
Abstract
The biological functions of the cellular prion protein remain poorly understood. In fact, numerous studies have aimed to determine specific functions for the different protein domains. Studies of cellular prion protein (PrPC) domains through in vivo expression of molecules carrying internal deletions in a mouse Prnp null background have provided helpful data on the implication of the protein in signalling cascades in affected neurons. Nevertheless, understanding of the mechanisms underlying the neurotoxicity induced by these PrPC deleted forms is far from complete. To better define the neurotoxic or neuroprotective potential of PrPC N-terminal domains, and to overcome the heterogeneity of results due to the lack of a standardized model, we used neuroblastoma cells to analyse the effects of overexpressing PrPC deleted forms. Results indicate that PrPC N-terminal deleted forms were properly processed through the secretory pathway. However, PrPΔF35 and PrPΔCD mutants led to death by different mechanisms sharing loss of alpha-cleavage and activation of caspase-3. Our data suggest that both gain-of-function and loss-of-function pathogenic mechanisms may be associated with N-terminal domains and may therefore contribute to neurotoxicity in prion disease. Dissecting the molecular response induced by PrPΔF35 may be the key to unravelling the physiological and pathological functions of the prion protein.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The cellular prion protein (PrPC) is a glycosylphosphatidylinositol (GPI)-anchored glycoprotein located in the detergent-resistant areas of the membrane of several neuronal and non-neuronal cells [1, 2]. Pathogenesis of prion diseases in animals is associated with misfolding of PrPC into an infectious, proteinase-K-resistant, β-sheet-enriched isoform (PrPSC) [2]. Mature sequence of PrPC can be divided into two structurally well-defined regions: a long, flexible N-terminal tail (approximately the first 100 residues) and a globular C-terminal domain containing three α-helices and two β-strands flanking the first α-helix [3]. The N-terminal tail also has distinctive features: an octarepeat (OR) region, and a central domain (CD), which in turn comprises a charge cluster (CC) and a hydrophobic region (HR) [4].
Transgenic expression of PrPC variants carrying specific point mutations, domain insertions and deletions triggers cell death in a mouse Prnp o/o background (see [5, 6] for reviews). Among others, deletion mutants such as CD-lacking PrPC mice (94-134) [7], N-terminal ΔF35 PrPC mice (32-121/32-134) [8], and mice lacking PrPC residues 105-125 (ΔCR mice) [9] experience early cell degeneration (generally associated with the cerebellum), ataxia, myelin deficits and premature death. Surprisingly, degenerating granule neurons from ΔCR mice display extensive DNA fragmentation, without caspase-3 or caspase-8 activation [5], in contrast to mice carrying other PrPC forms (e.g., [10]). All these pathological neuronal phenotypes can be partially or totally rescued either by co-expression of one to five copies of the PRNP gene (see [5, 6] for reviews) or by overexpression of antiapoptotic proteins under neuronal promoters [10, 11]. Taken together, these results suggest that the central regions of PrPC play an important role in its physiological function, and that the neuroprotective properties of PrPC may lie in a putative binding region for other partners [6]. However, antibody-directed binding to different PrPC domains yields different results [12, 13]. In fact, PrPC aggregation in lipid rafts using the SAF61 antibody increased NADPH-oxidase-dependent oxidative stress, leading to cell death [14, 15].
Indeed, more relevantly, there is no consensus regarding which mechanisms, signalling pathways, or putative ligands or membrane receptors modulate PrPC-mediated cell death or survival (e.g., [4, 6]). However, emerging evidence indicates that PrPC may participate in the assembly of complexes at the cell surface and that this is likely the basis for its neurotrophic functions (see [16] for a review). Indeed, some partners have been described as binding PrPC, leading to particular functions, such as the 37 kDa/67 kDa laminin receptor, which has two binding sites on PrPC for LRP, termed PrPLRPbd1 (aa. 144-179) and PrPLRPbd2 (aa. 53-93) (e.g., [17]); the low-density lipoprotein receptor-related protein 1 (LRP1), LRP1-binding domain on PrPC aa. 23-107 [18]; heparan sulphate-containing proteins, with three binding regions on the prion protein aa. 23-52, 53-93, and 110-128 [19]; and the stress inducible protein 1 (STI1), with the interaction site on PrPC mapped to residues 113-128 [20].
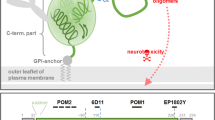
Thus, to determine the relative contribution of specific domains of PrPC to cell death or survival, we produced PRNP constructs containing the following deletions: PrPΔF35 (deleted residues 32-134), PrPΔCD (deleted residues 95-133), PrPΔCC (deleted residues 95-110), PrPΔHR (deleted residues 112-133), and PrPΔCR (deleted residues 105-125) (Fig. 1). These cDNAs were transfected in neuroblastoma (N2A) cells. Results indicated no detectable changes in protein trafficking and localization between constructs, or in glycosylation patterns of the deleted forms. However, PrPΔF35 and PrPΔCD truncated forms clearly induced cytotoxicity. This cytotoxic process was distinct in each truncated form, both of which correlated with a final increase in cleaved caspase-3. Surprisingly, these two mutants showed impaired α-cleavage processing. Our findings indicate that the absence of different PrPC domains triggers distinct cell death mechanisms.

