, 39]. In some cases, patients may also experience muscle stiffness, tremors, or seizures [40]. Neurological disorders in SAE are similar to the description of hypoactive and hyperactive delirium, which is considered a manifestation of cerebral dysfunction during sepsis [41].
Even though SAE has been considered a reversible syndrome, mild-to-moderate neurological symptoms, including memory changes, depression, anxiety, or cognitive disorders, can persist between 20 and 40% of patients 1 year after hospital discharge [16, 38, 42, 43].
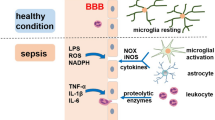
Numerous works address certain and new pathophysiological factors that are involved in cerebral dysfunction during sepsis, trying to understand its complexity. This is the exciting point of SAE, because new factors, resulting from sepsis, have been elucidated by researchers as causing or mediating SAE (Fig. 1). These pathophysiological factors are described below.

Sepsis is a systemic dysfunction caused by a host’s uncontrolled inflammatory response to an infection. Circulating proinflammatory mediators promote the expression of adhesion molecules in brain microvessel endothelial cells that facilitate the passage of inflammatory cells and proinflammatory cytokines to brain tissue. Inflammatory mediators cause cellular changes in brain tissue, such as the activation of astrocytes and microglia, which release pro-inflammatory cytokines. Neuroinflammation causes changes and damage to neural cells. The production of reactive oxygen species also contributes to brain tissue damage and injury. Injuries and damage to brain tissue during SAE are multifactorial, being accompanied and enhanced by changes in cerebral perfusion and reduced metabolism. BBB is the main selective barrier of neural tissue. During sepsis, encephalopathy can cause changes and dysfunctions in the integrity of the BBB, compromising its selectivity. These complications and interconnected processes, in addition to causing damage to the neural tissue, compromise synaptic interactions, causing cognitive and memory disorders, which can leave long-term sequelae
Pathophysiology of SAE
Neuroinflammation and Changes in Neural Cell Function and Signaling
Neuroinflammation is one of the main processes involved in SAE [20, 21], playing a crucial role in neural apoptosis and cognitive impairment during and after sepsis [22, 38]. The inflammatory process in SAE involves increased expression and release, by the activated cells of the immune system, of pro-inflammatory cytokines, such as nitric oxide, tumor necrosis factor alpha (TNF-α), and interleukins (IL) IL-1β and IL-6, increasing the inflammatory response [22, 44, 45]. In addition, circulating proinflammatory mediators promote the expression of adhesion molecules in brain endothelial cells of microvessels, which facilitate the passage of neurotoxic factors and inflammatory cells into the brain tissue [46]. The results of these phenomena are the activation of microglia, which acquires neurotoxic properties, more specifically by the release of nitric oxide, cytokines, reactive oxygen species, and glutamate, inducing neuronal injury and death in vulnerable brain areas [47].
Systemic inflammation also triggers the local production of cytokines. In the brain, this local production can cause neuroinflammation, which mediate neuronal dysfunction and, ultimately, cell death. In this scenario, TNF-α, IL-1β, and IL-6 seem to be the most relevant inflammatory mediators [24, 48]. Significant neuroinflammation induces neutrophil infiltration in brain tissue, activation of astrocytes through toll-like receptors, and overexpression of aquaporin 4. Moreover, it increases synthesis of prostaglandins and nitric oxide that activate the hypothalamus and adrenal axis [49,50,51]. This pro-inflammatory environment results in behavioral changes, fever, and severe neurological impairment, due to cerebral edema and neuronal apoptosis, contributing to both transitory and permanent cognitive dysfunction present in the survivors of sepsis [12, 16].
Alterations and activation of microglial and astroglial cells, orchestrated by the inflammatory process, are among the most relevant phenomena in SAE [20, 23,24,25]. The function of these cells is essential for cerebral homeostasis [23, 24]. They contribute to brain defense against infection by being closely involved in maintaining blood-brain and cerebrospinal fluid barriers [20, 24]. Also, astrocytes and microglia are involved in the modulation and maintenance of synapses, being closely related to the entire intracerebral communication network [20, 23, 24]. Therefore, the alterations in their activity can cause synaptic dysfunctions [24] and tissue injuries (apoptosis), affecting not only the structure of the organ but also its functions, such as memory, attention, cognition, and consciousness [20, 23, 25, 52]. Many researchers have raised the hypothesis that the activation of immune cells in the brain, and the entire inflammatory process of releasing pro-inflammatory cytokines, is crucial for the development of SAE [16, 53, 54].
The exact mechanism of acute brain dysfunction, caused by cerebral inflammation during sepsis, remains unclear. Nonetheless, a lot of efforts are being done to elucidate the role of this process in the development of SAE in order to find new treatments that can prevent the severe neurological injuries, dysfunctions, and deficits disorders in septic patients.
Oxidative Stress
Neuroinflammation increases according to metabolic and bioenergetic demands, resulting in oxidative stress and mitochondrial dysfunction [26, 27]. Mitochondrial dysfunction results in the production of reactive oxygen (ROS) and reactive nitrogen species (RNS) [27]. ROS includes not only oxygen-centered free radicals, such as superoxide (O2.) and hydroxyl (.OH) anions, but also some non-radical oxygen derivatives, such as hydrogen peroxide (H2O2). Peroxynitrite (ONOO−) and nitrogen dioxide (NO2.) are examples of RNS [54]. All those species can cause structural damage to the membrane and induce inflammation.
The formation of ONOO− by the scavenge of NO by ROS and RNS activates the inducible NO synthase, increasing the NO production and, hence, the yielding of more ONOO−. Donors and producers of NO and, subsequently, of ONOO−, induce a rapid decrease in oxygen consumption, inhibiting complexes I and IV of the electron transport chain in mitochondria and uncoupling oxidative phosphorylation, leading to neuronal bioenergetic failure [55,56,57]. NO reversibly inhibits mitochondrial respiration, competing with oxygen for the binding site in complex I, while ONOO− attenuates electron transport both in complex I [56, 58] and, to a lesser extent, in complex IV [55] through irreversible oxidative changes [56].
