
Abstract
Identification of molecular mechanisms underlying early-stage Alzheimer’s disease (AD) is important for the development of new therapies against and diagnosis of AD. In this study, gas chromatography time-of-flight mass spectrometry (GC-TOF-MS)-based metabolomics approach was employed to investigate the metabolic profiles in plasma and brain tissues harvested from 5-month-old APP/PS1 transgenic mice and their wildtype counterparts. Since different brain regions were expected to have their own distinct metabolic signals, four different brain regions, namely cortex, hippocampus, midbrain and cerebellum tissues, were dissected and had their metabolic profiles studied separately. Biochemical assays were also performed on plasma and brain cortex tissue of transgenic mice and wildtype mice, with a focus on mitochondrial health. Amyloid precursor protein and amyloid-β levels in plasma, brain cortex tissue and mitochondria fractions isolated from brain cortex were measured to assess the amyloid pathology. Our findings include the observation of extensive metabolic alterations in cortex and cerebellum of APP/PS1 mice, but not in their hippocampus, midbrain and plasma. The major pathways affected in cortex and cerebellum of APP/PS1 mice were closely related to impaired energy metabolism and perturbation of amino acid metabolism in these mice. APP/PS1 mice also exhibited higher amyloid-β40 and amyloid-β42 in their cortex, accumulation of mitochondria APP in their cortex, and presented an altered oxidative state in their brain. Treatment with the peroxisome proliferator-activated receptor gamma (PPARγ) agonist pioglitazone (PIO) successfully restored the energy metabolism, lowered amyloid-β levels and afforded the APP/PS1 mice a better antioxidative capacity in their cortex.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a progressive, irreversible neurodegenerative disorder that affects large areas of the cerebral cortex and hippocampus [1]. The disease is characterised by two key protein abnormalities, namely cerebral plaques laden with insoluble amyloid-β (Aβ) peptides and intraneuronal neurofibrillary tangles made up of hyperphosphorylated tau protein [2]. These amyloid plaques and neurofibrillary tangles spread through the brain as the disease progresses [3]. Because Aβ plays such a crucial role in the disease, it is little wonder that many pharmaceutical companies developing disease-modifying treatments for AD have chosen to focus on anti-Aβ therapeutics [4]. However, the evidence supporting amyloid-lowering approaches as clinically viable are waning. Several large phase 3 trials of anti-amyloid approaches in patients with mild-to-moderate AD have been published, albeit with disappointing results [5, 6]. Therefore, targeting Aβ alone may not be enough to achieve positive clinical outcomes, especially during late stages of the disease [7].
In order to unravel the pathological changes underlying AD pathogenesis and identify the drug targets that can effectively prevent or delay the disease progression, many researchers have recently been focusing their efforts on investigating and characterising early-stage AD phenotypes [8, 9]. Reliable early-stage AD diagnostic tools can also assist clinicians and researchers to position drug interventions at the right temporal therapeutic window for effective detection of treatment effects in AD clinical trials [10]. Therefore, tools that can facilitate investigation of early-stage AD would prove to be invaluable, and metabolic profiling presents itself as a promising approach in this aspect [11, 12]. Defined as the quantitative measurement of the dynamic multiparametric metabolic response of living systems to pathophysiological stimuli or genetic modification [13], metabolic profiling offers distinct advantages over other “-omics” approaches. Firstly, owing to their low molecular weights, metabolites are structurally conserved across species [14], unlike protein and gene expressions which usually involve different isoforms that often carry notably different properties. Secondly, since the metabolome lies downstream of genomes, transcriptomes and proteomes, metabolic signals have been shown to be amplified [15]. This allows metabolic profiling approach to detect subtle fluctuations in the system, and therefore represents a more sensitive platform for probing into early disease stages, as well as for detecting and monitoring the therapeutic response to drug treatment.
Many studies have shown that abnormalities in cerebral glucose metabolism are an early feature of AD [16, 17], suggesting that impaired energy metabolism in the brain may play an early and proximal, even if not the primary, role in the pathogenesis and progression of AD [18]. Because the brain relies almost exclusively on glucose as a substrate for energy production, cerebral function is directly dependent on the availability of glucose and its use. In the brain, free energy required to drive most cellular reactions is derived from phosphorylation of adenosine triphosphate (ATP), which is mostly produced in the mitochondria from the aerobic oxidation of glucose [19]. The peroxisome proliferator-activated receptors (PPARs) are a subfamily of nuclear receptors which serve as ligand-modulated transcription factors that regulate gene-expression programs of metabolic pathways [20]. Specifically, the isoform PPARγ is the most extensively studied and is well-documented to coordinate lipid, glucose and energy metabolism [21]. Moreover, it has been proposed as a potential therapeutic target to rescue mitochondrial function in neurological diseases [22]. The PPARγ agonist pioglitazone (PIO) has been studied extensively for AD therapy in both preclinical and clinical studies [23,24,25,26,27,28]. Despite substantial engagement of PPARγ agonists in AD clinical trials, the therapeutic effects of PPARγ agonists in the brain are still not fully understood [29]. Since metabolic profiling technique can potentially be used as a valuable tool to better understand the mechanisms of action of therapeutic compounds [30], we postulate that the metabolic profiling approach could be employed as a powerful study platform to elucidate the therapeutic effects of PPARγ agonists such as PIO in treating AD. Once validated, such findings will provide useful insights into the mechanisms of action for this class of compounds in AD therapy.
Applied on a carefully chosen disease model, metabolic profiling strategies could produce comprehensive data that allow further hypotheses to be generated and tested using the same model. One of the more commonly used disease models in AD research is the amyloid precursor protein (APP) expressing model, where cells (in vitro model) or animals (in vivo model, often mice are used as models) possessing the APP transgene would overexpress the transmembrane protein APP, proteolytic processing of which will lead to accumulation of neurotoxic amyloid-β deposits. To generate a more aggressive APP model, another transgene, presenilin-1 (PS1) could be added to amplify the toxicity induced by APP. PS1 mutation is responsible for increasing expression of PS1, a critical catalytic subunit which regulate the proteolytic activity of γ-secretase. Together with the increased expression of APP, the enhanced activity of γ-secretase will lead to accelerated generation of neurotoxic amyloid-β, therefore producing observable AD phenotypes in the disease models at a much earlier time point [31]. The APP/PS1 double transgenic mouse model has been widely used in the field of AD research to understand the disease better [32], and has also been used routinely as an in vivo screening model for therapeutic compounds [33].
In this study, we employed a gas chromatography time-of-flight mass spectrometry (GC-TOF-MS) setup to perform metabolic profiling on C57BL/6 mice that carry the APP and PS1 transgenes, with a similar focus on early-stage disease phenotypes. A thorough search among published literatures revealed relatively fewer studies that employed GC-MS-based metabolic profiling approach to study AD, as compared to studies that used NMR-based metabolic profiling technique. There is also a lack of study that employed GC-MS-based metabolic profiling technique to look at metabolic changes in different brain regions in an AD model [34]. On top of that, most GC-MS-based metabolic profiling studies utilised GC-quadrupole-MS [34, 35], which in general has a lower sensitivity than GC-TOF-MS due to its compromised duty cycle [36], and our TOF-MS could be more superior to a quadrupole-MS when employed in a non-targeted metabolomics setting. Therefore, a GC-TOF-MS-based metabolic profiling of an appropriately chosen APP model could generate useful findings that will complement existing knowledge and other NMR-based AD metabolic profiling studies to allow for a better understanding of AD pathophysiology. Utilising the GC-TOF-MS-based metabolic profiling approach, we also investigated the ability of PIO in minimising or reversing the metabolic alterations observed in APP/PS1 transgenic mice. Selected biochemical assays were then performed to follow up on findings generated from metabolic data in this study. Accumulation of APP in cortical mitochondria was also assessed in this study, since mitochondrial APP accumulation was found to be one of the main early-stage AD phenotypes observed in an in vitro APP model [37]. On top of that, we also attempted to examine the oxidative state in APP/PS1 transgenic mice by measuring activities of two major antioxidative enzymes, namely superoxide dismutase (SOD) and catalase, in their cortex and plasma tissue and compare against activities observed in non-transgenic wildtype mice.
Materials and Methods
Chemicals
A 2% methoxyamine hydrochloride in pyridine (MOX reagent) and N-methyl-N-(trimethylsilyl) trifluoroacetamide (MSTFA) with 1% trimethylchlorosilane (TMCS) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). PIO was purchased from Cell Molecular Pharmaceutical R&D (Xi’an, China). All other reagents used were of analytical grades.