Domain organization of murine PrPC and its derived truncated forms
Material and Methods
Plasmids and Construction of PrPC Deleted Forms
Mouse PrPC-encoding plasmid (pcDNA 3.1 backbone) and PrPC truncated form ΔF35 were provided by D. A. Harris (Boston University School of Medicine, Boston, MA, USA) and A. Aguzzi (University Hospital Zürich, Institute for Neuropathology, Switzerland), respectively [21, 8]. To generate deletion constructs ΔCD, ΔCC, ΔHR, and ΔCR, the PrPC-encoding plasmid was used as a template for inverse PCRs, and the inserts obtained were fused. Briefly, a primer set was designed for each construct in order to amplify the entire plasmid, except for the region of PRNP to be deleted, i.e., regions 95-133, 95-110, 112-133, and 105-125 for ΔCD, ΔCC, ΔHR, and ΔCR, respectively. Primers (Ecogen) are detailed as follows: CD (F: 5′-AGCAGGCCCATGATCCATTTTG-3′, R: 5′-ATGGGTACCCCCTCCTTGGC-3′); CC (F: 5′-GTGGCAGGGGCTGCGGCAG-3′, R: 5′-ATGGGTACCCCCTCCTTGGCC-3′); HR (F: 5′-AGCAGGCCCATGATCCATTTTG-3′, R: 5′-TGCCACATGCTTGAGGTTGG-3′); CR (F: 5′-TACATGCTGGGGAGCGCC-3′, R: 5′-TTTTGGTTTGCTGGGCTTGTTC-3′). After amplification (Accuprime Taq PolymeraseTM, Invitrogen), the blunt ends of the amplimers were phosphorylated using the T4 kinase reaction (Invitrogen) and then religated (Fast-Link Ligase®, Epicentre Biotech.). An aliquot of each ligation reaction was electroporated into Escherichia coli DH5α, and transformants were selected for ampicillin resistance. Twenty-five candidates were selected and screened by sequence analysis (Terminator Big Dye® v3.1, Applied Biosystems).
Cell Culture and Transfection
N2A neuroblastoma cells (ATCC CCL 131, American Type Culture Collection, Rockville, MD, USA) were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen), 10 % fetal bovine serum (FBS, Invitrogen), and 1 % penicillin/streptomycin (Invitrogen) in 75-cm2 culture bottles (Nunc) in a 5 % CO2 atmosphere at 37 °C. One day before transfection, cells were cultured in DMEM supplemented with 10 % FBS and without antibiotics, on poly-d-lysine (Sigma)-coated plates (Nunc). Transfection was performed using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions as indicated [22].
Immunocytochemical Procedures
One day before transfection, counted N2A cells were seeded onto poly-d-lysine (0.01 μg/μl)-coated glass coverslips (12 mm Ø). Twenty-four hours post-transfection (p.t.), cells were fixed in 4 % buffered paraformaldehyde (Sigma), then and permeabilized with 0.1 % Triton X-100 (Sigma) in 0.1 M PBS. Coverslips were blocked with 10 % FBS in 0.1 M PBS prior to incubation with primary antibodies. Wild-type PrPC and its deleted forms were detected using anti-mouse 6H4 (1:500, Prionics). The antibodies used to detect intracellular markers were anti-rabbit sec61α (1:500, Upstate), anti-rabbit EEA1 (1:500, Abcam), and anti-rabbit rab4A (1:500, Santa Cruz). After incubation with primary antibodies, cells were incubated with the pertinent Alexa Fluor-tagged secondary antibodies (Alexa-488 goat anti-mouse or Alexa-568 goat anti-rabbit) (Invitrogen). Finally, cells were stained with 0.1 μM DAPI (Sigma) diluted in 0.1 M PBS, mounted on Mowiol (Calbiochem), and viewed using an Olympus BX61 fluorescence and spectral microscope. For total internal reflection fluorescence microscopy (TIRFM), poly-d-lysine-coated fluorodish cell culture dishes were used (23.5 mm Ø, World Precision Instruments). After incubation with secondary antibodies, cells were kept in PBS at 4 °C until visualization. Cells were viewed using an Olympus Total Internal Reflection Microscope (TIRF) from the Advanced Digital Microscopy Core Facility of the Institute for Research in Biomedicine (IRB Barcelona).
Determination of Cell Death
Cell death was assessed using a propidium iodide (PI, Sigma-Aldrich) staining method following the protocol of Enguita and colleagues [23, 24]. Briefly, 5 hours after transfection in 24-well plates, 30 μM PI was added to each well. PI fluorescence was measured using a FL600 Microplate Fluorescence Reader (BioTeck). Spectrofluorometer analysis and settings were as follows: 530-nm excitation, 645-nm emission, and data recorded in relative fluorescence units. As an index of cell death that was not related to actual differences between samples, baseline fluorescence F 1 was measured 1 h after addition of PI. Subsequent fluorescence readings were taken at several time points (F n) after the onset of the experiment, keeping the cells in the incubator between measurements. Finally, cells were permeabilized for 10 min at 37 °C with 500-μM digitonin (Sigma Aldrich) to obtain the maximum fluorescence (100 % of cell death or F max). The percentage of cell death was calculated as follows: % cell death = 100 × (F n −F1)/(Fmax−F1) [24].
Isolation of Detergent-Resistant Membrane Domains
Detergent-resistant membrane (DRM) domains were isolated as previously described ([25]). Transfected cells from two 100-mm culture dishes were lysed on ice for 20 min in 1 % Triton X-100 in MNE buffer (25 mM MES pH 6.5, 150 mM NaCl, 2 mM EDTA) and subsequently homogenized. The homogenates were mixed with 90 % sucrose prepared in MNE buffer to reach a final concentration of 45 % sucrose in MNE buffer and then placed at the bottom of a centrifuge tube. The samples were then overlayed with 35 % sucrose and 5 % sucrose in MNE buffer, and then ultracentrifuged at 175,000g in an Optima L-90K Ultracentrifuge (Beckman Coulter) for 20 h at 4 °C. Fractions of 0.4 ml were collected from the top of the gradient and subjected to Western blot analysis.
Dihydroethidium (DHE) Staining
The reactive oxidative species probe DHE (Sigma) was prepared as a 10-μg/μl stock in DMSO, as previously described [26, 14], and used at a final concentration of 1 μg/ml. Cells were stained for 15 min at 37 °C and afterwards visualized in an inverted Olympus microscope IX-71 equipped with a CO2 and temperature cell culture chamber (LCi instruments) (see [27, 28] for technical details). After photodocumentation, fluorescence images were analysed using ImageJTM software.
