
Abstract
The cellular prion protein (PrPC), mainly known for its role in neurodegenerative diseases, is involved in several physiological processes including neuritogenesis. In addition, its ability to bind copper or zinc has been suggested for its role in metal homeostasis. Although PrPC has been known as a copper-binding molecule, little is known about how copper can affect PrPC physiological functions. By combining genomic approaches, cellular assays, and focal stimulation technique, we found that PrPC neuritogenesis function is directly influenced by N-terminal copper-binding amino acids. Several recombinant mouse PrP (recMoPrP) mutants at N-terminal copper-binding sites were produced, and primary hippocampal cultures were treated either in bulk or exposed near the hippocampal growth cones (GC) of single neurons in local stimulation manner. While focal stimulation of GC with wild-type recMoPrP induced neurite outgrowth and rapid GC turning toward the source, N-terminal mutants fail to support this effect. Indeed, disrupting all the copper-binding sites at the N-terminus of PrPC was toxic to neurons indicating that these regions are crucial for the protein function. Mutants at both octarepeat and non-octarepeat region abolished the neuritogenesis effect. Altogether, our findings indicate the crucial role of copper-binding sites in maintaining the neuritogenesis function in PrP, suggesting a potential link between loss-of-function of the protein and disease initiation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The cellular form of the prion protein (PrPC) is known for its central role in prion disease and related disorders [1, 2]. Recent research has focused on PrPC emerging role in the central nervous system (CNS) describing its involvement in various cellular processes such as neuritogenesis [3], cell adhesion [4, 5], memory and cognition [6], neuroprotection [7, 8], and metal homeostasis [9]. PrPC has two main copper-binding sites lying on the octarepeat (OR) region and on the non-OR region at N-terminal of the protein with the amino acid histidine as the key residue for copper coordination [10]. It has been suggested that PrPC acts as a copper-binding protein that buffers copper homeostasis and it cooperates with copper in the S-nitrosylation process to protect neuron from N-methyl-d-aspartate receptors (NMDAR)–mediated excitotoxicity [8]. However, the inter-dependence between PrPC and copper in other neuronal function has not yet been investigated.
Copper ions play essential roles in many biological processes and are found at high concentration in mammalian brain [11]. Due to its redox ability of changing between oxidized state (Cu2+) and reduced state (Cu+), copper is able to bind a wide range of proteins as a catalytic or structural cofactor [12]. Under physiological condition, copper concentration is strictly regulated. While copper deficiency or genetic mutation of copper-transporting protein could lead to developmental defects [11,12,13], excess copper load would increase toxic-free radicals and cause oxidative damage within the cell [13, 14]. There are several neurodegenerative diseases involved perturbation of copper homeostasis such as the Menkes disease, Wilson disease, amyotrophic lateral sclerosis, Alzheimer disease and prion disease [15].
In a previous study, we have shown that soluble form of PrP could act as a signaling cue to promote neurite outgrowth and growth cone (GC) guidance [16]. We also identified the functional region for that activity lying around the helix A and octarepeat region of PrP. Focusing on the unstructured N-terminal PrP, this region contains main copper/zinc-binding site which is also critical regions in prion endoproteolysis and prion conversion [17, 18]. Therefore, in this study, we hypothesize the role of copper binding in modulating function of PrPC in neuritogenesis. Substituting histidine residues with tyrosine at OR and non-OR region, we found that disruption of the copper-binding sites of PrPC perturbed its structural conformation and failed to induce neurite outgrowth signal. Moreover, a minimal change in copper coordination due to the mutation P101L (mouse homolog of Gerstmann–Sträussler–Scheinker (GSS)–linked mutation in human prion disease) was sufficient to prevent PrPC function, supporting a loss-of-function hypothesis in prion pathology. Overall, we provide evidence for PrPC and copper to regulate neuronal processes and suggesting potential link with disease pathogenesis in which alterations in PrPC function could lead to neurodegeneration.
Materials and Methods
Antibodies and Reagents
Monoclonal antibody (mAb) to β3-tubulin (2G10, Thermo Fisher Scientific) was used at 1:100 dilution for immunofluorescence. For western blot experiments, β3-tubulin rabbit polyclonal antibody (pAb) from Sigma was diluted at 1:5000. Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) rabbit mAb (197G2, Cell Signaling Technology) was prepared at 1:4000; p44/42 MAPK (Erk1/2) mouse mAb (3A7, Cell Signaling Technology) was diluted 1:3000. Horseradish peroxidase (HRP) - conjugated secondary antibody (1:1000) and fluorochrome - conjugated secondary antibody (1:100) were from DAKO. All antibodies used for detecting phosphorylated protein were prepared in TBST solution with 5% BSA. Most of chemical reagents were from Sigma, otherwise indicated.