The increased presence of ONOO−, driven by NO and free radicals, within the brain can be critical for neuronal function in sepsis [56], leading to a clinical condition that affects glial cells, neurons, and the BBB structure, which contribute considerably to brain injury related to SAE [27, 59, 60]. Probably the apoptosis of neurons leads to SAE during sepsis, because its association with cognitive dysfunction in patients with sepsis has been well documented [61, 62].
The serious damage caused by oxidative stress during sepsis, including injury and cell death in the neural tissue, demonstrates the need for advances in research to better understand its triggering processes. Studies are needed in order to develop treatments and therapies to mitigate and perhaps to prevent oxidative stress and its effects on SAE, in an attempt to preserve the integrity of brain tissue during sepsis.
Alterations in Cerebral Perfusion and Metabolism
The glucose supply for the continuous energy production is more imperative in the brain than in any other organ [61]. The brain consumes more than 20% of the oxidative fuels supplied by the body, and, even though it has only 2% of the body’s weight, it receives a generous cardiac supply to get nutrients (glucose and oxygen) to maintain the metabolism (growth and function) of its cells [62]. The energy costs of the brain are mainly supported by adenosine triphosphate (ATP) derived from glucose oxidation, which is the primary energy substrate for the brain, although other substrates may contribute to the production of ATP, such as ketone bodies and lipids [63].
A recent research showed a significant reduction in cerebral glycolytic metabolism in C57BL6 mice submitted to an experimental model of severe sepsis [34]. Nonetheless, the pathophysiologic mechanism of this reduced cerebral glycolytic metabolism during sepsis remains to be elucidated. Some studies reported the presence of ischemia [17, 64, 65] and dysfunctions in cerebral perfusion and its microcirculation, leading to cerebral hypoperfusion [66]. The impaired cerebral microcirculation during sepsis can result in inadequate cerebral perfusion and may be related to electrophysiological abnormalities and neurological modifications [17, 67, 68]. These metabolic and hemodynamic reductions precede cognitive impairment and structural changes in the brain, such as atrophy of white and gray matters [31,32,33]. Thus, the changes in the blood flow, along with the release of inflammatory molecules, can represent a key component to trigger encephalopathy during the pathological process of sepsis and may be related to changes in glucose uptake by neural cells [17, 69].
The lack of maintenance in the metabolic rate of glucose has been related to cognitive impairment in patients with diabetes in Alzheimer’s disease [70, 71]. Likewise, other studies have shown that neurovascular dysfunction is highly associated with an accelerated decline in linguistic ability, verbal memory, attention, and visuospatial skills [72, 73]. Once the maintenance of adequate cerebral metabolic and vascular integrity is quite important to cognitive ability and mental health [69], a better understanding of brain metabolic processes, such as glucose metabolism, is essential for a thorough comprehension of the pathogenesis of sepsis and for the development of new therapeutic and adjuvant treatments to add to conventional treatments, both for sepsis and SAE.
Modifications in the BBB
The blood-brain has a key role in the control and maintenance of cerebral homeostasis and, hence, to maintain adequate neural function [29, 74, 75]. BBB plays a unique role as a highly selective biological interface between the brain and its periphery. The preservation of the BBB’s integrity maintains and protects the healthy brain function, largely orchestrated by the ion concentration gradients and the availability of nutrients [30]. Several studies support a regulatory role of the BBB in the progression of acute and chronic brain dysfunction [30].
Through mechanisms yet poorly understood, sepsis can induce acute and chronic changes in the central nervous system, particularly in the BBB [30, 69]. The production and exacerbated release of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6; endotoxins such as LPS; and mediators such as ROS and NO act on the brain barriers, changing cellular functions and leading to the rupture of homeostasis and the consequent increase in permeability [76,77,78]. In addition, these mediators cause the activation of MMPs, MMP-2, and MMP-9, in the BBB, and MMP-8, in the blood-cerebrospinal fluid barrier; these proteins act by breaking the junctions between the cells that make up the brain barriers, allowing even greater permeability. These inflammatory cascades and oxidative stress are responsible for structural changes in the BBB [29].
IL-1β, one of the main cytokines involved in this pathophysiology, induces the astrocyte-activated release of vascular endothelial growth factor A (VEGF-A) and thymidine phosphorylase (TYMP, or endothelial cell growth factor 1, ECGF1), which contribute to the negative regulation of expression of the tight junction proteins in brain endothelial cells, thus promoting the loss of integrity of the BBB [77]. At the same time, astrocytes can attenuate microglia activation through secretion of TGF-β, to detain the inflammatory process [78]. IL-1β is also recognized as a potent inducer of astrocyte response. Stimulation of astrocytes with IL-1β increases mRNA and protein expressions of TNF-α and IL-6 [79]. The constitutive astroglia overexpression of IL-6 is associated with neurodegeneration, astrogliosis, and BBB breakdown [24].
The understanding of the determinants of the integrity of the BBB during sepsis is quite important for the comprehension of SAE and for the development of new treatment options, aiming a positive prognosis. Thus, experimental studies have been focused on BBB as part of the research for short to long term therapeutic strategies. Advances in experimental studies, focused on the BBB as a strategy for SAE treatment, are of great importance and relevance in the search for therapies that inhibit BBB dysfunction, thus possibly inhibiting brain alterations and dysfunction during sepsis. The maintenance of the normal BBB function during sepsis is crucial to suppress inflammation, reduce mortality, and improve neurological outcomes in sepsis survivors, in addition to reducing long-term sequelae resulting in a better quality of life after sepsis.
Experimental Studies
The main focus of the experimental studies on SAE is related to understand the role of microglial and astroglial cells and the pathways to control neuroinflammation by modulating the function of these cells.