Caspase-3 Activity and Western Blotting Techniques
Twenty-four hours after transfection, cells were scraped and lysed for 20 min on ice in cold lysis buffer (50 mM Hepes pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA pH 8, 10 % glycerol, 1 % Triton X-100) containing 1x protease inhibitor cocktail and phosphatase inhibitors. Lysates were centrifuged at 12000g for 5 min at 4 °C, and supernatants were collected. Protein concentration was determined using a BCA protein assay kit (Pierce). The caspase-3 activity assay, with Ac-Asp-Glu-Val-Asp-7-amino-4-trifluoromethylcoumarin (Ac-DEVD-AFC, Sigma) as a substrate, was performed as previously described [29]. Cell extracts were boiled in Laemli sample buffer at 100 °C for 10 min, and equal amounts of protein were resolved on 8–15 % sodium dodecyl sulfate-polyacrylamide gels, electrotransferred overnight at 4 °C to nitrocellulose membranes (Whatmann), and processed for immunoblotting. Primary antibodies used were anti-PrP 6H4 (1:5000, Prionics), anti-β-actin (1:20000, Cell Signalling), anti-flotillin-1 (1:1000, BD Transduction Laboratories), and anti-caspase-3 (1:750, Cell Signalling). The appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (DAKO) was used. Blots were visualized using the ECL-plus kit (Amersham-Pharmacia Biotech). Densitometric analysis was performed using ImageJTM software [39].
Mitochondrial Membrane Potential Assay
Mitochondrial membrane potential (MMP) was assessed using a JC-10 fluorometric assay (Sigma-Aldrich). Eighteen hours after transfection in 24-well plates, JC-10 dye loading solution was added to each well following the manufacturer’s instructions, and cells were incubated for 30 min at 37 °C. PI fluorescence was measured using a FL600 Microplate Fluorescence Reader (BioTeck). Spectrofluorometer analysis and settings were as follows: 540-nm excitation, 590-nm emission to detect red-fluorescence JC-10 aggregates, and 490-nm excitation, 525-nm emission to detect green fluorescence due to the MMP collapse. Data were recorded in relative fluorescence units and corrected with total protein determined using a BCA protein assay kit (Pierce). Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP; 20 μM) was used as a positive control and the ratio of 525/590 nm fluorescence intensity was used to determine mitochondrial membrane depolarization.
PNGase F Digestion
Cell lysates obtained as previously described were treated with PNGase F (New England Biolabs), following the manufacturer’s instructions. Briefly, 20 μg of total protein was denatured for 10 min at 100 °C, digested overnight at 37 °C using 1000 enzyme units in a final volume of 30 μl, and subsequently analysed with Western blotting using anti-PrP 6H4 antibody.
Statistical Analysis
All results are shown as mean ± SEM. One-way analysis of variance (ANOVA test) was used for statistical analysis of data using Statgraphics Plus for Windows software version 5.1 (Statpint Technologies Inc., Warrenton, VA, USA). Asterisks in the histograms indicate the following p values of significance: (*) p < 0.05 or (**) p < 0.01 was considered statistically significant.
Results
Characterization of PrPC Deleted Forms in N2A Cells
After cloning, the expression of all PrPC variants in N2A cells was tested with Western blotting (Fig. 2). Endogenous level of PRNP expression in neuroblastoma cells was low, and all PrPC-modified proteins were detectable. However, expression of both PrPΔF35 and PrPΔCD was markedly lower than the rest of deleted forms, representing less than 50 % of that of full-length PrPC (Fig. 2a, b). Wild-type PrPC displayed three major bands ranging from 25 to 35 kDa, representing the un-, mono-, and diglycosylated isoforms of the protein, respectively [30]. All PrPC forms showed a similar band pattern, suggesting that these proteins were also N-glycosylated. Upon PNGase F treatment, all deleted variants displayed enhanced electrophoretic motility, which supports the claim that each protein was glycosylated. Furthermore, similar to full-length PrPC, in PrPΔCC, PrPΔHR, and to a lesser extent PrPΔCR, a fast-migrating band of approximately 17 kDa was observed, which corresponded to the C1 fragment, produced physiologically by cleavage approximately at residue 110 [31, 32]. In contrast, PNGase F treatment of PrPΔF35 and PrPΔCD did not produce an equivalent fragment, which is consistent with the absence not only of the α-cleavage site in these proteins, but also of a large flanking region (Fig. 2c). Taken together, these results indicate that all the deleted forms of PrPC generated in this study are processed through the secretory pathway, although there is an abnormal endoproteolysis of PrPΔF35, and PrPΔCD that prevents the formation of biologically active C1 and N1 fragments.

Expression levels and PNGase F digestion of PrPC N-terminal deleted forms from transfected N2A cells. a N2A cells were transiently transfected for 24 hours. Cell lysates were obtained and normalized for total protein by BCA assay. b Histogram showing quantification of the expression levels of PrPC N-terminal deleted forms from transfected N2A cells. Bars represent the mean ± SEM of three independent experiments (*p < 0.05, **p < 0.01, versus PrPC transfected controls (ANOVA test)). c Samples were subsequently analysed directly or after PNGase F digestion with SDS-PAGE and immunoblotting with antibody 6H4 (recognizes a C-terminal domain epitope, human PrP: amino acids 144-152). Detected band resulting from PrPΔF35 PNGase F digestion corresponds to unglycosylated full-length protein. Detected bands of other PrPC forms correspond to C1 fragment. Blots are representative of three independent experiments
Cellular Localization of Transfected PrPC Deleted Variants in Lipid Rafts of N2A Cells
Since PrPC is known to associate with lipid rafts in cultured cells [33], we employed TIRFM to corroborate the localization of the transfected PrPC form using the monoclonal 6H4 antibody which recognizes mouse PrP, amino acids 144-152. All PrPC deleted forms exhibited, in two independent experiments, punctate labelling at the membrane (data not shown), similar to that of PrPC, which is typical of raft-associated, GPI-anchored proteins [34, 35]. See Fig. 3a, for PrPC, PrPΔF35, and PrPΔCD. To confirm these results, we isolated detergent-resistant membranes (DRM) of homogenates of transfected N2A cells with sucrose density gradient centrifugation. All PrPC variants, as well as PrPC, were enriched in fractions containing the lipid raft resident protein flotillin-1 (see Fig. 3b and Suppl. Fig. 2). To further characterize the intracellular distribution of the PrPC constructs, we developed double immunocytochemical detection of the transfected forms and intracellular organelle or compartment markers. We did not observe significant co-localization of either full-length PrPC or any of the PrP variants with sec61, a marker for the endoplasmic reticulum (Fig. 4a and Suppl. Fig. 3), EEA1, an early endosomal marker (Fig. 4b and Suppl. Fig. 4), or rab4, a marker for recycling endosomes (Fig. 4c and Suppl. Fig. 5). These results suggest that all constructs are intracellularly processed and that none of the deletions obtained in this study disturbed either the intracellular or cell surface distribution of PrPC in N2A cells.

