
Abstract
Prion diseases are caused by a misfolding of the cellular prion protein (PrP) to a pathogenic isoform named PrPSc. Prions exist as strains, which are characterized by specific pathological and biochemical properties likely encoded in the three-dimensional structure of PrPSc. However, whether cofactors determine these different PrPSc conformations and how this relates to their specific biological properties is largely unknown. To understand how different cofactors modulate prion strain generation and selection, Protein Misfolding Cyclic Amplification was used to create a diversity of infectious recombinant prion strains by propagation in the presence of brain homogenate. Brain homogenate is known to contain these mentioned cofactors, whose identity is only partially known, and which facilitate conversion of PrPC to PrPSc. We thus obtained a mix of distinguishable infectious prion strains. Subsequently, we replaced brain homogenate, by different polyanionic cofactors that were able to drive the evolution of mixed prion populations toward specific strains. Thus, our results show that a variety of infectious recombinant prions can be generated in vitro and that their specific type of conformation, i.e., the strain, is dependent on the cofactors available during the propagation process. These observations have significant implications for understanding the pathogenesis of prion diseases and their ability to replicate in different tissues and hosts. Importantly, these considerations might apply to other neurodegenerative diseases for which different conformations of misfolded proteins have been described.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A group of rare and fatal neurodegenerative disorders called transmissible spongiform encephalopathies (TSEs) or prion diseases, characterized by long incubation periods and neuronal loss, are well known due to their unusual causative agent. TSEs, as defined in the early eighties by the protein-only hypothesis, are caused by an aberrantly folded isoform (PrPSc) of the normal cellular prion protein (PrPC). Once formed, PrPSc induce neurodegeneration and have the ability to transform other PrPC into PrPSc [2].
Distinct prion diseases have been described in humans which affect different brain areas and result in clearly distinguishable clinical manifestations such as Creutzfeldt–Jakob disease (CJD), Gerstmann–Straussler–Scheinker syndrome (GSS) and fatal familial insomnia (FFI) [55]. Similarly, other mammalian species affected by TSEs have proven the existence of different types of prions that show not only different pathological properties but also distinct PrPSc biochemical features. These misfolded PrPs with identical amino acid sequences but causing clearly distinguishable pathologies are known as strains, and it is thought their strain properties are encoded in their three-dimensional structures [54]. The prion strain-specific properties include tropism for different brain regions, formation of morphologically distinct aggregates with different physicochemical properties and different self-templating and cross-seeding capacities [11]. The greatest strain variability has been generated and observed by experimental challenge with prion isolates in a wide number of animal models using different species [70]. Interestingly, the existence of different strains has also been proposed in other neurodegenerative diseases associated to protein misfolding, such as Alzheimer’s disease (AD) [33, 44, 58, 60], Parkinson’s disease (PD) [49] and Amyotrophic lateral sclerosis (ALS) [9].
The emergence of different prion strains is probably due to the conformational variability derived from the PrP-misfolding event, which gives rise to PrP isoforms that maintain their structure through the self-templated propagation mechanism [8]. At present, it is not possible to predict which PrP sequence may be suitable to propagate a specific prion strain or which may give rise to different isoforms. Experimental animal models have become the major source of different prion strains [45] but the techniques that allow prion propagation in vitro are the preferred way to study the molecular basis of prion transmission and strain diversity [12, 23, 35, 42, 62]. Recently, the phenomenon of strain diversity has been suggested to be due to the existence of prions as pools of intrinsically heterogeneous PrPSc isoforms named quasi-species. These pools of slightly different prion isoforms could be the basis of strain variation as changes in the prion propagation environment may provide selective advantage to some of the quasi-species leading to changes in strain properties through selection [18, 38].
The mechanism of spontaneous misfolding of prion proteins remains unknown. Although it may be favoured by certain mutations or polymorphisms [34, 69] and achieved in vitro [5], it is still not possible to predict the propensity of certain PrPs to misfold spontaneously. After a spontaneous misfolding of PrP, it is highly unlikely that a unique strain is generated in the light of the great strain diversity found for a single PrP sequence in some species, such as mouse [10]. Moreover, changes in the propagation environment of a given prion isolate can exert a selective pressure that leads to the emergence of new strain properties, such as the resistance to prion propagation inhibitors [31, 38] or alterations due to the presence of different cellular cofactors [32]. Thus, strain variation or mutation might be an intrinsic feature of spontaneously misfolded prions, although little is known about the molecular mechanisms underlying their origin or the factors that could define strain-specific characteristics.