Michels et al. [23] conducted a study on an animal model of severe sepsis by cecal ligation puncture (CLP) in rats, to determine the effects of microglial depletion, by clodronate-liposomes, on the systemic and brain inflammatory response. An increase in pro-inflammatory cytokines was observed in animals submitted to CLP, as expected. Administration of clodronate decreased the density of microglia in the hippocampus and increased the proinflammatory cytokines even further and also increased the expression of CD11b. The CD11b integrin is a surface receptor expressed by monocytes, macrophages, neutrophils, dendritic cells, and subsets of B lymphocytes that bind to several ligands, including members of the ICAM family, and to the complement factor iC3b. It is involved in essential immune and pro-inflammatory processes, including leukocyte extravasation and phagocytosis. Therefore, increased expression of CD11b may potentiate the neural inflammatory process during sepsis [23, 80]. Microglial restocking, by doxycycline, was able to reverse brain and systemic cytokine levels. The study suggests that depletion of microglia, during the development of severe sepsis, may be associated with exacerbation of cerebral and systemic inflammation, whereas the microglial restocking is able to reverse this condition [23].
Neutrophils and monocytes are recruited by chemokines during the inflammatory process in sepsis, being the CCR2+ receptor as one of the main receptors responsible for the recruitment of inflammatory response cells to the brain [81, 82]. A recent study demonstrated, for the first time, that the recruitment of monocytes by CCR2+ to the brain causes microglial activation, playing an important role in the long-term cognitive impairment resulting from SAE [19]. In this study, beyond observing human patients, the researchers also induced severe sepsis in mice, by pneumonia caused by Streptococcus pneumoniae, which also triggered encephalopathy with long-term cognitive sequelae in animals. They demonstrated that controlling the recruitment of inflammatory monocytes by CCR2+ could reduce microglial activation and neuroinflammation, preventing signs of cognitive impairment after sepsis. They suggest that using the control of monocyte recruitment by CCR2+ as a therapeutic target during sepsis could represent a new clinical intervention strategy to prevent the development of sequels and long-term cognitive deficiencies resulting from SAE [19].
A key factor involved in inflammation is the NF-κB. During sepsis, the activation of NF-κB increases the expression of pro-inflammatory cytokines. Studies have shown that the expression of the silent information regulator 1 (SIRT1) can neutralize the activation of NF-κB [22, 83, 84]. SIRT1 is an important nicotinamide adenine dinucleotide (NAD+)–dependent protein lysine deacetylase which regulates stress responses, inflammation, apoptosis, and cellular senescence, and its activation exerts protection against neurological disorders such as ischemic stroke and neurodegenerative disorders [85, 86]. SIRT1 activation is intensively connected with the SIRT1-mediated deacetylation of downstream signaling proteins such as FOXO1, p53, and NF-κB, thus exerting anti-apoptotic, antioxidant, and anti-inflammatory actions [87,88,89]. A recent study evaluated the effect of attractylone (Atr), a plant sesquiterpene, in an LPS-induced sepsis model in mice. Atr promoted the expression of the SIRT1 and suppressed the expression of NF-κB, thus attenuating cognitive dysfunction caused by microglia activation and also reducing neural apoptosis and neural inflammatory factors caused by sepsis [22]. Another study evaluated the activation of SIRT1 using butein (3,4,2′, 4′-tetrahydroxychalcone), a plant flavonoid, which has been described acting as neuroprotective agent reducing inflammation and oxidative stress in neurons [85, 90, 91]. In a CLP mice sepsis model, the treatment with butein upregulated the SIRT1 levels, resulting in a significant reduction in the expression of IL-6, TNF-α, and IL-1β and in a decreasing in acetylated NF-kB, FOXO1, and p53 (Ac-NF-κβ, Ac-FOXO1, and Ac-p53) levels. The treatment with butein had a protective effect on BBB integrity, attenuating the inflammatory process and reducing oxidative stress, thus reducing neurological dysfunctions caused by SAE [92].
Pyroptosis is a recently discovered type of programmed cell death, which is performed by the GSDMD-NT, the N-terminal of gasdermin-D protein (GSDMD). GSDMD-NT can bind to the lipids of the cell’s inner membrane, forming pores in the plasma membrane, resulting in cell lysis and release of inflammatory mediators such as IL-1β [93, 94]. Inflammatory mediators cause a rapid change in the microglia phenotype in the brain, and the activated microglia produces inflammatory mediators [93]. A study investigated the effect of caspase-1 inhibition, by administration of caspase-1 inhibitor VX765, as a protective treatment for the brain, aiming to suppress GSDMD expression and its cleavated form GSDMD-NT, to reduce pyroptosis in the brain during sepsis, in a CLP-induced sepsis animal model [93]. Inhibited caspase-1 suppressed GSDMD expression and its form of GSDMD-NT cleavage, resulting in reduced pyroptosis in brain tissue. The inhibition of caspase-1 also reduced the expression of IL-1β, MCP-1, and TNF-α in serum and brain tissue and prevented the interruption of the blood-brain barrier induced by sepsis and damage to brain ultrastructure. The authors defend the hypothesis that inflammatory mediators circulating in the peripheral blood, as a result of sepsis, access the brain through the damaged BBB and trigger pyroptosis in the neural tissue. This process would be followed by activation of microglia and neural dysfunction, leading then to cognitive impairments. The study demonstrated that inhibition of caspase-1 protected the brain’s ultrastructure, especially BBB, drastically reducing pyroptosis and reducing the release of inflammatory cytokines, resulting in preservation of cognitive functions in CLP-induced experimental sepsis in mice [93].
Wang and colleagues [94] evaluated a pro-apoptotic mitochondrial serine protease involved in caspase-dependent cell death, Omi/HtrA2. They described a specific Omi/HtrA2 inhibitor, UCF-101, and investigated its effect on brain cell injury and death caused by oxidative stress due to SAE, in an animal model of CLP-induced sepsis. The levels of caspase-3, caspase-9, and PARP (poly [ADP-ribose] polymerase) in the hippocampus of CLP animals were higher than levels in control animals, whereas Omi/HtrA2 levels were slight increased in the hippocampus of septic animals, with a translocation of Omi/HtrA2 from mitochondria to the cytosol. The treatment with UCF-101 prevented the translocation of Omi/HtrA2 from the mitochondria to the cytosol, decreased the cleaved expression of caspase-3, caspase-9, and PARP, as well as reduced apoptosis. The inhibition of Omi/HtrA2 by UCF-101 also reduced the levels of inflammatory cytokines (IL-6 and TNF-α) and the oxidative stress (CAT, GSH, and MDA) in the hippocampus of septic animals. Taken together, these data indicate that Omi/HtrA2 regulates a mitochondria-dependent apoptotic pathway and suggests that the inhibition of Omi/HtrA2 by UCF-101 could result in neuroprotection by inhibiting the cytosolic translation of Omi/HtrA2 and antagonizing a caspase-dependent apoptosis pathway [94].