Cellular localization of PrPC-deleted forms PrPΔF35 and PrPΔCD. a TIRFM image reveals a punctate distribution of PrPC and its variants in the cell surface region. Scale bar = 20 μm. b Fractions 1-12 of the sucrose density gradient were analysed with Western blotting. Flotillin (48-kDa) was used as a marker of lipid rafts. Non-buoyant PrP may indicate raft disruption or presence of the protein in other subcellular fractions. Blots are representative of three independent experiments

Neither full-length PrPC nor the N-terminal deleted forms PrPΔF35 and PrPΔCD co-localize with the ER, early or recycling endosomes. Representative micrographs of two independent experiments of transfected N2A cells labeled with specific markers for a ER (Sec61, red), b early endosomes (EEA1, red) or c recycling endosomes (Rab4, red) for co-localization with PrP forms (6H4-green). Scale bar = 50 μm
Neurotoxic Properties of the Truncated Forms of PrPC
The cell viability of transiently transfected N2A cells was analysed using a quantitative method, based on the measure of PI emission after nuclear DNA uptake. Damage to the cytoplasmic membrane permits the entry of PI, which yields bright red fluorescence after its interaction with nuclear DNA. Twenty-four hours post-transfection (p.t.), cells expressing PrPΔF35 showed a 3.5-fold increase in cell death (20.7 %) compared to cells over-expressing full-length PrPC (6 %). PrPΔCD, PrPΔHR and PrPΔCR provoked a 2-fold increase in cell death (13.9, 14.2 and 12.4 %, respectively), whereas no significant differences were observed in the case of PrPΔCC over-expression (7.5 %). Forty-eight hours p.t., N2A over-expressing PrPΔF35 reached 40 % of cell death. There were slight differences between the rest of the PrPC variants and PrPC (23 %), as the values of cell death ranged from 25 % (PrPΔCC) to 31 % (PrPΔCD) (Fig. 5a). An alternative analysis of the mitochondrial activity of cells 24 h p.t., WST-1 assay (Promega, Madison, MA, USA), confirmed the cell death results obtained (data not shown).

PrPΔF35 and PrPΔCD are cytotoxic for N2A cells. a IP histogram showing the percentage of non-viable N2A cells over time after transfection with the empty vector pcDNA3.1, wild-type PrPC, and PrP deleted variants. Bars represent the mean ± SEM of three independent experiments (*p < 0.05, **p < 0.01, versus PrPC transfected or non-transfected controls (ANOVA test)). b Analysis of caspase-3 activation in transfected N2A cultures, as determined with Western blotting. Densitometric values are standardized with empty vector transfected cells, and quantification of two independent experiments was represented as the average of fold change ± SEM. c Fluorescence and parallel phase contrast views of cultures incubated with dihydroethidium (1 μg/ml) after 24 h of transfection with the empty vector, full-length PrPC, PrPΔF35 and PrPΔCD. Scale bar = 200 μm. d Histogram illustrating the quantitative results of ROS production. Bars represent the mean ± SEM of three independent experiments (**p < 0.01, versus empty-vector transfected cells (ANOVA test))
To further analyse the cell death observed, we measured the level of activated (cleaved) caspase-3 24 h p.t. with Western blotting. As shown in Fig. 5b, only cells transfected with PrPΔF35 and PrPΔCD showed a statistically significant increase in the percentage of cleaved caspase-3 (43 and 33 %, respectively) (**p < 0.01, versus empty vector transfected cells (ANOVA test)). These results were confirmed by a fluorometric assay using DEVD-AFC as the caspase-3 substrate to evaluate this protease activity (Suppl. Fig. 1). Moreover, we performed an assay to analyse mitochondrial membrane potential (MMP) at 18 p.t. and we found a statistically significant loss (36 %) only in cells transfected with PrPΔCD (*p < 0.05, versus empty vector transfected cells (ANOVA test)), see Suppl. Fig 6.
Recently, several PrPC deleted forms and disease-associated mutations have been shown to induce spontaneous ionic currents produced by non-selective, cation-permeable channels in the cell membrane. These currents can be abolished with the application of exogenous glycosaminoglycans (GAGs) [36, 37]. Since these channels might alter the reactive oxygen species (ROS) balance in the cell, transfected N2A monolayers were incubated with the fluorescent probe DHE to monitor ROS generation 24 h p.t. Analysis of three independent experiments show that only PrPΔCD clearly increased ROS generation, which was assessed by the number of DHE-labelled nuclei. Furthermore, the addition of heparin (100 μg/ml, Sigma) to the culture medium reversed this ROS production (Fig. 5 c, d). These data suggest that the cell deaths induced by PrPΔF35 and PrPΔCD involve different pathways.
Discussion
Transgenic PrPC-null mice expressing deleted forms of PrPC have a neurodegenerative phenotype that is rescued by co-expression of full-length PrPC [8, 7, 9]. These animals show white matter degeneration in the brain and spinal cord, as well as degeneration of the cerebellar granule neurons. However, not all deleted mice showed a similar pattern of degeneration. In fact, ΔC4 mice do not display cerebellar degeneration in contrast to ΔF35 mice [8]. When the PrPC paralog Doppel (Dpl), a protein structurally similar to N-terminal deleted PrPC, is ectopically expressed in the brain of mice, it also causes a neurodegenerative phenotype that can be overcome by wild-type PrPC [38–40]. Thus, the observed toxicity has been attributed to the lack of the physiological neuroprotective function of PrPC in these mutants, since expression of wild-type PrPC reverses neurodegeneration in a dose-dependent manner.