Plasmid Construction and Protein Purification
Full-length MoPrP (23–231) DNA sequence was cloned into pET-11a plasmid (Novagen) respectively by restriction-free method. Histidine residues at octarepeat (OR) region (H60, H68, H76, H84) and at non-OR region (H95, H110) were mutated step by step to tyrosine residues and introduced into pET-11a::MoPrP (23–231) by mutagenesis kit (Stratagene). These plasmids were then used to produce three recombinant mutant prion proteins at the N-terminal copper-binding sites denoted as recMoPrP(H1234Y) with octarepeat H-to-Y mutations, recMoPrP(H56Y) with non-OR mutations, and recMoPrP(H123456Y) with all 6 histidine residues mutated. The mutation P101L was also introduced for producing of additional mutant. The recombinant proteins were expressed in Rosetta-GAMI E.Coli cells by 0.8 mM IPTG overnight (~ 16 h) induction. Inclusion bodies containing the protein were isolated after cell homogenization by Homogenizer Panda Plus 2000 (Gea Niro Soavi) at around 1500 bar and following centrifugation at 13,000g for 30 min, 4 °C. They were washed in 25 mM Tris-HCl pH 8.8, 5 mM EDTA pH 8, 0.8% Triton X100, and two times in bi-distilled water. Then, they were solubilized in 6 M GdnHCl and 25 mM Tris base pH 8 and shaken overnight, and the supernatant was collected for further purification. The samples with recMoPrP-wt and H56Y proteins were purified using 5 mL HisTrap column chromatography (GE Healthcare) whereas recMoPrP(H1234Y) and recMoPrP(H123456Y) were purified using size-exclusion chromatography (column HiLoad 26/60 Superdex 200 pg, GE Healthcare) because of lacking the histidine residues. The recMoPrP(P101L) was also produced by the same protocol as recMoPrP-wt. All the purified protein samples were in vitro refolded by dialysis against refolding buffer (20 mM sodium acetate, 0.005% NaN3, pH 5.5) using a Spectrapor-membrane (MWCO 3500) and exchanged to PBS (phosphate buffer saline) pH 7.4 buffer prior to treatments in cell cultures.
Circular Dichroic Measurement
Purified and refolded proteins were dialyzed in phosphate buffer pH 7.2 and diluted at concentration around 0.1 mg/mL for circular dichroic (CD) measurement by spectrophotometer JASCO J-810. Measurements were carried out at room temperature using 0.1 cm optical path length quartz cell. Spectra were obtained in the 190 to 260 nm far-UV region and accumulated at least three times. CD data were reconstructed by CDSSTR program with reference data set 7 (Sreerama and Woody 2000), NRMSD < 0.05 on DichroWeb server (http://dichroweb.cryst.bbk.ac.uk/).
Cell Culture
Primary hippocampal neurons were isolated from P2 FVB wild-type (wt) mouse brain as described previously [16]. P2 FVB wt mouse pups were sacrificed by decapitation in accordance with the guidelines of the Italian Animal Welfare Act. Dissociated cells were plated in an incubator with 5% CO2, 37 °C on the 0.1 mg/mL poly-ornithine (Sigma) coated coverslips in B27 supplemented neurobasal medium (Thermo Fisher Scientific) with around 2 × 104 cells per coverslip. About 22 h post-plating, the cells were used for local stimulation experiment or treated in bulk with 2 μM of the recombinant proteins or PBS control for further neurite outgrowth assay by immunofluorescence. For latter experiments, the dissociated cells were filtered through 40-μm cell strainer (Corning) prior to plating to obtain evenly distributed cell culture.
Immunofluorescence
After around 20 h of treatments with wt or mutant recMoPrP proteins, the hippocampal neurons were fixed by 4% paraformaldehyde (PFA) in 20 min, washed three times in PBS, and quenched with glycine 0.1 M. The fixed cells were blocked in 5% normal goat serum (NGS, Sigma) for 30 min, permeabilized in 0.3% Triton, and blocked again in 5% NGS solution. To appreciate the total neurite morphology, the cells were incubated with monoclonal antibody to β3-tubulin (Thermo Fisher Scientific) in blocking solution for 2 h at room temperature, washed three times with PBS, and incubated for additional 1 h at room temperature with the goat anti-mouse fluorochrome-conjugated secondary antibody in blocking solution. After three washes with PBS, nuclear staining was performed by incubated with 4′,6-diamidino-2-phenylindole (dapi) for 5 min. The coverslips were eventually mounted on slides with Vectashield mounting medium (Vector Laboratories). Fluorescent images were acquired at 40 × oil immersion objective by a Leica laser scanning confocal microscope with a Krypton-Argon and UV lasers. Five to six projections of six optical sections were randomly taken per coverslip at the region with cells evenly distributed and similar density between treatments, usually away from the edge and from the center of the coverslip. Total neurite length was measured using NeuriteTracer plugin for ImageJ [19]. Average neurite length per neurons is calculated by dividing total neurite length to the number of total neurons counted. Data were collected from at least three independent experiments per treatment and analyzed with ImageJ 1.49 software (http://rsb.info.nih.gov/ij/).