The treatment with intravenous immunoglobulin (IVIg) is an established modality for immunomodulation in neurological diseases [95], so it will be interesting to evaluate its use for brain protection to damage. Esen et al. [96] developed experimental tests with IVIg in an animal sepsis model. The results suggest that the administration of IVIg reduces brain damage caused by sepsis by inhibiting complement-mediated neuronal death. The findings indicate that IVIg can suppress the classic complement pathway, reducing C5a activity and the pro-apoptotic expressions of NF-κβ and Bax, thereby inhibiting large cascades of inflammation and apoptosis, reducing cell death by apoptosis, and, consequently, neuronal dysfunction and behavioral deficits [96].
The phosphatidylinositol-3-kinase (PI3K)/Akt pathway is a vital survival signaling pathway in neurons ([97]). A study reported that neuroglobin (Ngb) protects mice from the effects of SAE through a PI3K/Akt/Bax-dependent mechanism in vivo. Ngb is a species of globin expressed mainly in vertebrate neurons and works similarly to myoglobin, described by Burmester and colleagues [98]. Some studies have reported that Ngb has neuroprotective effects [99,100,101]. The study performed by Deng et al. [101] administered neuroglobin via intracerebroventricular injection of Ngb plasmids in rats with CLP-induced sepsis. The results showed that Ngb attenuated brain damage, by histological analysis of brain tissue, and demonstrated a protective effect on neurological dysfunction. The mechanism was observed by Western blot analyses, which demonstrated an increasing in Akt phosphorylation and a decreasing in the level of Bax protein, confirming the protective effect of Ngb in SAE through the PI3K/Akt/Bax-dependent mechanism [101].
During neuroinflammation, cytokine levels are high [22, 44, 45], which stimulate the formation of neurotoxic metabolites by the kynurenine (KYN) pathway [27, 102]. During inflammation, indoleamine 2,3-dioxigenase is activated in extrahepatic tissues, converting tryptophan to KYN [103]. This process is followed by the synthesis of enzymes that regulate the generation of neurotoxic metabolites, such as 3-hydroxykynurenine, 3-hydroxyanthranilic acid, and quinolinic acid, causing neurotoxicity through the formation of ROS [27]. Danielski et al. [27], in a study with a CLP-induced sepsis model in Wistar rats, evaluated the effect of vitamin B6 (vit B6) on survival, activation of the kynurenine pathway, acute neuroinflammation, and long-term cognitive dysfunction due to SAE. Vit B6 has anti-inflammatory and antioxidant properties and also acts as a cofactor for enzymes in the KYN pathway [104, 105]. They observed higher levels of tryptophan in septic animals treated with vit B6, when compared with septic animals without treatment, suggesting a reduction in the conversion of tryptophan to KYN. The results showed that vit B6 affects the activation of KYN pathway, being able to restore normal levels of tryptophan. The vit B6 administration led to an improvement in neurochemical and neuroinflammatory parameters, such as a reduction in IL-1β, IL-6, and TNF-α, especially in the hippocampus of septic animals that received the treatment. The administration of vitamin B6 also had a protective effect in preserving the integrity of the BBB. Consequently, there was a decrease in neuroinflammation and oxidative stress resulting from sepsis in brain tissue. Animals from the CLP group and CLP + vit B6 group were followed for 10 days to determine survival rate and cognitive functions. The CLP + vit B6 animals had better long-term memory and improved cognition when compared with animals with CLP alone. The results indicate that vitamin B6 has neuroprotective effects on the acute and long-term consequences of sepsis [27].
Bedirli and colleagues [28] evaluated the effects of sevoflurane, a volatile anesthetic, in the oxidative process and brain tissue injury during SAE, as well as on memory changes in an experimental model of CLP sepsis in rats. Sevoflurane treatment had a protective role against oxidative damage, significantly increasing the activities of GPx and SOD compared with CLP animals. The levels of IL-6, IL-1b, MDA, and of caspases 3, 8, and 9 were lower in the treated animals. Sevoflurane also decreased apoptosis in brain tissue and improved memory in this experimental model of CLP in rats. The study suggests that sevoflurane sedation in septic patients may have beneficial effects on sepsis-related brain damage and memory impairment [28].
Recently, our research group evaluated the effect of treatment with fructose-1,6-bisphosphate (FBP) on glucose metabolism and oxidative stress in brain tissue, in a model of severe sepsis in C57BL6 mice [106, 107]. Septic animals without treatment showed a strong and significant reduction in brain metabolism when compared with basal metabolism. Treatment with FBP had a significant protective effect on SAE, being able to preserve the cerebral glucose metabolism during severe sepsis, without significant changes when compared with the basal metabolism. FBP also reduced the activity of CAT and GPx in septic animals. These results suggest that fructose-1,6-bisphosphate may be a possible candidate for adjuvant treatment in SAE [34].
Plants used in traditional medicine, with anti-inflammatory properties, have been studied by researchers for SAE treatment. A study explored the neuroprotective effects of Ecballium elaterium (EE), a plant with anti-inflammatory properties from the cucurbitaceous family, during SAE in a CLP-induced sepsis model in rats [108, 109]. They reported a significant reduction in TNF-α levels in the brain of animals treated with EE. The treatment with EE also reduced neuronal damage, pericellular and perivascular edema, and infiltration of inflammatory cells in septic animals. These data suggest that EE contains components that have protective effects against SAE, reducing the accumulation of pro-inflammatory cytokines and attenuating the neural damage resulting from inflammation [108].