In the present study, we generated several PrPC-deleted constructs that affect the main domains characterizing the N-terminal region of the protein, in order to set up a culture system for their analysis and comparison, overcoming the differences observed in transgenic animals. We chose the N2A cell line as our culture model to dissect the N-terminal part of PrPC because it shows neuronal-like characteristics [41] and has been successfully used in in vitro prion propagation and signalling [42, 43, 22]. Deletion of domains CD, CC, HR, CR and even all the unstructured N-terminal domain of PrPC (PrPΔF35) does not seem to interfere with maturation or protein trafficking of the PrPC forms to the plasma membrane. All deleted forms were complex glycosylated and present in lipid rafts (Figs. 2 and 3b). In spite of this, PrPΔF35 caused a clear increase in cell death 24 h p.t., accompanied by an increment of cleaved caspase-3 (Fig. 5a, b). Conversely, PrPΔCD, which showed slight differences with PrPC in terms of cell death 24 h p.t., induced ROS production that also correlated with mitochondrial membrane depolarization and caspase-3 activation (Suppl. Fig. 6 and Fig. 5). The rest of the deleted forms did not induce substantial survival changes in our experiments.
Under physiological conditions, PrPC is cleaved in a region termed the α-cleavage site. This results in two fragments: the flexible N-terminal domain, or N1, and the globular C-terminal domain, or C1 [44, 32]. All the aforementioned PrPC N-terminal deleted variants that cause neurotoxicity in transgenic PrP-null mice lack a region that is relatively close to or embraces this cleavage site [8, 7, 9]. However, Oliveira-Martins and coworkers reported high tolerance to variation of the sequence surrounding the cleavage site [45]. Interestingly, only PrPΔF35 and PrPΔCD showed an absolute lack of a C1-like fragment when expressed in N2A cells (Fig. 2C). This suggests that α-cleavage might somehow be involved in the cytotoxicity conferred by these PrPC mutants. In fact, neuroprotective properties of N1 and the neurotoxic consequences of the absence of the α-cleavage have recently been reported [46].
PrPC can be divided into two structurally well-defined parts: an unstructured N-terminal domain and a globular C-terminal domain. Since the deletions obtained in this study affect only the N-terminal part of PrPC (Fig. 1), they are not likely to alter the overall folding of the resulting deleted proteins. Nor are differences in the expression level of the transgenes responsible for the observed effects, since Western blotting with antibody 6H4 shows that both PrPΔF35 and PrPΔCD are present at lower levels than the rest of the deleted forms and full-length PrPC (Fig. 2a, b). Furthermore, all deleted proteins seem to undergo correct sorting to the cell surface (Fig. 3). In this respect, the presence of PrPΔF35 and PrPΔCD in the detergent-resistant areas of the membrane suggests that their cytotoxicity could be linked in part to a putative receptor, inducing, blocking, or lacking a transduction signal due to competition with PrPC for this common ligand, as has been proposed for transgenic PrPC-null mice expressing PrPC deletion mutants [7, 9, 8]. In this respect, a wide variety of ligands for PrPC unstructured N-terminal region have been described, including metals, proteins, lipids, and glycosaminoglycans (reviewed in [47]), supporting the role of PrPC as a broad-spectrum molecular sensor at the cell surface. In this scenario, the authors describe the effects of subversion of this domain on lack of neuroprotective function through α-cleavage reduction [48] and/or specific ligand interaction and endocytosis [47]. In addition, the N-terminal interaction with Aβ oligomers [49] and PrPSC [50] may promote a switch in PrPC function, leading to neurotoxicity.
A recent study reported that removal of an N-terminal polybasic amino acid segment (i.e., residues 23-31) eliminated the toxicity of deleted forms of PrPC in vivo as well as in vitro, which suggests that this domain is responsible for the deleterious effects caused by altered forms of PrPC [51]. All PrPC deleted forms used in our study contained this N-terminal polybasic domain (PBD). However, only PrPΔF35 and PrPΔCD caused cytotoxicity in our culture model. Thus, although the N-terminal PBD may be of relevance, there is at least one other mechanism by which these molecules exert their cytotoxic effect. Consequently, our results suggest that the cell death observed when PrPΔF35 and PrPΔCD are expressed is caused by two different mechanisms. PrPΔF35 causes a high rate of cell death and caspase-3 activation but does not induce a substantial increase in ROS production. Conversely, PrPΔCD provokes minor cleavage of caspase-3 and raises ROS levels, an effect that is counteracted by the application of heparin as an exogenous GAG. Residues 23-31 constitute one of the three regions involved in GAG binding on PrPC [19]. Furthermore, it has previously been reported that pentosan polysulphate may abolish PrPΔCR-induced currents and hypersensitivity to certain classes of cationic drugs by binding to the PBD [36, 52]. Thus, the ROS imbalance observed when PrPΔCD is expressed might be a consequence of the PBD-induced cationic channels. Nevertheless, this effect was observed for PrPΔCD but not for the rest of the deleted forms assayed. This indicates that PrPC pore-forming activity may not be exclusively dependent on regions 23-31.
The distinct cytotoxicity elicited by PrPΔF35 indicates that there is at least one other pathway by which aberrant forms of PrPC might exert toxicity. In fact, the phenotype of mice that express either Dpl or PrPΔF35 [53, 39, 8] is partially rescued by Bcl-2 over-expression [10, 54] and Bax suppression [55, 11], which suggests that both apoptotic mechanisms and Bax-independent pathways are involved. In this sense, our analysis of MMP has shown a depolarization induced by PrPΔCD that precedes the activation of caspase-3, reinforcing the implication of an intrinsic pathway in this model. However, PrPΔF35 has not shown a significant change in MMP reduction compared with control, suggesting that the extrinsic pathway promotes the highest cell death detected with IP and the activation of caspase-3. At any rate, there may be a connection between the death-receptor (extrinsic) pathway and the mitochondrial (intrinsic) pathway that explains the implication of Bcl-2 family members.