Animal Husbandry
APP/PS1 transgenic male and female breeders were used to generate breeding colony to obtain APP/PS1 transgenic mice for experiments in this study. Transgenic breeders were generous gifts from Professor Gavin S. Dawe’s research group (National University of Singapore, Singapore). The mice were paired for breeding in separate cages and pups were weaned 4 weeks after birth. Mouse tail genotyping was used to confirm the presence of APP and PS1 transgene in offspring. To carry out the genotyping, PureLink Genomic DNA Mini kit (Life Technologies, Carlsbad, CA, USA) was first used to extract DNA from sampled mouse tails. DNA extractions were then subjected to PCR amplification with primers (separately for APP, PS1 and internal control) and GoTaq Green Master Mix (Promega, Madison, WI, USA). Mixtures were then resolved by DNA gel electrophoresis, whereas gel imaging was done using Molecular Imager Gel Doc XR and analysed using Image Lab Software (Bio-Rad, Hercules, CA, USA). APP/PS1 transgenic mice intended for experiments were housed in groups (maximum of 5 mice per cage) under standard conditions of humidity, temperature and 12-h light/dark cycle with ad libitum access to food and water. All mice were maintained under constant conditions for 4 days prior to experiments. All animal handling protocols were carried out in accordance with Singapore National Advisory Committee on Laboratory Animal Research (NACLAR) guidelines and approved by Institutional Animal care and Use Committee (IACUC), National University of Singapore.
Animal Experiment and Sample Collection
A total of 10 male APP/PS1 transgenic mice (18 weeks old) were split into two treatment groups (n = 5), where one group received PIO treatment and the other group received vehicle treatment as disease control. Another 5 male wildtype mice (18 weeks old, taken from the same breeding colony) were also given vehicle treatment and they serve as healthy control in this experiment. PIO was dissolved in DMSO and diluted 20 times with 1% (w/v) methyl cellulose to be administered via oral gavage (p.o.). PIO treatment was given as single p.o. administration every day for 14 days, and dosage used in this study was 30 mg/kg of PIO. Vehicle-treated APP/PS1 and wildtype mice were also given vehicle treatment for 14 days. Twenty-four hours after the last dose of the 14-day treatment regimen, all mice (20 weeks old) were sacrificed by CO2 euthanization and their blood samples were collected via cardiac puncture into eppendorf tubes supplemented with heparin. Transcardial perfusion with saline was then performed on the sacrificed mice to remove traces of blood from their organs before whole brain tissues were carefully removed from their skulls. Immediately after the whole brains were collected, different brain parts (cortex, hippocampus, cerebellum and midbrain) were dissected and transferred into separate clean eppendorf tubes. All biological samples were kept on ice immediately after collection. Blood samples were centrifuged at 4000g for 10 min and plasma samples were then aliquoted from the supernatants into clean eppendorf tubes. All plasma and brain part samples were stored at − 80 °C until analysis.
Sample Preparation for GC-TOF-MS-Based Metabolic Profiling Analysis
Plasma and brain part samples were thawed and kept on ice before sample preparation. About 20–25 mg of brain part samples (cortex, hippocampus, cerebellum and midbrain) were transferred into new eppendorf tubes, and 10 μL/mg of Milli-Q water was added to each tube containing the brain part sample before they were lysed with bead homogeniser. One hundred fifty microlitres of each tissue homogenate was then transferred into new tubes for subsequent sample preparation. One hundred microlitres of plasma samples were used without dilution for sample preparation. To prepare biological samples for GC-TOF-MS analyses, 950 μL of chilled methanol (containing 50 μM of myristic-d27 acid as internal standard) was first added to plasma and brain part homogenate samples for protein precipitation. All mixtures were vortex-mixed at high speed for 5 min, followed by centrifugation (14,000g) for 20 min at 4 °C to pellet the precipitated protein. For each sample, 900 μL of clear supernatant was transferred into pre-silanized glass tubes for drying and then evaporated to dryness at 50 °C under a gentle stream of nitrogen gas using TurboVap nitrogen evaporator (Calliper Life Science, Hopkinton, MA, USA). To ensure complete elimination of water which might interfere with the subsequent sample preparation steps, 100 μL of anhydrous toluene (stored with sodium sulphate) was added to each dry residue, vortex-mixed for 1 min and dried again at 50 °C under nitrogen gas. After completion of the drying process, 50 μL of MOX reagent was added to each dried extract, vortex-mixed for 2 min and incubated at 60 °C for 2 h as a methoximation step. Following the methoximation step, derivatisation of metabolites was then initiated by adding 100 μL of MSTFA (with 1% TMCS) to each sample, vortex-mixed for 2 min and incubated at 60 °C for 1 h. This step aimed to increase volatility and stability of metabolites present in the samples. Following the completion of derivatisation step, each sample was vortex-mixed again for 2 min and centrifuged at 3000g at room temperature for 5 min to spin down any insoluble solids. Supernatants were then carefully transferred into GC autosampler vials for subsequent injection into the GC-TOF-MS analytical instrument.
GC-TOF-MS Data Acquisition and Preprocessing
Brain part and plasma samples were analysed in batches separated according to the types of samples. Within each batch for one particular sample type, sample injections were randomised to minimise procedural artefact during subsequent analyses. Data acquisition for GC-TOF-MS analyses were carried out using an Agilent 7890A Gas Chromatography (Agilent Technologies, Santa Clara, CA) coupled to PEGASUS 4D Time-of-Flight Mass Spectrometer TOF-MS (LecoCorp., St. Joseph, MI). A DB-1 GC column (Agilent Technologies) with an altered length of 22.9 m (internal diameter of 250 μm and film thickness of 0.25 μm) was used as the primary column. The secondary column was removed and settings for secondary column were disabled. Helium was used as the carrier gas to facilitate separation at a constant flow rate of 1.5 mL/min. Injector split ratio was set to 1:10 and a total injection volume of 1 μL was used. The injector inlet and ion source temperatures were both maintained at 250 °C throughout the entire GC run. Oven temperature gradient was programmed as follows: firstly, the oven temperature was set to equilibrate at 70 °C for 0.5 min before sample injection was initiated; upon sample injection, oven temperature was kept at 70 °C for another 0.2 min and then ramped up at a rate 8 °C/min to 270 °C where it was held at that temperature for 5 min; finally, oven temperature was further increased by 40 °C/min to reach 310 °C and held for another 5 min. The MS detection was operated in EI mode (standard ionisation energy 70 eV) and detector voltage was set at 1800 V. Data were acquired using full scan mode, with mass range set at m/z 50–600, and an acquisition rate of 15 Hz was employed. Acquisition delay was set at 195 s to prevent the large solvent front peak from entering the mass analyser. GC chromatogram data acquisition, baseline correction, peak deconvolution, analyte alignment, peak area calculation as well as preliminary analyte identification by mass spectral searches (based on NIST, Fiehn Rtx5 and our own in-house libraries built using previous analyses of commercially available metabolite standards) were all performed using LECO ChromaTOF software version 4.21 before subjecting the metabolic data to multivariate data analysis. For preliminary metabolic peak identification, peaks with similarity index (SI) of 70% or more were assigned putative metabolite identities based on the mass spectral libraries matches. To ensure proper matching between sample spectrum and library spectrum, mass spectral from both sample peak and library peak were visually inspected, and retention time (RT) of libraries were checked using a mix of fatty acid methyl esters peak markers that were injected before sample injections. As for integration of area under each metabolite peak, baseline offset, minimum peak width, signal to noise (S/N) ratio and number of apexing masses were set at 0.5, 2.5 s, 100 and 3, respectively. Calculation of the area under peak for each metabolite was performed based on the unique mass selected from mass spectrum detected for each metabolic peak. Metabolic data matrix was generated using LECO ChromaTOF’s calibration feature as described in previous literature [38], with similarity threshold between analyte peak and reference peak set at 70% instead of 60%. The resulting metabolic data were processed by normalising peak area of each analyte based on peak area of myristic-d27 acid in their own respective sample, followed by another normalisation using total integral area of all included metabolic peaks. Normalised metabolic peak areas were then constructed into a data matrix for subsequent multivariate data analysis.