A fish oil (FO)-55–enriched lipid emulsion was orally administered to rats with CLP-induced sepsis to determine its effect as an important anti-inflammatory compound in brain dysfunction in septic rats. The lipid emulsion enriched with fish oil reduced BBB permeability in the prefrontal cortex and in the total cortex of septic rats, decreased levels of IL-1β and protein carbonylation in all brain structures, and also decreased activity of myeloperoxidase in the hippocampus and prefrontal cortex. The FO improved levels of neurotrophic factors derived from the brain in the hippocampus and prefrontal cortex and prevented cognitive impairment in the animals [109].
Conclusion
SAE’s pathophysiology is complex and multifactorial, combining intertwined processes, and is promoted by numerous changes and dysfunctions resulting from sepsis, such as inflammation, neuroinflammation, oxidative stress, reduced brain metabolism, and injuries to BBB integrity. These dysfunctions cause damage to the neural tissue, compromise synaptic interactions, leading to cognitive and memory disorders, in addition to leaving long-term sequelae. It is evident that in the absence of a precise understanding of its cause, the treatment is limited, such as patient sedation, being clearly unsatisfactory face to its complexity [19]. The variety of dysfunctions and also mental and brain symptoms have an impact on clinical strategies and may require different therapeutic approaches. Researchers are engaged in the task force of better understanding the trigger process and the aggravating factors, looking for new and effective approaches to treat and mitigate the effects of SAE during and after sepsis.
Data Availability
Not applicable.
Abbreviations
- SAE:
-
Sepsis-associated encephalopathy
- ICU:
-
Intensive care units
- CNS:
-
Central nervous system
- BBB:
-
Blood-brain barrier
- TNF-α:
-
Necrosis factor alpha
- IL:
-
Interleukins
- CSF:
-
Cerebrospinal fluid
- ROS:
-
Reactive oxygen species
- NO:
-
Nitric oxide
- RNS:
-
Reactive nitrogen species
- H2O2 :
-
Hydrogen peroxide
- O2 . :
-
Superoxide
- .OH:
-
Hydroxyl
- NO2 . :
-
Nitrogen dioxide
- ATP:
-
Adenosine triphosphate
- CLP:
-
Cecal ligation puncture procedure
- FBP:
-
Fructose-1,6-bisphosphate
- 18F-FDG:
-
18F-fluoro-2-deoxy-d-glucose
- GSDMD:
-
Gasdermin-D protein
- CAT:
-
Catalase
- GPx:
-
Glutathione peroxidase
- KYN:
-
Kynurenine
- SOD:
-
Superoxide dismutase
- EE:
-
Ecballium elaterium
- FBP:
-
Fructose-1,6-bisphosphate
- Ngb:
-
Neuroglobin
- FO:
-
Fish oil
- IVIg:
-
Immunoglobulin
References
Shankar-Hari M, Phillips GS, Levy ML, Seymour CW, Liu VX, Deutschman CS et al (2016) Developing a newdefinition and assessing newclinical criteria for Septic shock: for the third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA - J Am Med Assoc 315(8):775–787
Gotts JE, Matthay MA (2016) Sepsis: pathophysiology and clinical management. BMJ 353:1–20
Delano MJ, Ward PA (2016) Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest 126(1):23–31
Schulte W, Bernhagen J, Bucala R (2013) Cytokines in sepsis: potent immunoregulators and potential therapeutic targets - an updated view. Mediators Inflamm 2013.
Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM et al (2013) Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med 39(2):165–228
Shankar-Hari M, Phillips GS, Levy ML, Seymour CW, Liu VX, Deutschman CS et al (2016) Developing a new definition and assessing new clinical criteria for septic shock: for the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA - J Am Med Assoc 315(8):775–787
Cossart YE (2014) The rise and fall of infectious diseases: Australian perspectives, 1914-2014. Med J Aust 201(1 Suppl):11–14
Nedeva C, Menassa J, Puthalakath H (2019) Sepsis: inflammation is a necessary evil. Front Cell Dev Biol 7(JUN):1–12.
Fleischmann C, Scherag A, Adhikari NKJ, Hartog CS, Tsaganos T, Schlattmann P et al (2016) Assessment of global incidence and mortality of hospital-treated sepsis current estimates and limitations. Am J Respir Crit Care Med 193(3):259–272
Wang TL (2014) Prolonged neuroinflammation after lipopolysaccharide exposure in aged rats. PLoS One 9(8)
Michelon C, Michels M, Abatti M, Vieira A, Borges H, Dominguini D et al (2020) The role of secretase pathway in long-term brain inflammation and cognitive impairment in an animal model of severe sepsis. Mol Neurobiol 57(2):1159–1169
Sankowski R, Mader S, Valdés-Ferrer SI (2015) Systemic inflammation and the brain: novel roles of genetic, molecular, and environmental cues as drivers of neurodegeneration. Front Cell Neurosci 9(FEB):1–20.
Widmann CN, Heneka MT (2014) Long-term cerebral consequences of sepsis. Lancet Neurol 13(6):630–636
Sonneville R, Verdonk F, Rauturier C, Klein IF, Wolff M, Annane D (2013) Understanding brain dysfunction in sepsis. Ann Intensive Care 3(Figure 1):15.