Liposome Preparation and Focal Stimulation
The liposome kit used for microvesicle preparation was from Sigma (L4395). The liposome preparation protocol and optical manipulation setup were previously described [16, 20]. Briefly, the lipid solution was prepared at the concentration of 10 mg/mL in chloroform:methanol (2:1 v/v). The solution obtained was then saturated with nitrogen and stored at − 20 °C. The lipid solution was dried in a vacuum oven for 24 h and then the lipid film was rehydrated with a PBS solution containing desired concentration of recMoPrP and 100 mM sucrose which helps improve vesicle trapping. The vial containing the rehydrated lipid film was vortex well and incubated overnight at 4 °C. Liposomes with a diameter of 1–5 μm should be formed. Then, they were gently washed three times in PBS solution with 100 mM glucose by centrifuging at 5000 rpm for 3 min each time. Final liposome solution was then administered to the cell cultures on the day of the experiment.
In particular, the recombinant wild-type or mutant prion proteins were prepared in PBS at 4 μM and encapsulated in liposomes one night before the experiment. About 22 h post-plating, hippocampal single neurons with isolated growth cone (GC) were focally stimulated after photolyzing one trapped liposome containing the protein of interest at distance of 10–20 μm. The optical manipulation setup was customized from an inverted microscope (Nikon Eclipse TE-2000-E, Japan). The microscope was installed with custom IR optical tweezers used to trap the liposome and a UV laser-dissection system (MMI-cellCut Plus, Switzerland) used to photolyze the trapped one and deliver the protein molecules (Fig.S1). The behavior of the stimulated growth cone was recorded by time-lapse phase contrast imaging using a digital camera (Orca Flash 4.0, Hamamatsu, Japan) at a frame rate of 2 s and analyzed by our custom code developed in Matlab.
Western Blotting
Hippocampal neurons were plated at 4 × 105 cells/mL in a 6-well plate. At day 1 in vitro, each well was treated with mock control or 2 μM recMoPrP(wt) or recMoPrP(H56Y) for 25 min. Cells were then washed with ice-cold PBS 1X (pH 7.4) twice and lysed by lysis buffer (50 mM Tris HCl pH 7.5, 150 mM NaCl, 0.5% NP-40, and 0.5% sodium deoxycholate) supplemented with protease and phosphatase inhibitor cocktails (both from Roche). Cell extracts were collected after centrifugation at 14,000 × g, 4 °C, 15 min. Total protein amount was measured using BCA kit (Euroclone). The same amount of protein in each sample was loaded on SDS-PAGE gel 10%. Proteins were transferred onto polyvinylidene difluoride (PVDF) membrane (Immobilon-P, Milipore) at 300 mA, 4 °C, 150 min and then blocked in 5% BSA (for detecting phosphorylated protein) or skim milk in TBST (TBS with 0.5% Tween-20) for 30-45 min. Membranes were probed with primary antibody diluted in blocking solution overnight at 4 °C, washed two times (5 min each) with TBST, and then probed with HRP conjugated secondary antibody for 1 h. Immunoreactivity was detected by chemiluminescence HRP substrate (Milipore) and imaged with UVITEC Cambridge Imaging system. Densitometric analysis of band intensities was performed using Alliance v16.14 software.
Viability/Cytotoxicity Test
After incubation with recMoPrP proteins for about 22 h, neuronal cultures were subjected to cytotoxicity test by using the LIVE/DEAD Viability/Cytotoxicity Kit (Thermo Fisher Scientific). The staining protocol was applied from the manufacturer manual. Briefly, the dye mix was prepared in PBS 1X with 2 μM calcein AM for live cell staining and 4 μM EthD-1 for dead cell staining at final concentration. After being washed twice in warm PBS, the cells were incubated with the dye mix for 20 min at 37 °C. Then the coverslips containing the cells were proceeded for observation under 20 × objective of the Nikon confocal microscope at laser 488 for the signal of calcein AM and laser 561 for the signal of EthD-1. Numbers of live and dead cells from each image in each treatment were analyzed by the plugin Analyze Particles of ImageJ 1.49 software.
Statistical Analysis
Statistical data are presented as mean (SD) or ± SEM depending on data type. Statistics for multiple comparison were analyzed using one-way ANOVA with Games–Howell or Dunnett’s T3 post hoc test by IBM SPSS statistics 23. The minimum significance was set at p < 0.05.
Results
Structural Integrity of N-Terminal Copper-Binding Sites of PrPC Is Crucial for Its Function in Neuritogenesis
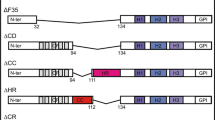
To investigate whether copper-binding sites play a role in the function of PrPC in neuritogenesis, we mutate the histidine residues of the N-terminal of PrP involved in copper coordination [10]. In addition, we produced the homologous GSS-linked mutation P101L (P102L in human) shown to alter copper coordination at the non-OR region [18] (Fig. 1a). The purified and folded proteins were assessed using circular dichroism spectra. As result, substitution of histidine to tyrosine at the OR denoted as H1234Y changed PrP secondary structure with very high α-helix-rich content comparing to the wild-type (wt) protein. The same change was observed in the construct were all 6 histidine residues were mutated to tyrosine (6-H toY mutant (H123456Y)). However, mutations at non-OR including H-to-Y substitutions or P101L had similar structural patterns where changes favor more β-sheet contents. (Fig. 1b, c).