In conclusion, different mechanisms whereby deleted mutant forms of PrPC cause neurotoxicity seem to coexist (Fig. 6), including a lack of interaction with different neurotrophic ligands or new interactions that activate neurotoxicity themselves. This could explain the different pathological phenotypes of deleted transgenic mice and prionopathies. In this context, it is important to note that the N-terminal domain might contribute to neurotoxicity, as proximity of this flexible tail (FT) to the plasma membrane triggers intracellular oxidative stress responses leading to cell death [56]. As well, protein misfolding in prion disease such as truncated forms of PrPC may be associated in this same context. And in this sense, Sonati et al. demonstrated the induction of similar cellular death cascades in two models: use of antibodies directed to the globular C-terminal domain of PrPC and prions [12]. Moreover, antibodies against the N-terminal domain of PrPC prevented this toxicity, suggesting a role for the FT in both models [56]. In addition to all the above, the CD of PrPC protein, which lacks both deleted forms associated with neurotoxicity, PrPΔF35 and PrPΔCD, exerts site- and/or cell-specific neurotrophic functions that might prevent neurotoxic effects of FT membrane interaction [6]. A similar situation could exist in PrPSC membrane interaction as a consequence of the misfolding in prionopathies [56].

Pathogenic mechanisms of gain-of-function and loss-of-function may contribute to neurotoxicity in prion diseases. Therefore, promoting neurotoxicity, independently of PrPC expression or loss of neuroprotective functions of PrPC, converges to generate the final pathogenic phenotype
In conclusion, PrPΔF35 and PrPΔCD mutants lead to death by at least two different mechanisms in the same cellular background, both sharing loss of alpha-cleavage and activation of caspase-3. Dissecting the molecular response induced by PrPΔF35 may be key to the study of the signalling pathways induced by PrPC or PrPSC in a physiological and pathological state. Furthermore, the advantages of a simple cell culture model compared to more complicated cell assays and/or transgenic mice mean that this study may pave the way to future research.
References
Gorodinsky A, Harris DA (1995) Glycolipid-anchored proteins in neuroblastoma cells form detergent-resistant complexes without caveolin. J Cell Biol 129(3):619–627
Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216(4542):136–144
Riek R, Hornemann S, Wider G, Glockshuber R, Wuthrich K (1997) NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231). FEBS Lett 413(2):282–288
Nicolas O, Gavin R, del Rio JA (2009) New insights into cellular prion protein (PrPc) functions: the "ying and yang" of a relevant protein. Brain Res Rev 61(2):170–184. doi:10.1016/j.brainresrev.2009.06.002
Solomon IH, Schepker JA, Harris DA (2010) Prion neurotoxicity: insights from prion protein mutants. Curr Issues Mol Biol 12(2):51–61
Baumann F, Pahnke J, Radovanovic I, Rulicke T, Bremer J, Tolnay M, Aguzzi A (2009) Functionally relevant domains of the prion protein identified in vivo. PLoS One 4(9), e6707. doi:10.1371/journal.pone.0006707
Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH, Heikenwalder M, Rulicke T et al (2007) Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J 26(2):538–547
Shmerling D, Hegyi I, Fischer M, Blattler T, Brandner S, Gotz J, Rulicke T, Flechsig E et al (1998) Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 93(2):203–214
Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA (2007) Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105-125. EMBO J 26(2):548–558
Nicolas O, Gavin R, Braun N, Urena JM, Fontana X, Soriano E, Aguzzi A, del Rio JA (2007) Bcl-2 overexpression delays caspase-3 activation and rescues cerebellar degeneration in prion-deficient mice that overexpress amino-terminally truncated prion. FASEB J 21(12):3107–3117. doi:10.1096/fj.06-7827com
Li A, Barmada SJ, Roth KA, Harris DA (2007) N-terminally deleted forms of the prion protein activate both Bax-dependent and Bax-independent neurotoxic pathways. J Neurosci 27(4):852–859
Sonati T, Reimann RR, Falsig J, Baral PK, O'Connor T, Hornemann S, Yaganoglu S, Li B et al (2013) The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 501(7465):102–106. doi:10.1038/nature12402nature12402
Klohn PC, Farmer M, Linehan JM, O'Malley C, Fernandez de Marco M, Taylor W, Farrow M, Khalili-Shirazi A et al (2012) PrP antibodies do not trigger mouse hippocampal neuron apoptosis. Science 335(6064):52. doi:10.1126/science.1215579335/6064/52
Gavin R, Braun N, Nicolas O, Parra B, Urena JM, Mingorance A, Soriano E, Torres JM et al (2005) PrP(106-126) activates neuronal intracellular kinases and Egr1 synthesis through activation of NADPH-oxidase independently of PrPc. FEBS Lett 579(19):4099–4106. doi:10.1016/j.febslet.2005.06.037
Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O (2000) Signal transduction through prion protein. Science 289(5486):1925–1928
Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR (2008) Physiology of the prion protein. Physiol Rev 88(2):673–728. doi:10.1152/physrev.00007.2007
Mbazima V, Da Costa Dias B, Omar A, Jovanovic K, Weiss SF (2010) Interactions between PrP(c) and other ligands with the 37-kDa/67-kDa laminin receptor. Front Biosci 15:1150–1163