Kozlov AV, Bahrami S, Redl H, Szabo C (2017) Alterations in nitric oxide homeostasis during traumatic brain injury. Biochim Biophys Acta - Mol Basis Dis 1863(10):2627–2632
Robba C, Crippa IA, Taccone FS (2018) Septic encephalopathy. Curr Neurol Neurosci Rep 18(82)
Taccone FS, Scolletta S, Franchi F, Donadello K, Oddo M (2013) Brain perfusion in sepsis. Curr Vasc Pharm 11(2):170–186
Young GB (2013) Encephalopathy of infection and systemic inflammation. J Clin Neurophysiol 30(5):454–461
Andonegui G, Zelinski EL, Schubert CL, Knight D, Craig LA, Winston BW et al (2018) Targeting inflammatory monocytes in sepsis-associated encephalopathy and long-term cognitive impairment. JCI insight 3(9):1–20
Mazeraud A, Pascal Q, Verdonk F, Heming N, Chrétien F, Sharshar T (2016) Neuroanatomy and physiology of brain dysfunction in sepsis. Clin Chest Med 37(2):333–345
Adam N, Kandelman S, Mantz J (2013) Sepsis-induced brain dysfunction. Expert Rev Anti Infect Ther 11(2):211–221
Tian M, Qingzhen L, Zhiyang Y, Chunlong C, Jiao D, Zhang L et al (2019) Attractylone attenuates sepsis-associated encephalopathy and cognitive dysfunction by inhibiting microglial activation and neuroinflammation. J Cell Biochem 120(5):7101–7108
Michels M, Ávila P, Pescador B, Vieira A, Abatti M, Cucker L et al (2019) Microglial cells depletion increases inflammation and modifies microglial phenotypes in an animal model of severe sepsis. Mol Neurobiol 56(11):7296–7304
Shulyatnikova T, Verkhratsky A (2019) Astroglia in sepsis associated encephalopathy. Neurochem Res 45(1):83–99
Hoogland ICM, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D (2015) Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflammation 12(1):1–13
Lemstra AW, Cm J, Hoozemans JJM, Van Haastert ES, Rozemuller AJM, Eikelenboom P et al (2007) Microglia activation in sepsis: a case-control study. J Neuroinflammation 15(4):1–8
Danielski LG, Giustina A Della, Goldim MP, Florentino D, Mathias K, Garbossa L, et al. (2018) Vitamin B6 reduces neurochemical and long-term cognitive alterations after polymicrobial sepsis: involvement of the kynurenine pathway modulation. Mol Neurobiol 55(6):5255–5268
Bedirli N, Bagriacik EU, Yilmaz G, Ozkose Z, Kavutçu M, Cavunt Bayraktar A et al (2018) Sevoflurane exerts brain-protective effects against sepsis-associated encephalopathy and memory impairment through caspase 3/9 and Bax/Bcl signaling pathway in a rat model of sepsis. J Int Med Res 46(7):2828–2842
Danielski LG, Giustina A Della, Badawy M, Barichello T, Quevedo J, Dal-Pizzol F, et al. (2018) Brain barrier breakdown as a cause and consequence of neuroinflammation in sepsis. Mol Neurobiol 55(2):1045–1053
Nwafor DC, Brichacek AL, Mohammad AS, Griffith J, Lucke-Wold BP, Benkovic SA et al (2019) Targeting the blood-brain barrier to prevent sepsis-associated cognitive impairment. J Cent Nerv Syst Dis 11:117957351984065
Bookheimer SY, Strojwas MH et al (2000) Patterns of brain activation in people at risk for Alzheimer’s disease. N Eng J Med 343(7):450–456
Reiman EM, Caselli RJ, Chen K, Alexander GE, Bandy D, Frost J (2001) Declining brain activity in cognitively normal apolipoprotein epsilon 4 heterozygotes: a foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer’s disease. Proc Natl Acad Sci USA 98(6)
Cunnane S, Ph D, Nugent S, Sc B, Roy M, Sc M, et al. (2011) Brain fuel metabolism , aging , and Alzheimer’s disease. Nutrition 27(1):3–20.
Catarina A V, Luft C, Greggio S, Venturin GT, Ferreira F, Marques EP, et al. (2018) Fructose-1,6-bisphosphate preserves glucose metabolism integrity and reduces reactive oxygen species in the brain during experimental sepsis. Brain Res 1698:54–61.
Dantzer R, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56
Garcia J, Kimeldorf DJ, Koelling RA (1955) Conditioned aversion to saccharin resulting from exposure to gamma radiation. Science 122:157–158
Gamache FW Jr DT (1982) Alterations in neurological function in head-injured patients experiencing major episodes of sepsis. Neurosurgery 10(2):468–472.
Calsavara AJC, Nobre V, Barichello T, Teixeira AL (2018) Post-sepsis cognitive impairment and associated risk factors: a systematic review. Aust Crit Care 31(4):242–253
Ebersoldt M, Sharshar T, Annane D (2007) Sepsis-associated delirium. Intensive Care Med 33(6):941–950
Leon A, Lepousé C, Floch T, Graftieaux J (2006) Agression cérébrale au cours du sepsis sévère Brain injury during severe sepsis. Ann Fr Anesth Reanim 25(8):863–867
Dittus RS, Thomason JWW, Jackson JC, Shintani ÃAK, Ely EW, Mph à (2006) Delirium and its motoric subtypes: a study of 614 critically ill patients. J Am Geriatr Soc 54(3):479–484.
Barichello T, Sayana P, Giridharan VV, Arumanayagam AS, Narendran B, Giustina A Della, et al. (2019) Long-term cognitive outcomes after sepsis: a translational systematic review. Mol Neurobiol 56(1):186–251
Semmler A, Widmann CN, Okulla T, Urbach H, Kaiser M, Widman G, et al. (2013) Persistent cognitive impairment , hippocampal atrophy and EEG changes in sepsis survivors. J Neurol Neurosurg Psychiatry 84(1):62–69.
Li W, Wang Y, Wang X, He Z, Liu F, Zhi W et al (2016) Esculin attenuates endotoxin shock induced by lipopolysaccharide in mouse and NO production in vitro through inhibition of NF-κB activation. Eur J Pharmacol 791(76):726–734
Qin X, Jiang X, Jiang X, Wang Y, Miao Z, He W (2016) Micheliolide inhibits LPS-induced inflammatory response and protects mice from LPS challenge. Sci Rep :1–13.