Assessment of secondary structure for recombinant wild-type (wt) and mutant prion proteins by circular dichroism (CD). a Point mutation positions shown on part of sequences of the recombinant mutant MoPrP proteins compared to the wt recMoPrP. b Superimposed CD spectra representative of the purified and refolded recombinant MoPrP proteins indicating that OR mutant (recMoPrP(H1234Y)) and recMoPrP(H123456Y) have completely different structure from the wt MoPrP whereas non-OR mutants including recMoPrP(H56Y) and recMoPrP(P101L) have minimal changes but mostly the beta-sheet content. (c) Detailed superimposed CD spectra indicating minimal changes in secondary structures between recMoPrP(wt) and non-OR mutants. CD data were reconstructed by CDSSTR program with reference data set 7 (Sreerama and Woody 2000), NRMSD < 0.05 on DichroWeb server (http://dichroweb.cryst.bbk.ac.uk/)
Recombinant PrP proteins are without GPI-anchoring residues to mimic the soluble form of PrP which may act as a signaling molecule for neuronal processes. The recombinant molecules were delivered to the neuronal cell culture following overnight incubation. Different concentrations of wt recMoPrP at 0.5 μM, 1 μM, or 2 μM were used to treat the P2 hippocampal neurons in bulk. After about 22 h of incubation in the presence or absence of recMoPrP, the cells were fixed for the immunostaining with β3-tubulin as a specifically neuronal marker. Neurites in cultures treated with 2 μM of wt recMoPrP sample showed significant outgrowth and connection than that of the mock control treatment though the increase in neurite growth could already be observed at 1-μM-treated condition (Fig. 2).

Culture of hippocampal neurons treated with 2 μM recMoPrP-wt showed significant neurite outgrowth and connections comparing to the mock control and 0.5 μM concentration. a Representative immunofluorescent images of β3-tubulin for P2 hippocampal mouse neuronal cultures after about 22 h of incubation with mock control or different concentration of recMoPrP(wt). Images were taken at 40 × oil immersion objective by the Leica confocal microscope; scale bar 50 μm, nuclear staining with dapi. b Quantitative analysis of average length of neurites in different treatments by ImageJ; data represent mean (SD) from at least three independent experiments with 28 to 88 images analyzed; statistical comparison by one-way ANOVA with Games–Howell post hoc test (****p < 0.0001; *p < 0.05)
We next investigate the effect of minimally structural change in non-OR copper-binding site on the neuritogenesis process using two mutants recMoPrP(H56Y) and recMoPrP(P101L). After the cells were plated for 1 day, the neuronal cultures were treated with either the mutant or wt recombinant proteins at 2 μM concentration. As a result, neither recMoPrP(H56Y) nor recMoPrP(P101L) could enhance neurite outgrowth comparing to wt recMoPrP treatment (Fig. 3a). The cultures treated with the non-OR mutants appeared to have shorter neurites than in the mock control despite the differences which were of non-statistical significance. Average length of neurites in wt recMoPrP treatment could reach almost 200 μm after 2 days in culture whereas, in recMoPrP(H56Y) and recMoPrP(P101L) mutant treatments, neurites were about 80 and 70 μm shorter respectively. (Fig. 3b).

Neuronal culture treated with recMoPrP-wt showed significant neurite outgrowth and connections whereas non-OR mutant PrP(s) failed to promote the neuritogenesis in those treated cultures. a Representative immunofluorescent images of β3-tubulin for P2 hippocampal mouse neuronal cultures after about 22 h of incubation with mock control or 2 μM recMoPrP(wt) or non-OR mutants including recMoPrP(H56Y) and recMoPrP(P101L). Images were taken at 40 × oil immersion objective by the Leica confocal microscope; scale bar 50 μm, nuclear staining with dapi. b Quantitative analysis of average length of neurites in different treatment by ImageJ; data represent mean (SD) from at least three independent experiments with 23 to 88 images analyzed; statistical comparison by one-way ANOVA with Games–Howell post hoc test (****p < 0.0001)
With dramatic changes in the protein structure, it is expected that the OR and 6-H toY mutant proteins would show altered PrP function. Indeed, while recMoPrP(H1234Y) failed to increase the neurite growth comparing to the mock control, recMoPrP(H123456Y) could surprisingly cause high toxicity to neurons (Fig. 4 and Fig.S2). This data suggests that copper binding may contribute to stabilize the unstructured N-terminal of PrP maintaining its proper conformation for the signaling function. Particularly, the toxic effect was observed only with the additional substitution at the non-OR region together with the OR-region. Hence, the total disruption of copper binding at N-terminal of PrP, especially at the non-OR region, leads the protein to become more vulnerable to structural alteration and conversion to toxic species.