Multivariate Data Analysis of Metabolic Data Matrix
Normalised data was subjected to multivariate data analysis (SIMCA-P software version 13.0, Umetrics, Umeå, Sweden) with separate analyses carried for different types of brain part samples (plasma, cortex, hippocampus, cerebellum and midbrain). Data were first mean-centred and unit-variance scaled before being subjected to principal component analysis (PCA). The purpose of PCA was to observe clustering trends among analysed samples, as well as to identify outliers in the data and exclude them with sufficient justifications based on observation of gross errors that might have been introduced during sample handling or GC-TOF-MS analytical runs. After the initial data overview using PCA, metabolic data were further subjected to partial least squares discriminant analysis (PLS-DA) to build a discriminant model, with one built separately for plasma and each type of brain part sample. Model validity and potential overfitting of both PLS-DA models were checked by performing 100 observation-dependent randomised permutation tests and visually inspected using model validation plot. As a rule of thumb, model validation plot for this permutation test is considered valid when (1) none of the cumulative Q2 values calculated for permuted datasets is higher than Q2 value of original dataset, and (12) the regression line of Q2 values of original and permuted datasets trend towards zero (or negative values) when the correlation between original dataset and permuted datasets is decreasing. Once PLS-DA model passed model validation step, the same dataset was then analysed using orthogonal partial least squares discriminant analysis (OPLS-DA), which affords a more straightforward identification of discriminant metabolites responsible for differentiation between APP/PS1 transgenic mice and wildtype mice (healthy control). To select from OPLS-DA model a list of potential discriminant metabolites for further analysis, variable importance in projection (VIP) cut-off value was set to 1.00 to generate a list of potential discriminant metabolites that could have contributed significantly to the separation between APP/PS1 and wildtype mice in the OPLS-DA model. To determine significance of difference in metabolite levels between APP/PS1 and wildtype mice, two-tailed independent t test with Welch’s correction was used for statistical comparison of these potential discriminant metabolites between the two groups and Bonferroni-adjusted P value was used to determine significance. Discriminant metabolites that have VIP ≥ 1.00 and P value lower than Bonferroni-adjusted significance levels were deemed to have contributed significantly to the differentiation of metabolic profiles between APP/PS1 and wildtype mice. Fold-change (FC) value for each discriminant metabolite with respect to the healthy control was calculated using the following Eq. 1, where x and y represent average metabolite levels calculated for APP/PS1 transgenic mice and non-transgenic wildtype mice, respectively.
Fold-change values were used to assess the degree of alterations for each discriminant metabolite. To interpret the biological meaning behind the observed metabolic alterations, information regarding discriminant metabolites was sourced from freely available online metabolite databases such as Human Metabolome Database (HMDB) [39] for information on the metabolite itself, Kyoto Encyclopedia of Genes and Genomes database (KEGG) [40] for metabolic pathway interpretations and relevant published literature.
Evaluating PIO’s Therapeutic Effects on Discriminant Metabolites
To assess the therapeutic potential of PIO on metabolic perturbation in plasma and different brain parts of APP/PS1 transgenic mice, we examined PIO’s effects on the list of discriminant metabolites identified from multivariate data analysis. Discriminant metabolites for APP/PS1 mice were regarded as being normalised or “treated” if their levels in PIO-treated transgenic mice were altered significantly when compared against vehicle-treated transgenic mice, and the direction of change is heading towards metabolite levels observed in non-transgenic wildtype mice (healthy control). Since different types of samples (plasma, cortex, hippocampus, cerebellum and midbrain) have their own list of discriminant metabolites, PIO’s treatment effects on each list were assessed separately. Statistical comparison of data for evaluation of treatment effects was performed using two-tailed independent t test with Welch’s correction, and P value < 0.05 will be defined as being “treated” to guide the discussion in this paper.
Measurement of Amyloid-β40 and Amyloid-β42 Levels in Cortex and Plasma
Amyloid-β40 and 42 levels in cortex and plasma samples of vehicle-treated APP/PS1 mice, PIO-treated APP/PS1 mice and non-transgenic wildtype mice were quantitatively measured using colorimetric sandwich ELISA kits, according to the protocols provided by the manufacturer (#SIG-38954 and #SIG-38956 from Covance, Princeton, NJ). Cortex tissue samples from all mice were homogenised with PBS supplemented with 1× protease inhibitor cocktail in a ratio of 1:10 before loading into coated plates that came with the ELISA kits. Plasma samples were diluted 30 times with PBS for their amyloid-β40 and 42 measurements to be taken using this method. Total protein analysis measured using micro BCA protein assay kit (#23235 from Pierce Biotechnology, Rockford, IL) indicated that cortex tissue harvested from APP/PS1 mice consistently displayed higher level of total protein expression than non-transgenic wildtype mice. Therefore, non-normalised cortical amyloid-β40 and 42 data were used for comparison between the three groups of mice, and data were made comparable by loading equal tissue mass into plate before measurements were taken. All absorbance readings were measured using Tecan Infinite M200 microplate reader (Tecan, Switzerland). Data means were statistically compared using two-tailed independent t test with Welch’s correction, with significance level set at P < 0.05.
Measurement of APP Levels in Cortex and Cortical Mitochondrial Fractions
APP levels were assessed in both cortex samples and mitochondria fractions extracted from cortex tissue of APP/PS1 mice, APP/PS1 mice treated with PIO and non-transgenic wildtype mice. Cortex tissue samples were homogenised in a ration of 1:10 with PBS supplemented with 1× protease inhibitor cocktail. To extract mitochondria fractions, cortex tissue samples were first homogenised on ice using a pre-chilled dounce homogeniser and differential centrifugation procedure detailed in a commercially available kit was used for mitochondria extraction (#89874 from Thermo Fisher Scientific, Waltham, MA). It should be noted that, following the manufacturer’s recommendation, mitochondrial pellets were obtained using centrifugation at 3000g (instead of 12,000g) to improve purity of extraction. To assess the purity of mitochondrial fraction, a western blot analysis was used to detect for presence of different organelles in extracted fractions and compared against whole tissue homogenate. Presence of different organelles was measured using antibody cocktail suitable for organelle detection (ab133989 from Abcam, Cambridge, UK). Antibodies targeting four different organelle markers were used in the western blot analysis, namely anti-sodium potassium ATPase antibody (plasma membrane), anti-ATP5A antibody (mitochondria), anti-GAPDH antibody (cytosol) and anti-histone H3 antibody (nucleus). Following extraction, the purified mitochondrial fractions were further homogenised using a probe sonicator to completely lyse the mitochondria for analysis of APP using colorimetric sandwich ELISA kit (#KHB0051 from Life Technologies, Carlsbad, CA). Cortical APP data were not normalised for the same reason stated in previous section, whereas mitochondrial APP data were normalised based on total protein level present in each mitochondrial fraction (measured using micro BCA protein assay kit). All absorbance readings were measured using Tecan Infinite M200 microplate reader. Means between different groups were compared using two-tailed independent t test with Welch’s correction, with P < 0.05 to be considered as a significant mean difference.
Measurement of Lactate Dehydrogenase (LDH) and Citrate Synthase Activities
Comparison between metabolic data for APP/PS1 and non-transgenic wildtype mice showed that two major discriminant metabolites between these two groups are citric acid and lactic acid (results shown below). Among these two discriminant metabolites, PIO exerted treatment effect on lactic acid but not citric acid. Therefore, we assessed the activities of LDH and citrate synthase in cortex samples taken from APP/PS1 mice, PIO-treated APP/PS1 mice and non-transgenic wildtype mice. LDH activities were measured using a commercially available colorimetric assay kit (#K726-500, BioVision, Milpitas, CA, USA), where LDH’s ability to reduce NAD to NADH was assessed in each sample, and colorimetric probe was used to measure the activity of this reaction. Citrate synthase activities in cortex samples harvested from all three groups of mice were also measured using a commercially available kit (#K318-100, BioVision, Milpitas, CA, USA), where citrate synthase activities for all samples were measured based on their ability to convert acetyl-CoA and oxaloacetate into intermediates which would then develop into coloured products for detection. All absorbance readings were measured using Tecan Infinite M200 microplate reader. Means between different groups were compared using two-tailed independent t test with Welch’s correction, with P < 0.05 to be considered as a significant mean difference.
Measurement of SOD and Catalase Activities
Since oxidative stress is one pathological phenotype common in most AD models, we attempted to assess the oxidative capacities in cortex and plasma samples of vehicle-treated APP/PS1 mice and wildtype mice as well as PIO-treated APP/PS1 mice by measuring activities of two major antioxidative enzymes. A commercially available kit (#706002, Cayman Chemical, Ann Arbor, MI, USA) was used to measure SOD activity, where a mixture of xanthine oxidase and hypoxanthine was used to generate superoxide radicals which would be scavenged by SOD present in samples. Unreacted superoxide radicals would react with tetrazolium salt to form a detectable coloured dye, thus producing an inverse relationship between amounts of coloured dye formed and SOD activities in samples. Catalase activity was also assessed using a commercially available kit (#707002, Cayman Chemical, Ann Arbor, MI, USA), where reaction of catalase with methanol in presence of H2O2 to produce formaldehyde was monitored colorimetrically using a triazole dye. All absorbance readings were measured using Tecan Infinite M200 microplate reader. Means between different groups were compared using two-tailed independent t test with Welch’s correction, with P < 0.05 to be considered as a significant mean difference.