He H, Geng T, Chen P, Wang M, Hu J, Kang L et al (2016) OPEN NK cells promote neutrophil recruitment in the brain during sepsis-induced neuroinflammation. Sci Rep 6:1–14
Sharshar T, Gray F, Lorin G, Grandmaison D, Hopkinson NS, Ross E et al (2003) Mechanisms of disease Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet 362(9398):1799–1805
Alexander JJ, Jacob A et al (2008) TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1. Neurochem int 52(3):447–456
Rorato R, Menezes AM, Giusti-paiva A, De Castro M (2009) Prostaglandin mediates endotoxaemia-induced hypophagia by activation of pro-opiomelanocortin and corticotrophin-releasing factor neurons in rats. Exp Physiol 94(3):371–379
Rump K, Adamzik M (2018) Function of aquaporins in sepsis: a systematic review. Cell Biosci 8:1–7
Takatani Y, Ono K (2018) Inducible nitric oxide synthase during the late phase of sepsis is associated with hypothermia and immune cell migration. Lab Investig 98(5):629–639
Cunningham C, Maclullich AMJ (2013) At the extreme end of the psychoneuroimmunological spectrum: delirium as a maladaptive sickness behaviour response. Brain Behav Immun 28:1–13
Westhoff D, Engelen-Lee JY, Hoogland ICM, Aronica EMA, Van Westerloo DJ, Van De Beek D et al (2019) Systemic infection and microglia activation: a prospective postmortem study in sepsis patients. Immun Ageing 16(1):1–10
Halliwell B (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97(6):1634–1658
Brookes PS, Bolan JP, Y SJRH (1999) The assumption that nitric oxide inhibits mitochondrial ATP synthesis is correct. FEBS Lett 446(2–3):261–263.
Berg RMG, Møller K, Bailey DM (2011) Neuro-oxidative-nitrosative stress in sepsis. J Cereb Blood Flow Metab 31(7):1532–1544
Cassina A, Radi R (1996) Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys 328(2):309–316
Tang G, Yang H, Chen J, Shi M, Ge L (2017) Metformin ameliorates sepsis-induced brain injury by inhibiting apoptosis, oxidative stress and neuroinflammation via the PI3K/Akt signaling pathway. Oncotarget 8(58):97977–97989.
Zhu S, Huang W, Huang L, Han Y, Han Q (2016) Huperzine A protects sepsis associated encephalopathy by promoting the deficient cholinergic nervous function. Neurosci Lett 631:70–78
Semmler A, Okulla T, Sastre M, Dumitrescu-ozimek L, Heneka MT (2005) Systemic inflammation induces apoptosis with variable vulnerability of different brain regions. J Chem Neuroanat 30:144–157
Brain T The expensive-tissue. 36(2).
Tsacopoulos M, Magistretti PJ (1996) Metabolic coupling between glia and neurons. J Neurosci. 16(3):877–885
Shulman RG, Hyder F, Rothman DL (2001) Cerebral energetics and the glycogen shunt: neurochemical basis of functional imaging. Proc Natl Acad Sci USA 98(11):6417–6422
Gofton TE, Young GB (2012) Sepsis-associated encephalopathy. Nat Rev Neurol 8(October):557–566
Oddo M, Taccone FS (2015) How to monitor the brain in septic patients? Minerva Anestesiol 81(7):776–788
Taccone FS, Castanares-Zapater, et al. (2010) Cerebral autoregulation is influenced by carbon dioxide levels in patients with septic shock. Neurocrit Care. 12(1):35–42
Semmler A, Hermann S (2008) Sepsis causes neuroinflammation and concomitant decrease of cerebral metabolism. J Neuroinflammation 5:38
Everson-Rose SA, Ryan JP (2015) Diabetes, obesity, and the brain: new developments in biobehavioral medicine. Psychosom Med 77(6):612–615
Lin A-L, Parikh I, Hoffman JD, Ma D (2017) Neuroimaging biomarkers of caloric restriction on brain metabolic and vascular functions. Curr Nutr Rep 6(1):41–48
Bangen KJ, Beiser A, Delano-Wood L, Nation DA, Lamar M, Libon DJ et al (2013) APOE genotype modifies the relationship between midlife vascular risk factors and later cognitive decline. J Stroke Cerebrovasc Dis 22(8):1361–1369
Cunnane S, Nugent S, Roy M, Courchesne-Loyer A, Croteau E, Tremblay S et al (2012) Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 27(1):3–20
Keaney J, Campbell M (2015) The dynamic blood–brain barrier. FEBS J 282(21):4067–4079.
Engelhardt B, Sorokin L (2009) The blood–brain and the blood–cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol 31(4):497–511.
Weighardt H, Holzmann B (2008) Role of Toll-like receptor responses for sepsis pathogenesis. Immunobiology 212(9–10):715–722
Janeway CA, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20(2):197–216
Junior CAJ (2001) How the immune system protects the host from infection. Microbes Infect 3(13):1167–1171
Chapouly C, Argaw AT, Horng S, Castro K, Zhang J, Asp L, et al. (2015) Astrocytic TYMP and VEGFA drive blood–brain barrier opening in inflammatory central nervous system lesions. Brain 138(6):1548–1567.
Vincent VAM, Tilders FJH, Dam AVAN (1997) Inhibition of endotoxin-induced nitric oxide synthase production in microglial cells by the presence of astroglial cells: a role for transforming growth factor. Glia 19(3):190–198.
Lee SC (1993) Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 beta. J Immunol 150(7):2659–2667
Geissmann F, Markus G, Manz SJ, Sieweke MH, Merad M, Ley K (2010) Development of monocytes, macrophages, and dendritic cells. Science 327(5966):656–661
Opal SM, Ellis JL, Suri V, Freudenberg JM, Vlasuk GP, Li Y et al (2016) Pharmacological SIRT1 activation improves mortality and markedly alters transcriptional profiles that accompany experimental sepsis. Shock 45(4):411–418
Zhao L, An R, Yang Y, Yang X, Liu H, Yue L et al (2015) Melatonin alleviates brain injury in mice subjected to cecal ligation and puncture via attenuating in fl ammation, apoptosis, and oxidative stress: the role of SIRT1 signaling. J Pineal Res 59(2):230–239
Hernández-Jiménez M, Hurtado O, Cuartero MI, Ballesteros I, Moraga A, Pradillo JM et al (2013) Silent information regulator 1 protects the brain against cerebral ischemic damage. Stroke 44(8):2333–2337
Bai XZ, He T, Gao JX, Liu Y, Liu JQ, Han SC et al (2016) Melatonin prevents acute kidney injury in severely burned rats via the activation of SIRT1. Sci Rep 6(August):1–13
Zhu Y, Wang K, Ma Z, Liu D, Yang Y, Sun M et al (2019) SIRT1 activation by butein attenuates sepsis-induced brain injury in mice subjected to cecal ligation and puncture via alleviating inflammatory and oxidative stress. Toxicol Appl Pharmacol 363:34–46
Lee D, Jeong G (2016) Butein provides neuroprotective and anti-neuroin fl ammatory effects through expression by activating the PI3K/Akt pathway Tables of Links. Br J Pharmacol 173(19):2894–2909.