Parkyn CJ, Vermeulen EG, Mootoosamy RC, Sunyach C, Jacobsen C, Oxvig C, Moestrup S, Liu Q et al (2008) LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J Cell Sci 121(Pt 6):773–783. doi:10.1242/jcs.021816
Warner RG, Hundt C, Weiss S, Turnbull JE (2002) Identification of the heparan sulfate binding sites in the cellular prion protein. J Biol Chem 277(21):18421–18430. doi:10.1074/jbc.M110406200M110406200
Arantes C, Nomizo R, Lopes MH, Hajj GN, Lima FR, Martins VR (2009) Prion protein and its ligand stress inducible protein 1 regulate astrocyte development. Glia 57(13):1439–1449. doi:10.1002/glia.20861
Fioriti L, Dossena S, Stewart LR, Stewart RS, Harris DA, Forloni G, Chiesa R (2005) Cytosolic prion protein (PrP) is not toxic in N2a cells and primary neurons expressing pathogenic PrP mutations. J Biol Chem 280(12):11320–11328. doi:10.1074/jbc.M412441200
Llorens F, Carulla P, Villa A, Torres JM, Fortes P, Ferrer I, del Rio JA (2013) PrP(C) regulates epidermal growth factor receptor function and cell shape dynamics in Neuro2a cells. J Neurochem 127(1):124–138. doi:10.1111/jnc.12283
Enguita M, DeGregorio-Rocasolano N, Abad A, Trullas R (2005) Glycogen synthase kinase 3 activity mediates neuronal pentraxin 1 expression and cell death induced by potassium deprivation in cerebellar granule cells. Mol Pharmacol 67(4):1237–1246. doi:10.1124/mol.104.007062
Vilches S, Vergara C, Nicolas O, Sanclimens G, Merino S, Varon S, Acosta GA, Albericio F et al (2013) Neurotoxicity of prion peptides mimicking the central domain of the cellular prion protein. PLoS One 8(8):e70881. doi:10.1371/journal.pone.0070881PONE-D-13-04002
Legler DF, Doucey MA, Schneider P, Chapatte L, Bender FC, Bron C (2005) Differential insertion of GPI-anchored GFPs into lipid rafts of live cells. FASEB J 19(1):73–75. doi:10.1096/fj.03-1338fje
Bindokas VP, Jordan J, Lee CC, Miller RJ (1996) Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci 16(4):1324–1336
Gil V, del Rio JA (2012) Analysis of axonal growth and cell migration in 3D hydrogel cultures of embryonic mouse CNS tissue. Nat Protoc 7(2):268–280. doi:10.1038/nprot.2011.445
Nocentini S, Reginensi D, Garcia S, Carulla P, Moreno-Flores MT, Wandosell F, Trepat X, Bribian A et al (2012) Myelin-associated proteins block the migration of olfactory ensheathing cells: an in vitro study using single-cell tracking and traction force microscopy. Cell Mol Life Sci 69(10):1689–1703. doi:10.1007/s00018-011-0893-1
Gurtu V, Kain SR, Zhang G (1997) Fluorometric and colorimetric detection of caspase activity associated with apoptosis. Anal Biochem 251(1):98–102. doi:10.1006/abio.1997.2220
Pan T, Li R, Wong BS, Liu T, Gambetti P, Sy MS (2002) Heterogeneity of normal prion protein in two-dimensional immunoblot: presence of various glycosylated and truncated forms. J Neurochem 81(5):1092–1101
Harris DA, Huber MT, van Dijken P, Shyng SL, Chait BT, Wang R (1993) Processing of a cellular prion protein: identification of N- and C-terminal cleavage sites. Biochemistry 32(4):1009–1016
Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio-Gambetti L (1995) Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem 270(32):19173–19180
Naslavsky N, Stein R, Yanai A, Friedlander G, Taraboulos A (1997) Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J Biol Chem 272(10):6324–6331
Kwiatkowska K, Frey J, Sobota A (2003) Phosphorylation of FcgammaRIIA is required for the receptor-induced actin rearrangement and capping: the role of membrane rafts. J Cell Sci 116(Pt 3):537–550
Taylor DR, Watt NT, Perera WS, Hooper NM (2005) Assigning functions to distinct regions of the N-terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J Cell Sci 118(Pt 21):5141–5153. doi:10.1242/jcs.02627
Solomon IH, Huettner JE, Harris DA (2010) Neurotoxic mutants of the prion protein induce spontaneous ionic currents in cultured cells. J Biol Chem 285(34):26719–26726. doi:10.1074/jbc.M110.134619M110.134619
Solomon IH, Khatri N, Biasini E, Massignan T, Huettner JE, Harris DA (2011) An N-terminal polybasic domain and cell surface localization are required for mutant prion protein toxicity. J Biol Chem 286(16):14724–14736. doi:10.1074/jbc.M110.214973
Rossi D, Cozzio A, Flechsig E, Klein MA, Rulicke T, Aguzzi A, Weissmann C (2001) Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J 20(4):694–702
Sakaguchi S, Katamine S, Nishida N, Moriuchi R, Shigematsu K, Sugimoto T, Nakatani A, Kataoka Y et al (1996) Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature 380(6574):528–531
Moore RC, Mastrangelo P, Bouzamondo E, Heinrich C, Legname G, Prusiner SB, Hood L, Westaway D et al (2001) Doppel-induced cerebellar degeneration in transgenic mice. Proc Natl Acad Sci U S A 98(26):15288–15293
Schubert D, Humphreys S, Baroni C, Cohn M (1969) In vitro differentiation of a mouse neuroblastoma. Proc Natl Acad Sci U S A 64(1):316–323
Alais S, Simoes S, Baas D, Lehmann S, Raposo G, Darlix JL, Leblanc P (2008) Mouse neuroblastoma cells release prion infectivity associated with exosomal vesicles. Biol Cell 100(10):603–615. doi:10.1042/BC20080025
Rangel A, Burgaya F, Gavin R, Soriano E, Aguzzi A, Del Rio JA (2007) Enhanced susceptibility of Prnp-deficient mice to kainate-induced seizures, neuronal apoptosis, and death: Role of AMPA/kainate receptors. J Neurosci Res 85(12):2741–2755. doi:10.1002/jnr.21215
Mange A, Beranger F, Peoc'h K, Onodera T, Frobert Y, Lehmann S (2004) Alpha- and beta- cleavages of the amino-terminus of the cellular prion protein. Biol Cell 96(2):125–132. doi:10.1016/j.biolcel.2003.11.007S0248490004000061