Results
GC-TOF-MS-Based Metabolic Profiling of Plasma Samples
PCA for plasma samples did not display any visible clustering trend on scores plot, suggesting only slight or no difference between the plasma metabolites from the two groups of mice (Fig. 1a). A PLS-DA model generated using the same metabolic data did not pass the model validation plot as randomly permuted datasets gave higher Q2 values than the Q2 value calculated for original dataset (Fig. 1b), which indicates high risk of model over-fitting and little interpretability for the dataset. OPLS-DA model constructed using the same data is presented in Fig. 1c (1-predictive and 1-orthogonal components, R2(Y) and Q2(cum) were 0.932 and 0.194, respectively). The small Q2(cum) value indicates that separation of plasma metabolic profiles between the two groups of mice was not clear. Nevertheless, a list of 25 potential discriminant metabolites (VIP ≥ 1.00) was generated based on the OPLS-DA model (data not shown), and only two metabolites (l-threonine and l-valine) have P < 0.05. Interestingly, one of them (l-threonine, + 28.6%) has P value lower than Bonferroni-adjusted significance level (0.002).

(a) PCA of plasma samples from wildtype and APP/PS1 transgenic mice; (b) Y-permuted model validation plot for PLS-DA of same dataset; (c) OPLS-DA of their plasma metabolic profiles (R2(Y) = 0.932, Q2(cum) = 0.194)
GC-TOF-MS-Based Metabolic Profiling of Cortex Samples
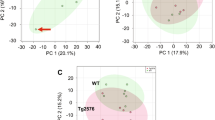
PCA for metabolic profiles in cortex samples displayed a clear clustering between APP/PS1 mice and non-transgenic wildtype mice (Fig. 2a). Model validation plot for PLS-DA model generated using the same dataset showed a valid model, as all Q2 values calculated for randomly permuted datasets were lower than actual Q2 value itself, and the regression line of Q2 values intersected y-axis below zero (Fig. 2b). OPLS-DA model constructed using the same data is presented in Fig. 2c (1-predictive and 1-orthogonal components, R2(Y) and Q2(cum) were 0.986 and 0.863, respectively). The high Q2(cum) value indicates that separation of cortical metabolic profiles between the two groups of mice was robust. Based on this OPLS-DA model, a list of 22 potential discriminant metabolites (VIP ≥ 1.00) that contributed to separation was identified and presented in Table 1. Ten out of these 22 metabolites achieved significance when means were compared between APP/PS1 and non-transgenic wildtype mice, as determined by P < 0.0023 (Bonferroni-adjusted significance level).

(a) PCA of cortex metabolic profiles for wildtype and APP/PS1 transgenic mice; (b) Y-permuted model validation plot for PLS-DA of cortex metabolic data; (c) OPLS-DA of cortex metabolic data (R2(Y) = 0.986, Q2(cum) = 0.863)
GC-TOF-MS-Based Metabolic Profiling of Hippocampus Samples
Similar to cortical metabolic profiles, PCA for hippocampus samples displayed a clear grouping that separates APP/PS1 mice from non-transgenic wildtype mice (Fig. 3a). Y-permuted validation plot shows that the PLS-DA model built using this dataset is valid (Fig. 3b). OPLS-DA model (1-predictive and 1-orthogonal components) constructed using the same data gave R2(Y) and Q2(cum) values of 0.986 and 0.914, respectively, which indicate that the separation between APP/PS1 mice and wildtype mice based on their hippocampal metabolic profiles is significant (Fig. 3c). Based on this OPLS-DA model, a list of 21 potential discriminant metabolites (VIP ≥ 1.00) that contributed to separation was identified and presented in Table 2. Only 1 out of these 21 metabolites achieved significance when means were compared between APP/PS1 and non-transgenic wildtype mice, as determined by P < 0.0024 (Bonferroni-adjusted significance level).

(a) PCA of hippocampus metabolic profiles for wildtype and APP/PS1 transgenic mice; (b) Y-permuted model validation plot for PLS-DA of hippocampus metabolic data; (c) OPLS-DA of hippocampus metabolic data (R2(Y) = 0.986, Q2(cum) = 0.914)
GC-TOF-MS-Based Metabolic Profiling of Cerebellum Samples
PCA for metabolic profiles of cerebellum samples also showed a clear separation between APP/PS1 mice and non-transgenic wildtype mice (Fig. 4a). Y-permuted validation plot shows that the PLS-DA model built using this dataset is valid (Fig. 4b). OPLS-DA model (1-predictive and 1-orthogonal components) constructed using the same data showed a robust separation between the two groups of mice, as reflected by their R2(Y) and Q2(cum) values of 0.966 and 0.818, respectively (Fig. 4c). Based on this OPLS-DA model, a list of 16 potential discriminant metabolites (VIP ≥ 1.00) that contributed to separation was identified and presented in Table 3. Five out of these 16 metabolites achieved significance when means were compared between APP/PS1 and non-transgenic wildtype mice, as determined by P < 0.003 (Bonferroni-adjusted significance level).

(a) PCA of cerebellum metabolic profiles for wildtype and APP/PS1 transgenic mice; (b) Y-permuted model validation plot for PLS-DA of cerebellum metabolic data; (c) OPLS-DA of cerebellum metabolic data (R2(Y) = 0.966, Q2(cum) = 0.818)
GC-TOF-MS-Based Metabolic Profiling of Midbrain Samples
Similar to our observations for the preceding three brain regions, PCA for midbrain samples produced a distinct clustering trend that separates APP/PS1 mice from non-transgenic wildtype mice (Fig. 5a). Validity of PLS-DA model for this metabolic dataset was checked using Y-permuted validation plot (Fig. 5b), and OPLS-DA model (1-predictive and 1-orthogonal components) constructed using the same dataset gave R2(Y) and Q2(cum) values of 0.939 and 0.680, respectively, which indicate that there is also a separation between midbrain metabolic profiles of APP/PS1 mice and wildtype mice (Fig. 5c). A list of 18 potential discriminant metabolites (VIP ≥ 1.00) was constructed based on this OPLS-DA model, and 3 out of these 18 metabolites achieved significance as determined by P < 0.0028 (Bonferroni-adjusted significance level). The list of discriminant metabolites for midbrain samples is summarised in Table 4.

(a) PCA of midbrain metabolic profiles for wildtype and APP/PS1 transgenic mice; (b) Y-permuted model validation plot for PLS-DA of midbrain metabolic data; (c) OPLS-DA of midbrain metabolic data (R2(Y) = 0.939, Q2(cum) = 0.680)
Evaluating PIO’s Therapeutic Effects on Discriminant Metabolites
To assess therapeutic potential of PIO in the AD model used in this study, we investigated PIO’s ability to alleviate or reverse the discriminant metabolites identified in plasma and different brain parts of APP/PS1 mice. PIO was observed to exert no therapeutic effect on discriminant metabolites that were identified in plasma (l-threonine), hippocampus tissue (lactic acid) and midbrain tissue (urea and uracil). On the other hand, PIO successfully treated 4 out of 10 discriminant metabolites (lactic acid, l-valine, glucose and glucose-6-phosphate) which characterised cortex tissue of APP/PS1 mice. On top of that, out of the 5 discriminant metabolites that differentiated cerebellum tissue of APP/PS1 mice from their wildtype counterparts, PIO successfully treated 4 of them (ribose-5-posphate, glucose-6-phosphate, l-glutamic acid and N-acetyl-l-aspartic acid). It is also worth noting that among the “treated” discriminant metabolites, those that are associated with energy metabolism (lactic acid, glucose, glucose-6-phosphate and ribose-5-phosphate) were completely rescued by treating APP/PS1 mice with PIO. Treatment effects exerted by PIO on discriminant metabolites are graphically summarised in Fig. 6.

(a) Discriminant metabolites in cortex tissue that were treated by PIO administration; (b) discriminant metabolites in cerebellum tissue that were treated by PIO administration
Measurement of Amyloid-β (40 and 42) Levels in Cortex and Plasma
Amyloid-β40 and amyloid-β42 levels in plasma of APP/PS1 mice and their non-transgenic wildtype counterparts did not show any significant difference, and PIO treatment had no effect on both amyloid-β in plasma of APP/PS1 mice. On the other hand, amyloid-β40 was observed to increase by 19% in cortex tissue of APP/PS1 mice as compared to non-transgenic wildtype mice, which was reversed by PIO treatment. Remarkably, amyloid-β42 levels in cortex tissue of APP/PS1 mice increased by as much as 364% when compared against non-transgenic wildtype mice. The increase in cortical amyloid-β42 was partially reduced by PIO treatment in the transgenic mice. All data for amyloid-β40 and amyloid-β42 in cortex and plasma samples are graphically summarised in Fig. 7.