Padmavathi G, Kishor N, Bordoloi D, Arfuso F, Mishra S, Sethi G, et al. (2017) Phytomedicine butein in health and disease: a comprehensive review. Phytomedicine 25:118–127.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H et al (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 562:660–665
Kayagaki N, Stowe IB, Lee BL, Rourke KO, Anderson K, Warming S et al (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526:666–671
Khan M, Shah SA, Kim MO (2018) 17β-estradiol via SIRT1/acetyl-p53/NF-kB signaling pathway rescued postnatal rat brain against acute ethanol intoxication. Mol Neurobiol 55(4):3067–3078
Kou D-Q, Jiang Y-L, Qin J-H, Huang Y-H (2017) Magnolol attenuates the inflammation and apoptosis through the activation of SIRT1 in experimental stroke rats. Pharmacol Rep 69(4):642–647
Al. P. BA. Z et (2016) Orexin activation counteracts decreases in nonexercise activity thermogenesis (NEAT) caused by high fat diet. Physiol Behav 176(1):139–148.
Xu X e., Liu L, Wang Y chang, Wang C tao, Zheng Q, Liu Q xin, et al. (2019) Caspase-1 inhibitor exerts brain-protective effects against sepsis-associated encephalopathy and cognitive impairments in a mouse model of sepsis. Brain Behav Immun 80(January):859–870
Wang P, Hu Y, Yao D, Li Y (2018) Omi/HtrA2 regulates a mitochondria-dependent apoptotic pathway in a murine model of septic encephalopathy. Cell Physiol Biochem 49(6):2163–2173
Lacroix-desmazes S, Kazatchkine MD, Kaveri SV (2005) Intravenous immunoglobulin in neurological disorders: a mechanistic perspective. J Neurol 252:1–6
Esen F, Ozcan PE, Tuzun E, Boone MD (2018) Mechanisms of action of intravenous immunoglobulin in septic encephalopathy. Rev Neurosci 29(4):417–423
Brunet A, Datta SR, Greenberg ME (2001) Transcription-dependent and -independent control of neuronal survival by the PI3K–Akt signaling pathway. Curr Opin Neurobiol 11(3):297–305.
Burmester T, Weich B, Reinhardt S (2000) A vertebrate globin expressed in the brain. 407(September):1998–2001.
Raida Z, Hundahl CA, Nyengaard JR, Hay-schmidt A (2013) Neuroglobin over expressing mice: expression pattern and effect on brain ischemic infarct size. PLoS One 8(10):e 76565.
Zhang LN, Ai YH, Gong H, Guo QL, Huang L, Liu ZY YB (2014) Expression and role of neuroglobin in rats with sepsis-associated encephalopathy*. Crit Care Med 42(1):e 12-21.
Deng S, Ai Y, Gong H, Chen C, Peng Q, Huang L et al (2017) Neuroglobin protects rats from sepsis-associated encephalopathy via a PI3K/Akt/Bax-dependent mechanism. J Mol Neurosci 63(1):1–8
Campbell BM, Charych E, Lee AW, Möller T, Dantzer R (2014) Kynurenines in CNS disease: regulation by inflammatory cytokines. Front Neurosci 8(February):1–22
Maddison DC, Giorgini F (2015) The kynurenine pathway and neurodegenerative disease. Semin Cell Dev Biol 40:134–141
Bordignon Nunes F, Simões Pires MG, Alves Filho JCF, Wächter PH, De Oliveira JR (2002) Physiopathological studies in septic rats and the use of fructose 1,6-bisphosphate as cellular protection. Crit Care Med 30(9):2069–2074
Pedrazza L, Lunardelli A, Luft C, Cruz CU, De Mesquita FC, Bitencourt S et al (2014) Mesenchymal stem cells decrease splenocytes apoptosis in a sepsis experimental model. Inflamm Res 63(9):719–728
Moataz E, El B, Chalupov M, Pra G, Suchý P (2015) Hepatoprotective and proapoptotic effect of Ecballium elaterium on CCl4-induced hepatotoxicity in rats. Asian Pac J Trop Med 8(7):526–531
Uslu C, Karasen RM, Sahin F (2006) Effect of aqueous extracts of Ecballium elaterium rich, in the rabbit model of rhinosinusitis. Int J Pediatr Otorhinolaryngol 70(3):515–518
Arslan D, Ekinci A, Arici A, Bozdemir E, Akil E, Ozdemir HH (2017) Effects of Ecballium elaterium on brain in a rat model of sepsis-associated encephalopathy. Libyan J Med 12(1)
Sc DM, Della A, Ph G, Pereira M, Ph G, Eduarda M et al (2020) Fish oil À rich lipid emulsion modulates neuroin fl ammation and prevents long-term cognitive dysfunction after sepsis. Nutrition 70:1–9
Funding
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—finance code 001.
Author information
Authors and Affiliations
Contributions
All authors have been involved in drafting the manuscript or revising it critically for important intellectual content.
Corresponding author
Ethics declarations
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Consent to Participate
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Catarina, A.V., Branchini, G., Bettoni, L. et al. Sepsis-Associated Encephalopathy: from Pathophysiology to Progress in Experimental Studies. Mol Neurobiol 58, 2770–2779 (2021). https://doi.org/10.1007/s12035-021-02303-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02303-2