Neuronal cultures treated with the OR mutant (H1234Y) and all 6 His to Tyr (H123456Y) mutant recMoPrP that folded into completely different structures from the wild-type protein also failed to promote the neuritogenesis. Especially, the latter induced lethal toxicity to neurons. a Representative immunofluorescent images are shown as β3-tubulin (green) and nuclear staining dapi (blue) for P2 hippocampal mouse neuronal cultures after about 22 h of incubation with mock control or 2 μM or mutant proteins. Images were taken at 40 × oil immersion objective by the Leica confocal microscope; scale bar 50 μm. b Quantitative analysis of average length of neurites in treatments with control and OR-mutant by ImageJ; data represent mean (SD) from at least three independent experiments with 30 to 88 images analyzed. Images from cultures treated with recMoPrP(H123456Y) could not be analyzed due to neuronal death
Mutant Prion Proteins at the N-Terminal Copper-Binding Site Fail to Induce Neuronal Growth Cone Polarity and Protrusion
After assessing the overall effect of the above PrP mutants to the growth of the neuronal network in cultures, we moved to evaluate single neuronal focal stimulation to understand how the mutants could affect the cell dynamics leading to the reduction in neuritogenesis. To this end, we applied an established neuronal guidance assay based on optical tweezer technique [16] to monitor the growth cone polarity and protrusion-retraction movement after the stimuli delivery. Briefly, recombinant PrP proteins were encapsulated in liposomes at concentration of 4 μM. With the help of optical tweezer composed of two focused IR laser beams, the liposome was trapped and placed near a dynamic growth cone at a distance around 10-20 μm. The proteins were released after photolysis by the UV (ultraviolet) pulse laser and diffused toward the growth cone. The usage of IR and UV laser in this assay was shown not to alter the protein conformation [16].
Upon stimulation with wt recMoPrP, the GC trace (red line) showed that it protruded forward and turned toward the protein source (Fig. 5) which replicated our previous finding about the function of wt PrP in promoting fast GC navigation [16]. As for the OR and the non-OR mutants as well as the PBS control, GC(s) have only spontaneous movements around the original positions showed clearly from the rose diagrams with angle distributions covering 0–180° (Fig. 6).

Trajectories (red) of representative growth cones’ responses monitored for about 15 min after the deliveries of recMoPrP(wt), OR (H1234Y), or non-OR (H56Y & P101L) mutants by photolysis of the encapsulating liposomes. Only the growth cone stimulated with the recMoPrP(wt) protruded dynamically toward the protein source. Blue arrows indicate original positions of the chosen liposomes before photolysis. Red lines indicate traces of the growth cone movement every 25 s analyzed by our custom code developed in Matlab

Rose diagrams showing angle α distribution between the target GC(s) and the liposome positions (protein delivery points) during different stimulation conditions. 0 < α < 90 indicates the GCs are attracted toward the protein source; vice versa 90 < α < 1800 indicates the GCs are repulsed from the protein source (see also Fig.S3). The GC responses to recMoPrP(wt) show a clear polarity toward the signaling cue whereas, in the other conditions, the GCs turn to the opposite direction many times. The angles are analyzed every 50 frames from all the experiments for each treatment by our custom code developed in Matlab
Stimulation with recMoPrP(P101L) showed only few GC(s) moving toward the source, and the mutant protein failed to significantly promote GC polarity (Fig. 5, Fig. 6, supplementary video 1). As far as neurite growth was concerned, wt recMoPrP stimulated GC outgrowth immediately following vesicle photolysis and sprouted after 3 min whereas all other cases started decreasing at the same time or maintained minimal changes in length (Fig. 7b). Wt PrP could increase the GC maximum growth up to 12 μm within a few minutes, threefold more than the control and fourfold than the H56Y mutant (Fig. 7a). On average, GC stimulated with the non-OR mutant (H56Y) did not increase growth and, in some cases, even retracted indicating the protein may have inhibitory effect (Fig. 7b, supplementary video 2). Interestingly, the difference in behavior of GC upon stimulation with H56Y and P101L mutants infer that the direct blocking of copper binding has a strong impact on PrP function in neurite outgrowth than the putative conformational changes at the non-OR region.

The OR mutant recMoPrP(H1234Y) (n = 9) and non-OR recMoPrP(H56Y) (n = 18) and (P101L) (n = 8) failed to enhance growth-cone protrusion and neurite outgrowth as well as the mock control (n = 8) whereas the wild-type protein (n = 6) could increase remarkably the neurite growth. a Maximum growth of neurites measured from each stimulation conditions; growth-inhibiting effect of the non-OR mutant recMoPrP(H56Y) is most pronounced indicating the importance of copper binding in this region on growth-promoting function. Data represent mean (SD), one-way ANOVA with Gabriel post hoc test (*p < 0.05, ***p < 0.001, and ****p < 0.0001). b Superimposition of GC protrusion from all the experiments of different stimulation conditions during 10 min after liposome photolysis. Data represent mean ± SEM every 25 s
Non-OR Copper-Binding Site Is Essential for PrP Trans-signaling in Neuritogenesis
Data from the previous neuritogenesis assays showed that non-OR mutant recMoPrP(H56Y) treated culture had shorter average neurite length (~ 117 ± 46.7 μM) comparing to control (~ 147 ± 49.6 μM) (Fig. 3b) and GC protrusion dynamic was even significantly lower than the one stimulated by other non-OR mutant recMoPrP(P101L) (Fig. 7a). This data set confirms that copper coordination mediated by both His95 and His110 (mouse numbering) could be relevant for PrP function. Hence, recMoPrP(H56Y) is chosen for further experiment to understand how non-OR copper-binding site affects PrP trans-signaling in neuritogenesis. Our previous studies implied that wt recMoPrP could promote growth signaling through NCAM-Fyn-ERK pathway [16]. In the present study, we could show direct activation of this pathway by detecting phosphorylated form of ERK in the hippocampal cultures after treatment with the recMoPrP (Fig. 8a). Mitogen-activated protein kinases (MAPK) are activated by phosphorylation cascade. p44/42 MAPK-ERK1 and 2 are phosphorylated by MEK 1 and 2 at Thr202/Tyr204 and Thr185/Tyr187 respectively which eventually activate downstream transcription factors [21] such as CREB for neuritogenesis [22].