(a) Amyloid-β40 levels in cortex samples; (b) Amyloid-β40 levels in plasma samples; (c) amyloid-β42 levels in cortex samples; (d) Amyloid-β42 levels in plasma samples; *P < 0.05 when compared against non-transgenic wildtype mice; **P < 0.05 when compared against non-treated APP/PS1 mice; all error bars represent one SD
Measurement of APP Levels in Cortex and Cortical Mitochondria
In this study, we measured the amount of APP present in cortex tissue and mitochondrial extracts of cortex tissue taken from APP/PS1 mice, PIO-treated APP/PS1 mice and non-transgenic wildtype mice. Cortical APP was observed to be higher in APP/PS1 mice by 33%, although this increase did not achieve significance. Interestingly, PIO treatment significantly reduced the cortical APP in APP/PS1 mice (Fig. 8a). APP was also observed to have accumulated significantly (+ 31%) in mitochondria extracted from cortex tissue of APP/PS1 mice. However, PIO treatment did not have any obvious effect on the accumulation of mitochondrial APP in transgenic mice (Fig. 8b). To ensure that there was no contaminant of plasma membrane (a considerable source of APP) in the mitochondrial extracts, a western blot analysis was used to assess the purity of extracted mitochondria fractions. Extracted mitochondrial fractions for cortex tissue showed an intense band at 55 kDa (mitochondrial marker) and a very faint band at 15 kDa (nuclear marker), whereas analysis of tissue homogenate gave intense bands at both 100 kDa (plasma marker) and 55 kDa (mitochondrial marker) as well as a faint band 15 kDa (nuclear marker) (Fig. 8c). Our western blot analysis demonstrated excellent mitochondrial purity in extracted fractions and ruled out the possibility of contamination from plasma membrane APP in mitochondrial APP measurements.

(a) APP levels in cortex samples harvested from all three groups of mice; (b) APP levels in extracted mitochondrial fractions; *P < 0.05 when compared against non-transgenic wildtype mice; **P < 0.05 when compared against non-treated APP/PS1 mice; all error bars represent one SD; (c) Western blot analysis of 4 organelle markers (100 kDa, plasma membrane; 55 kDa, mitochondria; 36 kDa, cytosol; 15 kDa, nucleus) performed on extracted mitochondrial fractions and cortex tissue homogenate
Measurement of LDH and Citrate Synthase Activities
Citrate synthase activities in cortex samples of all three groups of mice (APP/PS1 mice, PIO-treated APP/PS1 mice and non-transgenic wildtype mice) did not show any significant difference. On the other hand, activities of LDH were observed to have decreased significantly in cortex tissue of APP/PS1 mice when compared against their non-transgenic wildtype mice, and PIO treatment was observed to have reversed this reduction in LDH activities in APP/PS1 mice. Data for measurement of LDH activities in cortex samples from all three groups of mice are graphically summarised in Fig. 9.

LDH activities in cortex samples taken from all three groups of mice; *P < 0.05 when compared against non-transgenic wildtype mice; **P < 0.05 when compared against non-treated APP/PS1 mice; all error bars represent one SD
Measurement of SOD and Catalase Activities
SOD activities were observed to have dropped significantly in cortex samples of APP/PS1 mice when compared against wildtype mice, and PIO treatment successfully reversed this reduction of SOD activities in cortex tissue (Fig. 10a). There was no reduction in SOD activities in plasma samples, but PIO treatment was observed to have an up-regulating effect on SOD activities in plasma of APP/PS1 mice (Fig. 10b). On the other hand, no significant difference could be observed in catalase activities in cortex tissue of all three groups of mice (Fig. 10c), while catalase activities were observed to have increased significantly in plasma of APP/PS1 mice, which was also completely rescued by PIO treatment in the transgenic mice (Fig. 10d).

(a) SOD activities in cortex samples taken from all three groups of mice; (b) SOD activities in plasma samples; (c) catalase activities in cortex samples; (d) catalase activities in plasma samples; *P < 0.05 when compared against non-transgenic wildtype mice; **P < 0.05 when compared against non-treated APP/PS1 mice; all error bars represent one SD
Discussion
To capture the disease phenotypes associated with early-stage AD in this in vivo AD model, the sampling time point had to be carefully chosen to take a biological snapshot that allows us to meaningfully interpret our findings based on this snapshot. The sampling time point that is the most suitable for this study is where AD in these mouse models had progressed enough to elicit pathophysiological alterations when compared against non-transgenic wildtype mice, but had not progressed so much that these mice started to suffer from build-up of Aβ plaques in their brain tissue, which is associated more with moderate to late-stage AD. In this study, we sacrificed the APP/PS1 mice and harvested biological samples for investigation when they were 20 weeks old, which is a time that is slightly before these mice start to develop sparse amyloid plaques in their brain tissue [41, 42]. By placing our investigation in this time window, we hoped to capture early-stage AD signals in this model before amyloid plaques wreak havoc in their brains, which would then be heavily laden with extensive oxidative damages and neuronal cell death.
Metabolic Profiling of Plasma and Brain Tissue Samples
To gather a more comprehensive metabolic data using biological samples harvested from APP/PS1 transgenic mice and wildtype mice, we studied the metabolic profiles of plasma and four different brain parts (cortex, hippocampus, cerebellum and midbrain) of APP/PS1 mice and compared their metabolic profiles against non-transgenic wildtype mice. PCA of plasma metabolic profiles did not show any clear sign of separation between these two groups of mice, and the subsequent OPLS-DA did not produce any strong separation between them. This suggests that plasma in APP/PS1 mice at 20 weeks of age is not a sample matrix that could yield a lot of useful information about the pathological processes occurring in this disease model. Previously, a longitudinal study had reported it was more difficult to distinguish the plasma metabolome of APP/PS1 mice from their wildtype counterparts even at 6 months old [43]. In fact, metabolic markers detected in peripheral plasma of this transgenic model were usually reported in animals at ≥ 6 months of age [44, 45]. Female APP/PS1 mice were also reported to exhibit more intensive metabolic alterations in their plasma as compared to male AD and wildtype mice [46]. Nevertheless, we proceeded with mean comparison between the metabolites with VIP ≥ 1.00, which surprisingly gave one discriminant metabolite that differentiated plasma profiles of APP/PS1 mice from wildtype mice significantly (Bonferroni-adjusted). l-threonine was observed to be significantly higher in plasma of APP/PS1 mice, which was inversely related to level of l-threonine in their brains where lower levels of l-threonine were seen in cortex tissues of APP/PS1 mice. This indicates a clear compartmentalisation of metabolic activities between brain parts and plasma. It was reported that comparably fewer metabolites were observed in APP/PS1 mouse plasma than in brain extracts [47], which is not unexpected as brain is the immediate site of AD. Moreover, metabolic flux occurring in the brain was found to precede that of the peripheral plasma [43]. Higher plasma level of l-threonine had also been reported in AD patients before [48], but no clear explanation was given to explain this observation. While the GC-MS-based approach employed in our study tend to focus on lower molecular weight metabolites, lipid-based biomarkers had also been identified in the plasma of this animal model using liquid chromatography-mass spectrometry (LC-MS) analytical platform [49].
When sampling and analysing brain tissues harvested from APP/PS1 mice, we made the additional effort to look into metabolic profiles in different brain regions separately, as it had been reported that different brain regions exhibit distinct metabolic signals of their own when placed under stresses of APP-induced or age-induced toxicities [50, 51]. In this study, we investigated the metabolic alterations in four brain regions of APP/PS1 mice, namely cortex, hippocampus, cerebellum and midbrain. As expected, we observed different metabolic alterations in these four brain parts, with cortex tissue being the most informational among these four as judged by the highest number of discriminant metabolites observed in cortex tissue (citric acid, glycine, inosine, l-threonine, lactic acid, l-tyrosine, l-valine, glucose, l-aspartic acid and glucose-6-phosphate). Cerebellum and midbrain sample matrices also yielded considerable metabolic information, with cerebellum giving 5 discriminant metabolites (ribose-5-phosphate, glucose-6-phosphate, l-glutamic acid, N-acetyl-l-aspartic acid and l-aspartic acid) and midbrain giving three discriminant metabolites (urea, uracil and l-tyrosine). Another brain part that we have investigated is the hippocampus, but this particular sample matrix only produced one discriminant metabolite (lactic acid) in our analysis, suggesting that the metabolic alterations in this particular brain region may not be extensive in this mouse model at the sampling time point employed in this study (20 weeks of age). Another study that looked into metabolic changes in hippocampus tissue of APP/PS1 mice (16 weeks old) reported the P values for their top 30 discriminant metabolites they detected using univariate analysis [34]. However, none of them achieved significance if Bonferroni correction for multiple testing was applied on this set of 30 metabolites, suggesting that the metabolic changes were very limited in the hippocampus tissue of APP/PS1 mice, which is in agreement with our observations. Moreover, the extent of alterations in metabolic signatures revealed by PLS-DA was smaller in male APP/PS1 mice compared to female mice [34], suggesting that metabolic alterations in the hippocampus may be more obvious if female mice were used for our study instead. Although hippocampus was one of the main brain regions affected in AD, this was not reflected in APP/PS1 transgenic mice sampled at the time point used in this study. Studies which employed APP/PS1 mice at 6 months of age were able to detect lower molecular weight metabolites as potential markers for discrimination between transgenic mice and non-transgenic controls [52, 53], although it should be noted that both male and female animals were used in these studies. This highlights the caveats of employing a disease model to understand a disease, and caution should be exercised when interpreting the data generated based on this disease model.