Protein Misfolding Cyclic Amplification (PMCA) has been used to propagate mammalian prions in vitro for more than 10 years [29] and has been adapted to allow the use of bacterially expressed recombinant PrPs (rec-PrP) as a source of PrP in the propagation substrate [30]. Despite its novelty, the recombinant PrP-based PMCA (recPMCA) has already generated several different recombinant human prions [25]. The propagation of recombinant misfolded proteins with infectious properties started with the first generation of synthetic recombinant prions [37] and was further developed through a series of seminal studies [16, 20, 36, 41, 66], all of them giving rise to recombinant prions with different degrees of infectivity; from those showing low infectivity that required a second passage in a highly susceptible animal model to cause disease [37], to the one that showed high infectivity, comparable with those of natural mammalian prion isolates [66]. The protein preparations and in vitro misfolding techniques used in these studies were slightly different suggesting that a great diversity of strains were generated, each with its particular unique infective property. The idea that specific cofactors, such as certain lipids or polyanions, might be integral components of infectious recombinant prions was proposed [23] although readily discarded by the generation of the first recombinant infectious prion in the absence of any cofactors [36]. However, the role of certain compounds in driving and maintaining the conformation and differential properties of synthetic prion strains is well supported by the generation in vitro of different misfolded rec-PrPs with particular characteristics (reviewed in [59]). The in vitro propagation procedures used before apply slightly different sonication/incubation parameters, as well as conversion buffers with distinct detergents and concentrations. However, the main difference compared to the technique used here lies on the use of brain homogenate of animals devoid of PrP to complement the recombinant protein, which supplies all the cofactors that are present in vivo [3, 36]. However, the cofactor-complemented substrates devoid of brain homogenate were also employed in the present study to explore how specific cofactors influence the conformational selection of certain misfolded rec-PrPs. The notion that cofactors can play a key role in driving the conformational selection of prions has several implications for understanding the pathogenesis of prion diseases and their ability to propagate in different tissues or hosts [28]. Importantly, these considerations may apply to other neurodegenerative disorders in which different phenotypes have been associated to conformationally distinct misfolded proteins [9, 33, 44, 49, 58, 60].
This study investigates further the still largely unknown properties of recombinant infectious PrP generation, focusing on the strain features of the generated products and the effect of the propagation environment on the strain diversity and selection/mutation. Herein, we prove that recombinant bank vole PrP can be misfolded in vitro to become an infectious isoform, even in the absence of cofactors, despite the fact that an in vivo strain could not be faithfully reproduced on rec-PrP. Still, our results strongly suggest that a variety of infectious prion strains can be generated using rec-PrP in a test tube, most likely in the form of a mixture of quasi-species, in a process that seems largely independent from seeding with pre-formed prions. We also demonstrate that the quantity/type of conformations/strains formed is strongly dependent on the available cofactors.
Materials and methods
Preparation of purified recombinant PrP
Bacterial expression of bank vole recombinant PrP (amino acids 23–231) (rec-PrP) was performed as previously described [25]. Briefly, pOPIN E expression vector containing the wild-type I109 bank vole PRNP gene was prepared by standard molecular biology techniques using the oligonucleotides 5′ AGGAGATATACCATGAAGAAGCGGCCAAAGCCTGG3′ and 5′ GTGATGGTGATGTTTGGAACTTCTCCCTTCGTAGTA3′. E. coli Rosetta™ (DE3) Competent Cells (EMD Millipore) were transformed with the expression vector using standard molecular biology procedures allowing the expression of the recombinant protein. Although the protein does not contain His-tag, purification of the protein was performed with a histidine affinity column taking advantage of the natural His present in the PrP. A total of 11 different protein batches were prepared and their quality and purity were assessed by Coomassie blue staining after electrophoresis in SDS-PAGE gels.
Preparation of PMCA substrates
Brain-PMCA was performed using whole brain homogenates from transgenic mice expressing bank vole PrP I109 (TgVole) and brain homogenates from the non-transgenic natural host (bank vole I109). The TgVole mouse line expresses around four times the normal amount of bank vole I109 PrP under the control of the murine PrP promoter in a murine Prnp 0/0 background and was generated and characterized in a similar way as previously described [13].