Non-OR mutant recMoPrP(H56Y) did not increase activation of ERK pathway observed in treatment with the wt protein. a Representative western blot of phosphorylated ERK 1/2, total ERK 1/2, and β-tubulin as loading control. Around 18 μg of proteins were loaded in each well. b Ratio of phosphorylated ERK normalized to total ERK calculated from densitometric volume of each blotted signal; ratios of the control samples were set to 1 and data are presented as fold of control. Only recMoPrP(wt) treatment could significantly enhance phospho-p44/42 MAPK (Erk1/2) indicating elevated activation of the cascade whereas the recMoPrP(H56Y) treated culture had a reduction of the ERK phosphorylation level; *p < 0.05, **p < 0.01 one-way ANOVA with Dunnett’s T3 multiple comparison. c Ratio of ERK densitometric signal normalized to β-tubulin amount; PrP treated cultures appeared to have higher expression of ERK in spite of non-statistical significance. Data represents mean (SD), n = 4 (b and c)
To detect phosphorylation of ERK, the hippocampal neurons after 1 day of plating were treated with 2 μM wt recMoPrP or recMoPrP(H56Y) for 25 min prior to cell lysis. As a result, wt recMoPrP could promote significantly phosphorylation of p44/42 ERK 1/2 up to almost 20% (1.2-fold) comparing to the mock control (Fig. 8b). The mutation H56Y failed to induce this signaling event resulting in about 40% less phosphorylated ERK ratio comparing to the wt recMoPrP sample (Fig. 8a, b). The total expression of ERK after being normalized to the loading control β-tubulin in the recMoPrP treated samples was higher than that in the control although not statistically significant (Fig. 8c). Interestingly, while the possible upregulation of ERK could lead to more activation of the kinase by MEK in case of wt recMoPrP treated culture, the phosphorylating cascade was much less efficient in the cultures treated with mutants. Mutant PrP however could act through other unknown mechanism to increase ERK expression. Spontaneous activity of the pathway was still present since phosphorylation of ERK was not diminished indicating other growth factors could compensate the normal growth in the cells. As a conclusion, the non-OR copper-binding site in PrP seems to play a major role in inducing outgrowth signaling in hippocampal neurites.
Discussion
In the last few years, many attempts to investigate the physiological role of PrPC and its partners have been intensively carried out. In the CNS, different proposed functions of PrP have been proposed: synaptic regulation [23, 24], neuroprotection [8, 25], neurogenesis [25, 26], and neuritogenesis [3, 27]. The latter may account for PrPC role in memory and cognition [6]. PrPC and copper ions are found abundant at synaptic clefts [28,29,30], and some activity of PrPC at synapses has been proposed to act as a copper-binding protein to regulate Ca2+ influx signaling [6]. In addition, PrPC and copper cooperate in regulating NMDA receptor activity to protect neurons from glutamate excitotoxicity [8, 31]. Interestingly, PrPC expression increases proliferation and differentiation at the subventricular zone (SVZ) and dentate gyrus (DG) of the hippocampus where neurogenesis constitutively occurs in both developing and adult CNS [26]. Copper is also found at high concentration in SVZ than in other brain regions and its increased concentration during aging could reduce neurogenesis [32]. On the other hand, moderate copper deficiency at early developmental stage can affect dentate gyrus and hippocampal maturation [33]. Since either high or low copper concentration can disturb neurogenesis, the involvement of PrPC in neurogenesis should relate with its regulating activity in metal homeostasis.
PrPC has been also considered as a neurotrophic factor that facilitates neuritogenesis process either through cis or trans interactions between PrPC and other soluble ligands (laminin, vibronectin, STI1) and transmembrane receptors (NCAM, integrin) [3, 16, 24, 34]. However, most of these studies neglect to assess the role of copper binding in PrPC activity, although it has been shown that extracellular copper can regulate PrPC expression and release of its GPI anchorless form [35]. Following the observations about co-existence and co-function between copper ions and PrPC, we have attempted to study the role of copper binding in regulating PrP signaling. Altogether, in this study, we could provide evidence for the involvement of copper coordination in PrP signaling in neuritogenesis using specific point mutations to disrupt copper-binding sites on the protein. Our data shows that copper binding stabilizes the unstructured part on PrP. While disrupting copper binding at OR region may change the protein structure, altered copper coordination at non-OR cause minimal but sufficient alteration in PrP structure that could prevent it from inducing neuritogenesis signaling. Cu2+ has high affinity to PrP with a Kd of 10−14 M for OR region and 4 × 10−14 M for non-OR region with His 96 and 111 (as human numbering) [36]. Therefore, higher affinity for copper at non-OR may explain its vulnerability in structural alteration due to different copper coordination.