Impaired Energy Metabolism in Cortex and Cerebellum of APP/PS1 Mice
Analyses of metabolic data harvested from cortex, cerebellum and midbrain tissues generated some interesting findings. Among the discriminant metabolites that described cortex tissue of APP/PS1 mice, compounds that are closely linked in the energy metabolism and glycolysis pathway (KEGG, map00010), namely lactic acid, glucose and glucose-6-phosphate were observed to have increased considerably. In cerebellum of APP/PS1 mice, glucose-6-phosphate and ribose-5-phosphate also shot up significantly. The higher levels of monosaccharides observed in cortex and cerebellum of APP/PS1 mice were strikingly similar to the observations in our previously published in vitro study, whereby an excessively high level of extracellular glucose was detected for an AD cell model [37]. On the other hand, citric acid, another component closely associated with energy metabolism, was found to have dropped by as much as 35% in the cortex tissue of APP/PS1 mice when compared against wildtype control. An opposite trend was observed for citric acid in in vitro metabolic profiling experiments [37], but this could be due to the very dissimilar physiological conditions between interstitial fluid and culture medium. This illustrates clearly the differences between in vitro and in vivo experiments, highlighting the caveats when carrying out data interpretation for such experiments. Increased lactic acid in cortex of APP/PS1 mice could be explained by the reduction in their cortical LDH activities, which was measured in this study using LDH assay that assessed their rates of converting NAD+ to NADH. This observation was well-corroborated by another study which reported increased lactic acid in cortex of APP/PS1 mice which were harvested at 9 months of age [54], and by another study which reported high brain lactic acid as a biomarker for ageing [55]. Taken altogether, there is a clear signal of impaired energy metabolism in cortex tissue of 20-week-old APP/PS1 mice, making them a good model for therapeutic intervention which focuses on these particular brain region and disease mechanism.
Dysregulated Amino Acid Metabolism in Cortex and Cerebellum of APP/PS1 Mice
Looking at amino acids (glycine, l-threonine, l-tyrosine, l-valine and l-aspartic acid) that discriminated cortex tissue of APP/PS1 mice against wildtype control, we noticed that they were all lowered in APP/PS1 mice. This trend was similar to the observations in our previous in vitro metabolic profiling experiments [37]. Both could be similarly explained by the possibility of brain cells (or AD cell model) using up amino acids as compensatory fuel to sustain growth, since they all suffered from impaired energy metabolism. Among these amino acids, glycine and l-threonine are closely linked in the glycine, serine and threonine metabolism pathway (KEGG, map00260). Reduction in both glycine and l-threonine indicates that this pathway was severely affected by the APP and PS1 transgenes. It should be noted that l-serine was detected in this study, but their levels were not significantly different between cortex tissue of APP/PS1 and wildtype mice. Interestingly, we observed an inverse relation between l-aspartic acid and N-acetyl-l-aspartic acid, two discriminant metabolites detected in cerebellum tissue of APP/PS1 mice. l-aspartic acid was observed to have decreased in cerebellum of APP/PS1 mice, while the opposite is true for N-acetyl-l-aspartic acid. The relation between these two discriminant metabolites suggests that the enzyme that link them both, aspartate N-acetyltransferase (KEGG, K18309), could have been affected by the APP or PS1 transgene insertion. In agreement with what we had observed, lower levels of N-acetyl-aspartic acid in cerebellum of APP transgenic mice had been reported before [56]. Similar trend was observed in cortex tissue of APP/PS1 mice in our study as well, although N-acetyl-l-aspartic acid did not achieve significance after Bonferroni’s correction was applied. However, since N-acetyl-l-aspartic acid was frequently reported to be reduced in brains of AD patients [57, 58], the interpretability for our observations is limited. The differences between observations in disease models and clinical cases help to highlight the limitations of APP/PS1 mice as AD model, which should be employed with careful considerations for such differences.
PIO Exerted Treatment Effects in Cortex and Cerebellum Tissues
One interesting discovery in this study was the specificity of PIO’s treatment effects to cortex and cerebellum of APP/PS1 mice. In the cortex tissue of APP/PS1 mice, PIO administration for 14 days completely rescued the metabolic alterations related to impaired energy metabolism, namely lactic acid, glucose, and glucose-6-phosphate. Similar effects were also observed in cerebellum of APP/PS1 mice, where pathological changes in ribose-5-phosphate and glucose-6-phosphate were completely reversed by treatment of APP/PS1 mice with PIO. Treatment effect of PIO on cortical lactic acid could be explained by PIO’s therapeutic efficacy in rescuing LDH activities on cortex tissue of APP/PS1 mice. These findings were also supported by previous reports, where one reported normalisation of glucose metabolism with PIO treatment [24], and another study reported PIO’s therapeutic benefits on glucose metabolism and LDH activities [59]. More importantly, this is the first study to demonstrate specificity of PIO’s treatment effects on metabolic alterations in two particular brain regions, namely the cortex and cerebellum. PIO exerted no treatment effects in hippocampus and midbrain tissue of APP/PS1 mice used in this study, but this could be due to the occurrence of limited metabolic alterations in these two brain regions when mice were 20 weeks old. PIO’s treatment effects on metabolic fluctuations in cortex and cerebellum of APP/PS1 mice shed light on therapeutic mechanisms of PIO and one such example in this study was PIO’s reversing effect on the impaired cortical LDH in APP/PS1 mice. This information could prove to be useful for interpretation of PIO’s treatment effects observed in other studies and contribute to the field of therapeutic research for PPARγ agonist in AD.
Measurements of Amyloid-β40, Amyloid-β42, Cortical APP and Mitochondrial APP
At 20 weeks of age, amyloid-β40 and amyloid-β42 levels in plasma of APP/PS1 mice did not increase significantly as compared to their wildtype counterparts. On the other hand, both amyloid-β peptides, in particular amyloid-β42, had increased considerably in their cortex tissue. This has an important implication as the soluble form of amyloid-β40 and amyloid-β42 had been discussed to better correlate with severity of cognitive symptoms in AD [60], making APP/PS1 transgenic mice a suitable model for researching on these soluble amyloid-β peptides. PIO administration was observed to have successfully treated the pathological increase in amyloid-β40 and amyloid-β42 levels in the cortex of APP/PS1 mice. This amyloid-lowering effect of PIO had been reported before [61], which lends support to our findings in this study.
Because one of the main findings based on our metabolic profiling study of an in vitro AD model was the accumulation of APP in mitochondria [37], in this in vivo study we also measured the APP levels in cortex tissue and mitochondrial extracts from cortex tissues harvested from APP/PS1 mice and compared them against wildtype control. Cortical APP was observed to be higher in APP/PS1 transgenic mice, but this increase did not achieve significance. Interestingly, PIO administration appeared to have suppressed APP expression in cortex tissue of APP/PS1 mice, an observation that had been reported in previous study [61]. A higher level of mitochondrial APP was detected in cortex tissue of APP/PS1 mice, although PIO treatment did not appear to have a significant treatment effect on this increase. A western blot analysis was carried out to confirm the purity of mitochondria in the isolated extracts, which gave satisfying results and showed that there is little possibility of contamination from plasma APP in mitochondrial APP readings. Therefore, findings for mitochondrial APP in this in vivo study validates our earlier in vitro findings [37], suggesting that mitochondrial APP could potentially be a marker for assessing AD in its early disease stage.
Assessing the Oxidation State in APP/PS1 Mice
Neuroinflammation had been reported to be a common observation in several preclinical and clinical studies of AD [62, 63]. In this study, we assessed the oxidation state in cortex tissue of APP/PS1 mice, by measuring the activities of two major antioxidant enzymes in their cortical and plasma tissue samples, namely SOD and catalase. SOD activities were found to have decreased in cortex of APP/PS1 mice, though it remained unchanged in their plasma. PIO treatment enhanced the activities of SOD in both cortex and plasma of APP/PS1 mice, which could have given the APP/PS1 mice more antioxidative power in these two tissue compartments to resist the oxidative stress coming from all the destructive biochemical processes that occurred in this AD mouse model. On the other hand, catalase activities increased in both cortex and plasma of APP/PS1 mice, although this increase did not achieve significance in their cortex tissue. Once again, PIO treatment normalised activities of catalase in plasma of APP/PS1 mice, bringing catalase activities to a level similar to non-transgenic wildtype mice. Although the data in this study is insufficient to pinpoint the actual cause for fluctuations in SOD and catalase activities, a previous study had demonstrated the protective role of SOD and cytotoxic role of catalase in a human cell model of Parkinson’s disease [64]. All these observations indicate that oxidative stress had already been exhibited in APP/PS1 at 20 weeks of age, and PIO had effectively exerted its treatment effects by regulating the activities of these two major antioxidant enzymes.