Oliveira-Martins JB, Yusa S, Calella AM, Bridel C, Baumann F, Dametto P, Aguzzi A (2010) Unexpected tolerance of alpha-cleavage of the prion protein to sequence variations. PLoS One 5(2), e9107. doi:10.1371/journal.pone.0009107
McDonald AJ, Millhauser GL (2014) PrP overdrive: does inhibition of alpha-cleavage contribute to PrP(C) toxicity and prion disease? Prion 8 (2). doi:28796 [pii]
Beland M, Roucou X (2012) The prion protein unstructured N-terminal region is a broad-spectrum molecular sensor with diverse and contrasting potential functions. J Neurochem 120(6):853–868. doi:10.1111/j.1471-4159.2011.07613.x
Guillot-Sestier MV, Sunyach C, Druon C, Scarzello S, Checler F (2009) The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J Biol Chem 284(51):35973–35986. doi:10.1074/jbc.M109.051086M109.051086
Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457(7233):1128–1132. doi:10.1038/nature07761
Solforosi L, Bellon A, Schaller M, Cruite JT, Abalos GC, Williamson RA (2007) Toward molecular dissection of PrPC-PrPSc interactions. J Biol Chem 282(10):7465–7471. doi:10.1074/jbc.M610051200
Westergard L, Turnbaugh JA, Harris DA (2011) A nine amino acid domain is essential for mutant prion protein toxicity. J Neurosci 31(39):14005–14017. doi:10.1523/JNEUROSCI.1243-11.2011
Massignan T, Biasini E, Harris DA (2011) A drug-based cellular assay (DBCA) for studying cytotoxic and cytoprotective activities of the prion protein: a practical guide. Methods 53(3):214–219. doi:10.1016/j.ymeth.2010.11.005
Katamine S, Nishida N, Sugimoto T, Noda T, Sakaguchi S, Shigematsu K, Kataoka Y, Nakatani A et al (1998) Impaired motor coordination in mice lacking prion protein. Cell Mol Neurobiol 18(6):731–742
Heitz S, Gautheron V, Lutz Y, Rodeau JL, Zanjani HS, Sugihara I, Bombarde G, Richard F et al (2008) BCL-2 counteracts Doppel-induced apoptosis of prion-protein-deficient Purkinje cells in the Ngsk Prnp(0/0) mouse. Dev Neurobiol 68(3):332–348
Heitz S, Lutz Y, Rodeau JL, Zanjani H, Gautheron V, Bombarde G, Richard F, Fuchs JP et al (2007) BAX contributes to Doppel-induced apoptosis of prion-protein-deficient Purkinje cells. Dev Neurobiol 67(5):670–686
Herrmann US, Sonati T, Falsig J, Reimann RR, Dametto P, O'Connor T, Li B, Lau A et al (2015) Prion infections and anti-PrP antibodies trigger converging neurotoxic pathways. PLoS Pathog 11(2), e1004662. doi:10.1371/journal.ppat.1004662PPATHOGENS-D-14-01363
Acknowledgments
The authors thank Prof. D. Harris (University of Boston) for kindly providing pcDNA-PrP plasmid and Prof. A. Aguzzi (University Hospital of Zurich) for pcDNA-ΔF35 plasmid. The authors thank Tom Yohannan for the editorial advice, M. Martínez-Vicente from M. Vila’s Lab for her guidance in MMP experiments, and M. Segura for the technical assistance. This research was supported by the Spanish Ministry of Science and Innovation (BFU2012-32617), FP7-PRIORITY, the Generalitat de Catalunya (SGR2014-1218), CIBERNED (PI2014/02-4 and PRY-14-114), La Caixa Obra Social Foundation, La Marató de TV3, and the Basque Foundation of Health and Innovation Research (BIO12/AL/004) to JADR. R.G. was supported by Instituto de Salud Carlos Tercero (FIS, PI11-00075). C.V, S.V., and A.M. were supported by the Spanish Ministry of Science and Innovation. O.N. was supported by Fundación Ramón Areces.
Author information
Authors and Affiliations
Corresponding authors
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Suppl Fig. 1
Quantitative assay of caspase-3 activity in transiently transfected N2A cells. Bars represent the mean ± SEM of three independent experiments (* p < 0.05, ** p < 0.01, versus empty-vector transfected cells). (JPEG 147 kb)
Suppl Fig. 2
Separation of PrPC-deleted forms PrPΔCC, PrPΔHR, and PrPΔCR in sucrose gradient. Fractions 1-12 of the sucrose density gradient were analysed with Western blotting. Flotillin (48-kDa) was used as a marker of lipid rafts. Blots are representative of three independent experiments. (JPEG 113 kb)
Suppl Fig. 3
Micrographs of transiently transfected N2A cells with N-terminal deleted forms PrPΔCC, PrPΔHR, PrPΔCR, and the empty vector pCDNA labeled with specific marker for ER (Sec61, red) for co-localization with PrP forms (6H4-green). (JPEG 186 kb)
Suppl Fig. 4
Micrographs of transiently transfected N2A cells with N-terminal deleted forms PrPΔCC, PrPΔHR, PrPΔCR, and the empty vector pCDNA labeled with specific marker for early endosomes (EEA1, red) for co-localization with PrP forms (6H4-green). (JPEG 161 kb)
Suppl Fig. 5
Micrographs of transiently transfected N2A cells with N-terminal deleted forms PrPΔCC, PrPΔHR, PrPΔCR, and the empty vector pCDNA labeled with specific marker for recycling endosomes (Rab4, red) for co-localization with PrP forms (6H4-green). (JPEG 168 kb)
Suppl Fig. 6
Quantitative assay of mitochondrial membrane depolarization in transiently transfected N2A cells. Bars represent the mean ± SEM of two independent experiments (* p < 0.05 versus empty-vector transfected cells). (JPEG 53 kb)
Rights and permissions
About this article

Cite this article
Vilches, S., Vergara, C., Nicolás, O. et al. Domain-Specific Activation of Death-Associated Intracellular Signalling Cascades by the Cellular Prion Protein in Neuroblastoma Cells. Mol Neurobiol 53, 4438–4448 (2016). https://doi.org/10.1007/s12035-015-9360-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9360-6