Intriguingly, the GSS-linked mutation P102L (P101L as mouse numbering) seems to cause dysfunction of the protein. This result supports the loss-of-function hypothesis for prion pathology, particularly in the case of GSS syndrome. It has been shown in previous studies that this mutant protein could alter copper coordination at non-OR region making the His96 unable to participate in the interaction with copper at both neutral and acidic pH [18]. This result suggests that copper coordination at non-OR region may be critical to ensure correct conformation for this region, for correct interaction of PrPC with its partners in signaling complexes. Indeed, the non-OR mutant tested in our study failed to activate the ERK signaling cascade—the central pathway for neurite outgrowth [16, 37, 38]—comparing to the wt PrP protein. Moreover, this region has been suggested as a key site for prion conversion [18]. Thus, conformational changes in this region that would prevent copper binding could affect either PrPC function or increase its susceptibility to pathologic prion conversion.
Overall, our data provide evidence about the critical role of copper-binding residues in regulating PrPC function in neuritogenesis. All copper-binding sites are important to preserve the functional conformation of PrPC. The ability of PrPC in binding copper is key for maintaining the protein function and preventing its conversion to the prion state. Indeed, minimal structural changes at a non-OR copper-binding site may be sufficient to alter PrPC conformation and function, which may modulate its propensity to prion conversion. Our study may contribute to shed light between PrPC physiology and prion pathology.
References
Cisse M, Mucke L (2009) A prion protein connection. Nature 457:1090–1091
del Río JA, Ferrer I, Gavín R (2018) Role of cellular prion protein in interneuronal amyloid transmission. Prog Neurobiol
Martins VR, Beraldo FH, Hajj GN, Lopes MH, Lee KS, Prado MA, Linden R (2010) Prion protein: orchestrating neurotrophic activities. Curr Issues Mol Biol 12(2):63–86
Westergard L, Christensen HM, Harris DA (2007) The cellular prion protein (PrP(C)): its physiological function and role in disease. Biochim Biophys Acta 1772(6):629–644
Málaga-Trillo E, Solis GP, Schrock Y, Geiss C, Luncz L, Thomanetz V, Stuermer CAO (2009) Regulation of embryonic cell adhesion by the prion protein. PLoS Biol 7(3):e1000055
Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR (2008) Physiology of the prion protein. Physiol Rev 88(2):673–728
Roucou X, Gains M, LeBlanc AC (2004) Neuroprotective functions of prion protein. J Neurosci Res 75(2):153–161
Gasperini L, Meneghetti E, Pastore B, Benetti F, Legname G (2015) Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid Redox Signal 22(9):772–784
Pushie MJ, Pickering IJ, Martin GR, Tsutsui S, Jirik FR, George GN (2011) Prion protein expression level alters regional copper, iron and zinc content in the mouse brain. Metallomics 3(2):206–214
Walter ED, Stevens DJ, Spevacek AR, Visconte MP, Rossi AD, Millhauser GL (2009) Copper binding extrinsic to the octarepeat region in the prion protein. Curr Protein Pept Sci 10(5):529–535
Zatta P, Frank A (2007) Copper deficiency and neurological disorders in man and animals. Brain Res Rev 54(1):19–33
Turski ML, Thiele DJ (2009) New roles for copper metabolism in cell proliferation, signaling, and disease. J Biol Chem 284(2):717–721
Schlief ML, Gitlin JD (2006) Copper homeostasis in the CNS. Mol Neurobiol 33(2):81–90
Harris ZL, Gitlin JD (1996) Genetic and molecular basis for copper toxicity. Am J Clin Nutr 63(5):836S–841S
Waggoner DJ, Bartnikas TB, Gitlin JD (1999) The role of copper in neurodegenerative disease. Neurobiol Dis 6(4):221–230
Amin L, Nguyen XTA, Rolle IG, Este E, Giachin G, Tran TH, Šerbec VČ, Cojoc D, et al. Characterization of prion protein function by focal neurite stimulation. J Cell Sci, 2016.