Conclusion
In this study, we employed GC-TOF-MS-based metabolic profiling in studying the metabolic alterations in plasma and brain part samples (cortex, hippocampus, cerebellum and midbrain) of APP/PS1 mice and compared their metabolic profiles against wildtype control. At 20 weeks old, their plasma, hippocampus and midbrain tissue do not yield a lot of useful metabolic profiling data, indicating the limitation in using this model to study the metabolic alterations in these particular sample matrices and sampling time point. On the other hand, their cortex and cerebellum samples displayed extensive metabolic changes, which were all closely associated with impaired energy metabolism and a perturbed amino acid metabolism in both brain regions. PIO was observed to have exerted its treatment effects on metabolic alterations occurring in both cortex and cerebellum of APP/PS1 mice. PIO administration helped to normalise glucose metabolism, as well as their lactic acid levels by enhancing LDH activities in cortex of APP/PS1 mice. Selected biochemical assays were also conducted to assess other AD-related parameters in these mice. In brief, we observed that amyloid-β40 and amyloid-β42 levels were higher in cortex of APP/PS1 mice, which were both treated by PIO administration. APP was also observed to have accumulated in the cortical mitochondria of APP/PS1 mice, and PIO treatment did not have any effect on accumulation of mitochondrial APP. SOD activities were also observed to have been lowered in cortex of APP/PS1 mice, which were also normalised by PIO treatment. Although the number of animals used was relatively modest in this pilot investigation and a larger sample size would confer greater statistical power for metabolite measurements, our results nonetheless suggest the potential of PIO administration to exert a range of therapeutic effects in this AD model and present this drug as a promising therapeutic compound to be developed further for AD therapy. In addition, our study serves as an informative platform for planning of future experiments aimed at pinpointing the affected metabolic pathways in early-stage AD.
References
Masters CL, Bateman R, Blennow K et al (2015) Alzheimer’s disease. Nat Rev Dis Primers 1:15056. https://doi.org/10.1038/nrdp.2015.56
Querfurth HW, LaFerla FM (2010) Alzheimer’s disease. N Engl J Med 362:329–344. https://doi.org/10.1056/NEJMra0909142
Braak H, Braak E (1997) Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18:351–357
Qian X, Hamad B, Dias-Lalcaca G (2015) The Alzheimer disease market. Nat Rev Drug Discov 14:675–676. https://doi.org/10.1038/nrd4749
Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS et al (2014) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370:322–333. https://doi.org/10.1056/NEJMoa1304839
Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R et al (2014) Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med 370:311–321. https://doi.org/10.1056/NEJMoa1312889
Crous-Bou M, Minguillón C, Gramunt N, Molinuevo JL (2017) Alzheimer’s disease prevention: from risk factors to early intervention. Alzheimers Res Ther 9(71):71. https://doi.org/10.1186/s13195-017-0297-z
Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR Jr et al (2011) Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7:280–292. https://doi.org/10.1016/j.jalz.2011.03.003
Sperling R, Johnson K (2013) Biomarkers of Alzheimer disease: current and future applications to diagnostic criteria. Continuum (Minneap Minn) 19:325–338. https://doi.org/10.1212/01.CON.0000429181.60095.99
Peters KR, Lynn Beattie B, Feldman HH, Illes J (2013) A conceptual framework and ethics analysis for prevention trials of Alzheimer disease. Prog Neurobiol 110:114–123. https://doi.org/10.1016/j.pneurobio.2012.12.001
Xu X-H, Huang Y, Wang G, Chen S-D (2012) Metabolomics: a novel approach to identify potential diagnostic biomarkers and pathogenesis in Alzheimer’s disease. Neurosci Bull 28:641–648. https://doi.org/10.1007/s12264-012-1272-0
Wilkins JM, Trushina E (2017) Application of metabolomics in Alzheimer’s disease. Front Neurol 8:719. https://doi.org/10.3389/fneur.2017.00719
Nicholson JK, Lindon JC, Holmes E (1999) “Metabonomics”: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 29:1181–1189. https://doi.org/10.1080/004982599238047
Bundy JG, Spurgeon DJ, Svendsen C, Hankard PK, Osborn D, Lindon JC, Nicholson JK (2002) Earthworm species of the genus Eisenia can be phenotypically differentiated by metabolic profiling. FEBS Lett 521:115–120
Urbanczyk-Wochniak E, Luedemann A, Kopka J, Selbig J, Roessner-Tunali U, Willmitzer L, Fernie AR (2003) Parallel analysis of transcript and metabolic profiles: a new approach in systems biology. EMBO Rep 4:989–993. https://doi.org/10.1038/sj.embor.embor944
Mosconi L, Pupi A, De Leon MJ (2008) Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci 1147:180–195. https://doi.org/10.1196/annals.1427.007
Mosconi L, Berti V, Glodzik L, Pupi A, de Santi S, de Leon MJ (2010) Pre-clinical detection of Alzheimer’s disease using FDG-PET, with or without amyloid imaging. J Alzheimers Dis 20:843–854. https://doi.org/10.3233/JAD-2010-091504
Ferreira IL, Resende R, Ferreiro E et al (2010) Multiple defects in energy metabolism in Alzheimer’s disease. Curr Drug Targets 11:1193–1206
Mergenthaler P, Lindauer U, Dienel GA, Meisel A (2013) Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci 36:587–597. https://doi.org/10.1016/j.tins.2013.07.001
Heneka MT, Reyes-Irisarri E, Hüll M, Kummer MP (2011) Impact and therapeutic potential of PPARs in Alzheimer’s disease. Curr Neuropharmacol 9:643–650. https://doi.org/10.2174/157015911798376325
Berger J, Moller DE (2002) The mechanisms of action of PPARs. Annu Rev Med 53:409–435. https://doi.org/10.1146/annurev.med.53.082901.104018
Corona JC, Duchen MR (2016) PPARγ as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radic Biol Med 100:153–163. https://doi.org/10.1016/j.freeradbiomed.2016.06.023
Searcy JL, Phelps JT, Pancani T, Kadish I, Popovic J, Anderson KL, Beckett TL, Murphy MP et al (2012) Long-term pioglitazone treatment improves learning and attenuates pathological markers in a mouse model of Alzheimer’s disease. J Alzheimers Dis 30:943–961. https://doi.org/10.3233/JAD-2012-111661
Papadopoulos P, Rosa-Neto P, Rochford J, Hamel E (2013) Pioglitazone improves reversal learning and exerts mixed cerebrovascular effects in a mouse model of Alzheimer’s disease with combined amyloid-β and cerebrovascular pathology. PLoS One 8:e68612. https://doi.org/10.1371/journal.pone.0068612
Galimberti D, Scarpini E (2017) Pioglitazone for the treatment of Alzheimer’s disease. Expert Opin Investig Drugs 26:97–101. https://doi.org/10.1080/13543784.2017.1265504
Sato T, Hanyu H, Hirao K, Kanetaka H, Sakurai H, Iwamoto T (2011) Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease. Neurobiol Aging 32:1626–1633. https://doi.org/10.1016/j.neurobiolaging.2009.10.009
Geldmacher DS, Fritsch T, McClendon MJ, Landreth G (2011) A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. Arch Neurol 68:45–50. https://doi.org/10.1001/archneurol.2010.229
Schneider LS, Mangialasche F, Andreasen N, Feldman H, Giacobini E, Jones R, Mantua V, Mecocci P et al (2014) Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. J Intern Med 275:251–283. https://doi.org/10.1111/joim.12191
Landreth G, Jiang Q, Mandrekar S, Heneka M (2008) PPARgamma agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics 5:481–489. https://doi.org/10.1016/j.nurt.2008.05.003
Kaddurah-Daouk R, McEvoy J, Baillie RA, Lee D, Yao JK, Doraiswamy PM, Krishnan KRR (2007) Metabolomic mapping of atypical antipsychotic effects in schizophrenia. Mol Psychiatry 12:934–945. https://doi.org/10.1038/sj.mp.4002000
Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I et al (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4:97–100
Duyckaerts C, Potier M-C, Delatour B (2008) Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol 115:5–38. https://doi.org/10.1007/s00401-007-0312-8