Lau A, McDonald A, Daude N, Mays CE, Walter ED, Aglietti R, Mercer RCC, Wohlgemuth S et al (2015) Octarepeat region flexibility impacts prion function, endoproteolysis and disease manifestation. EMBO Mol Med 7(3):339–356
Giachin G, Mai PT, Tran TH, Salzano G, Benetti F, Migliorati V, Arcovito A, Longa SD et al (2015) The non-octarepeat copper binding site of the prion protein is a key regulator of prion conversion. Sci Rep 5:15253
Pool M, Thiemann J, Bar-Or A, Fournier AE (2008) NeuriteTracer: a novel ImageJ plugin for automated quantification of neurite outgrowth. J Neurosci Methods 168(1):134–139
Pinato G, Cojoc D, Lien LT, Ansuini A, Ban J, D’Este E, Torre V (2012) Less than 5 Netrin-1 molecules initiate attraction but 200 Sema3A molecules are necessary for repulsion. Sci Rep 2:675
Pearson G, Robinson F, Beers Gibson T, Xu B-e, Karandikar M, Berman K, Cobb MH (2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22(2):153–183
Ulla J, Vera N, Nina P, Palle S, Vladimir B, Elisabeth B (2001) The transcription factors CREB and c-Fos play key roles in NCAM-mediated neuritogenesis in PC12-E2 cells. J Neurochem 79(6):1149–1160
Caiati MD, Safiulina VF, Fattorini G, Sivakumaran S, Legname G, Cherubini E (2013) PrPC controls via protein kinase A the direction of synaptic plasticity in the immature hippocampus. J Neurosci 33(7):2973–2983
Kanaani J, Prusiner SB, Diacovo J, Baekkeskov S, Legname G (2005) Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. J Neurochem 95(5):1373–1386
Wulf M-A, Senatore A, Aguzzi A (2017) The biological function of the cellular prion protein: an update. BMC Biol 15:34
Steele AD, Emsley JG, Özdinler PH, Lindquist S, Macklis JD (2006) Prion protein (PrP<sup>c</sup>) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc Natl Acad Sci U S A 103(9):3416–3421
Santuccione A, Sytnyk V, Leshchyns’ka I, Schachner M (2005) Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59<sup>fyn</sup> and to enhance neurite outgrowth. J Cell Biol 169(2):341–354
Herms J, Tings T, Gall S, Madlung A, Giese A, Siebert H, Schürmann P, Windl O et al (1999) Evidence of presynaptic location and function of the prion protein. J Neurosci 19(20):8866–8875
Haeberlé AM, Ribaut-Barassin C, Bombarde G, Mariani J, Hunsmann G, Grassi J, Bailly Y (2000) Synaptic prion protein immuno-reactivity in the rodent cerebellum. Microsc Res Tech 50(1):66–75
Kardos J, Kovács I, Hajós F, Kálmán M, Simonyi M (1989) Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci Lett 103(2):139–144
SP K, Haitao Y, ZG W (2012) Copper-dependent regulation of NMDA receptors by cellular prion protein: implications for neurodegenerative disorders. J Physiol 590(6):1357–1368
Pushkar Y, Robison G, Sullivan B, Zheng W, Fu SX, Kohne M, Jiang W, Rohr S et al (2013) Aging results in copper accumulations in GFAP-positive cells in the subventricular zone. Aging Cell 12(5):823–832
Hunt CD, Idso JP (1995) Moderate copper deprivation during gestation and lactation affects dentate gyrus and hippocampal maturation in immature male rats. J Nutr 125(10):2700–2710
Chen S, Mange A, Dong L, Lehmann S, Schachner M (2003) Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol Cell Neurosci 22(2):227–233
Toni M, Massimino ML, Griffoni C, Salvato B, Tomasi V, Spisni E (2005) Extracellular copper ions regulate cellular prion protein (PrPC) expression and metabolism in neuronal cells. FEBS Lett 579(3):741–744
Jackson GS, Murray I, Hosszu LLP, Gibbs N, Waltho JP, Clarke AR, Collinge J (2001) Location and properties of metal-binding sites on the human prion protein. Proc Natl Acad Sci 98(15):8531–8535
Perron JC, Bixby JL (1999) Distinct neurite outgrowth signaling pathways converge on ERK activation. Mol Cell Neurosci 13(5):362–378
Cohen MR, Johnson WM, Pilat JM, Kiselar J, DeFrancesco-Lisowitz A, Zigmond RE, Moiseenkova-Bell VY (2015) NGF regulates TRPV2 via ERK signaling to enhance neurite outgrowth in developing neurons. Mol Cell Biol 35:4238–4252
Acknowledgments
This work was funded by the International School for Advanced Studies (SISSA) intramural funding to GL.
Author information
Authors and Affiliations
Contributions
XTAN and GL designed the experiments. DC developed the local delivery setup. XTAN performed practical work and analyzed the data. THT designed the plasmids and participated in protein purification and western blotting. XTAN and GL wrote the paper. All authors revised and approved the final manuscript.
Corresponding authors
Ethics declarations
All experiments were performed in accordance with European regulations (European Community Council Directive, November 24, 1986 [86/609/EEC]). Experimental procedures were notified to and approved by the Italian Ministry of Health, Directorate General for Animal Health. All experiments were approved by the local authority veterinary service of Trieste, Italy, and by the Ethics Committee of the Scuola Internazionale Superiore di Studi Avanzati (SISSA), Trieste. All efforts were made to minimize animal suffering and to reduce the number of animals used.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 353 kb)
Rights and permissions
About this article

Cite this article
Nguyen, X.T.A., Tran, T.H., Cojoc, D. et al. Copper Binding Regulates Cellular Prion Protein Function. Mol Neurobiol 56, 6121–6133 (2019). https://doi.org/10.1007/s12035-019-1510-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-019-1510-9