Howlett DR (2011) APP transgenic mice and their application to drug discovery. Histol Histopathol 26:1611–1632. https://doi.org/10.14670/HH-26.1611
Trushina E, Nemutlu E, Zhang S, Christensen T, Camp J, Mesa J, Siddiqui A, Tamura Y et al (2012) Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS One 7:e32737. https://doi.org/10.1371/journal.pone.0032737
Hu Z-P, Browne ER, Liu T, Angel TE, Ho PC, Chan ECY (2012) Metabonomic profiling of TASTPM transgenic Alzheimer’s disease mouse model. J Proteome Res 11:5903–5913. https://doi.org/10.1021/pr300666p
Chang KL, Ho PC (2014) Gas chromatography time-of-flight mass spectrometry (GC-TOF-MS)-based metabolomics for comparison of caffeinated and decaffeinated coffee and its implications for Alzheimer’s disease. PLoS One 9:e104621. https://doi.org/10.1371/journal.pone.0104621
Chang KL, Pee HN, Tan WP, Dawe GS, Holmes E, Nicholson JK, Chan ECY, Ho PC (2015) Metabolic profiling of CHO-AβPP695 cells revealed mitochondrial dysfunction prior to amyloid-β pathology and potential therapeutic effects of both PPARγ and PPARα Agonisms for Alzheimer’s disease. J Alzheimers Dis 44:215–231. https://doi.org/10.3233/JAD-140429
Chan ECY, Pasikanti KK, Nicholson JK (2011) Global urinary metabolic profiling procedures using gas chromatography-mass spectrometry. Nat Protoc 6:1483–1499. https://doi.org/10.1038/nprot.2011.375
Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, Djoumbou Y, Mandal R et al (2013) HMDB 3.0--the human metabolome database in 2013. Nucleic Acids Res 41:D801–D807. https://doi.org/10.1093/nar/gks1065
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40:D109–D114. https://doi.org/10.1093/nar/gkr988
Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, Purcell SM, Betensky RA, Raju S, Prada C, Greenberg SM et al (2006) Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis 24:516–524. https://doi.org/10.1016/j.nbd.2006.08.017
van Groen T, Kiliaan AJ, Kadish I (2006) Deposition of mouse amyloid beta in human APP/PS1 double and single AD model transgenic mice. Neurobiol Dis 23:653–662. https://doi.org/10.1016/j.nbd.2006.05.010
Pan X, Nasaruddin MB, Elliott CT, McGuinness B, Passmore AP, Kehoe PG, Hölscher C, McClean PL et al (2016) Alzheimer’s disease-like pathology has transient effects on the brain and blood metabolome. Neurobiol Aging 38:151–163. https://doi.org/10.1016/j.neurobiolaging.2015.11.014
González-Domínguez R, García-Barrera T, Vitorica J, Gómez-Ariza JL (2015) Application of metabolomics based on direct mass spectrometry analysis for the elucidation of altered metabolic pathways in serum from the APP/PS1 transgenic model of Alzheimer’s disease. J Pharm Biomed Anal 107:378–385. https://doi.org/10.1016/j.jpba.2015.01.025
González-Domínguez R, García-Barrera T, Vitorica J, Gómez-Ariza JL (2015) Metabolomic research on the role of interleukin-4 in Alzheimer’s disease. Metabolomics 11:1175–1183. https://doi.org/10.1007/s11306-015-0773-z
Wu J, Fu B, Lei H, Tang H, Wang Y (2016) Gender differences of peripheral plasma and liver metabolic profiling in APP/PS1 transgenic AD mice. Neuroscience 332:160–169. https://doi.org/10.1016/j.neuroscience.2016.06.049
Graham SF, Holscher C, McClean P, Elliott CT, Green BD (2013) 1H NMR metabolomics investigation of an Alzheimer’s disease (AD) mouse model pinpoints important biochemical disturbances in brain and plasma. Metabolomics 9:974–983. https://doi.org/10.1007/s11306-013-0516-y
Molina JA, Jiménez-Jiménez FJ, Vargas C et al (1998) Cerebrospinal fluid levels of non-neurotransmitter amino acids in patients with Alzheimer’s disease. J Neural Transm (Vienna) 105:279–286. https://doi.org/10.1007/s007020050057
González-Domínguez R, García-Barrera T, Vitorica J, Gómez-Ariza JL (2015) Deciphering metabolic abnormalities associated with Alzheimer’s disease in the APP/PS1 mouse model using integrated metabolomic approaches. Biochimie 110:119–128. https://doi.org/10.1016/j.biochi.2015.01.005
Zhang X, Liu H, Wu J, Zhang X, Liu M, Wang Y (2009) Metabonomic alterations in hippocampus, temporal and prefrontal cortex with age in rats. Neurochem Int 54:481–487. https://doi.org/10.1016/j.neuint.2009.02.004
Lalande J, Halley H, Balayssac S, Gilard V, Déjean S, Martino R, Francés B, Lassalle JM et al (2014) 1H NMR metabolomic signatures in five brain regions of the AβPPswe Tg2576 mouse model of Alzheimer’s disease at four ages. J Alzheimers Dis 39:121–143. https://doi.org/10.3233/JAD-130023
González-Domínguez R, García-Barrera T, Vitorica J, Gómez-Ariza JL (2014) Region-specific metabolic alterations in the brain of the APP/PS1 transgenic mice of Alzheimer’s disease. Biochim Biophys Acta 1842:2395–2402. https://doi.org/10.1016/j.bbadis.2014.09.014
González-Domínguez R, García-Barrera T, Vitorica J, Gómez-Ariza JL (2015) Metabolomic screening of regional brain alterations in the APP/PS1 transgenic model of Alzheimer’s disease by direct infusion mass spectrometry. J Pharm Biomed Anal 102:425–435. https://doi.org/10.1016/j.jpba.2014.10.009
Paban V, Manrique C, Filali M, Maunoir-Regimbal S, Fauvelle F, Alescio-Lautier B (2014) Therapeutic and preventive effects of methylene blue on Alzheimer’s disease pathology in a transgenic mouse model. Neuropharmacology 76 Pt A:68–79. https://doi.org/10.1016/j.neuropharm.2013.06.033
Ross JM, Öberg J, Brené S et al (2010) High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc Natl Acad Sci U S A 107:20087–20092. https://doi.org/10.1073/pnas.1008189107
Salek RM, Xia J, Innes A, Sweatman BC, Adalbert R, Randle S, McGowan E, Emson PC et al (2010) A metabolomic study of the CRND8 transgenic mouse model of Alzheimer’s disease. Neurochem Int 56:937–947. https://doi.org/10.1016/j.neuint.2010.04.001
Jessen F, Lewczuk P, Gür O, Block W, Ende G, Frölich L, Hammen T, Arlt S et al (2011) Association of N-acetylaspartate and cerebrospinal fluid Aβ42 in dementia. J Alzheimers Dis 27:393–399. https://doi.org/10.3233/JAD-2011-110398
Bittner DM, Heinze H-J, Kaufmann J (2013) Association of 1H-MR spectroscopy and cerebrospinal fluid biomarkers in Alzheimer’s disease: diverging behavior at three different brain regions. J Alzheimers Dis 36:155–163. https://doi.org/10.3233/JAD-120778
Masciopinto F, Di Pietro N, Corona C et al (2012) Effects of long-term treatment with pioglitazone on cognition and glucose metabolism of PS1-KI, 3xTg-AD, and wild-type mice. Cell Death Dis 3:e448. https://doi.org/10.1038/cddis.2012.189
Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 8:101–112. https://doi.org/10.1038/nrm2101
Liu L, Yan T, Jiang L, Hu W, Hu M, Wang C, Zhang Q, Long Y et al (2013) Pioglitazone ameliorates memory deficits in streptozotocin-induced diabetic mice by reducing brain β-amyloid through PPARγ activation. Acta Pharmacol Sin 34:455–463. https://doi.org/10.1038/aps.2013.11
Cameron B, Landreth GE (2010) Inflammation, microglia, and Alzheimer’s disease. Neurobiol Dis 37:503–509. https://doi.org/10.1016/j.nbd.2009.10.006
Sochocka M, Koutsouraki ES, Gasiorowski K, Leszek J (2013) Vascular oxidative stress and mitochondrial failure in the pathobiology of Alzheimer’s disease: a new approach to therapy. CNS Neurol Disord Drug Targets 12:870–881
Iglesias-González J, Sánchez-Iglesias S, Méndez-Álvarez E, Rose S, Hikima A, Jenner P, Soto-Otero R (2012) Differential toxicity of 6-hydroxydopamine in SH-SY5Y human neuroblastoma cells and rat brain mitochondria: protective role of catalase and superoxide dismutase. Neurochem Res 37:2150–2160. https://doi.org/10.1007/s11064-012-0838-6
Funding
This work was supported by research grant from the Academic Research Funding, National University of Singapore (R148-000-180-112).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chang, K.L., Wong, L.R., Pee, H.N. et al. Reverting Metabolic Dysfunction in Cortex and Cerebellum of APP/PS1 Mice, a Model for Alzheimer’s Disease by Pioglitazone, a Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) Agonist. Mol Neurobiol 56, 7267–7283 (2019). https://doi.org/10.1007/s12035-019-1586-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-019-1586-2