Recombinant PMCA (recPMCA) as previously described [25] was performed initially using substrates based on purified recombinant proteins supplemented with brain homogenates from Prnp 0/0 transgenic mice [43]. For each of the 11 protein batches prepared, some were used in several repetitions, being each of the substrates used in these repetitions, different dialysis and preparations of the same protein batch. In the case of non-cofactor substrate, the dialyzed recombinant PrP was mixed solely with conversion buffer to a final concentration of 50–100 ng/µl. For cofactor-complemented substrates, dextran sulfate sodium salt from Leuconostoc spp. with molecular weights from 6500 to 10,000 (Sigma-Aldrich) was added to a final concentration of 0.5% (w/v); mRNA extracted from Prnp 0/0-transgenic mouse liver with RNAzol RT (Sigma-Aldrich) was added to a final concentration of 150 ng/ml; and plasmid DNA was added to a final concentration of 100 μg/ml. In all the cases, specific cofactor-complemented substrates were devoid of Prnp 0/0 mouse brain homogenate, containing just rec-PrP, conversion buffer and the specific cofactor. The concentration of each compound was selected based on different reasons. Dextran sulfate and plasmid DNA concentrations were chosen from previous studies in which the optimum concentrations of each of them for efficient rec-PrPres propagation were determined, while RNA concentration was defined based on published data [66].
In vitro propagation of prions by PMCA
Seeded PMCA
In all the serial PMCA procedures in which brain-derived or recombinant seeds were used, they were added at 1:10 dilution to the substrate only in the first PMCA round. The three brain-derived seeds used initially for in vitro generation of recombinant prions (seeds 1, 2 and 3) were: (1) A natural CWD isolate adapted to bank vole I109 through three serial in vivo passages (I109CWD) homogenized at 10% (w/v) in PBS with protease inhibitor cocktail (Roche). (2) The same I109CWD homogenate adapted to the in vitro propagation (brain-PMCA) using first a bank vole I109 brain homogenate as substrate to propagate it by 15 serial PMCA rounds and then a TgVole (transgenic mouse expressing bank vole I109 PrP with 4X overexpression with respect to bank vole) brain homogenate for other 15 serial rounds (I109CWD-VolePMCA-TgVolePMCA) and (3) The same I109CWD homogenate adapted to the in vitro propagation (brain-PMCA) using directly the TgVole brain homogenate for 15 serial PMCA rounds (I109CWD-TgVolePMCA).
Unseeded PMCA
For PMCA reactions aiming to obtain spontaneously misfolded PrP, no seed was added in the first PMCA round. Just fresh PMCA substrate was used with the same volume as the sum of substrates and seeds used for seeded PMCA reactions. Thereon, the same 1:10 dilution of the product from the previous PMCA round was added to fresh substrate for both seeded and unseeded serial PMCA reactions.
The seeded and unseeded in vitro prion propagation studies were performed based on modified versions of the PMCA described previously [15, 52, 53].
RecPMCA
Q-700 Misonix sonicator with microplate system (Qsonica) was used with incubation cycles of 30 min, followed by sonication pulses of 15–20 s at 60–80% power. The whole process was performed at an average temperature of 37–39 °C regulated by a circulating water bath.
Brain-PMCA
S-4000 Misonix sonicator with microplate system (Qsonica) was used with incubation cycles of 30 min, followed by sonication pulses of 20 s at 80% power. The whole process was performed at an average temperature of 37 °C regulated by a circulating water bath. To avoid any cros-contamination risk, all the PMCA tubes were sealed with plastic film (Parafilm) prior to their introduction in the bath sonicator impeding accidental opening. After each PMCA round, the outside of all the tubes was thoroughly cleaned with sodium hypochlorite and tubes containing different seeds or from different experiments were treated separately. Unseeded controls were included together with all the seeded samples submitted to brain PMCA.
Prion amplification by serial dilutions
Serial dilutions of the seed were subjected to one round of PMCA (24–48 h) and the PK-resistant signal obtained by Western blot was compared to the non-amplified seed. Reproducibility of the results was favoured using 1 mm zirconia/silica beads (BioSpec Products) [29].
Generation of misfolded proteins
The misfolding studies (seeded or unseeded) using recombinant PrP were assessed by serial recPMCA. Successive rounds of recPMCA, where prion strains were diluted 1:10 (or without adding any seed) in the corresponding substrates, were performed. After 24-h PMCA round, a sample from the first round was diluted 1:10 in fresh substrate and the process was repeated over 10–45 rounds of recPMCA.
Biochemical characterization of in vitro- and in vivo-generated prion strains
Protease resistance assay
recPMCA-treated samples were incubated with 85 µg/ml of PK (Roche) for 1 h at 42 °C with agitation at 450 rpm as previously described [25]. In exceptional cases, other PK concentrations were used (listed individually).
PK-resistant PrP detection
Protein inmunodetection by Western blotting was performed as previously described [25].
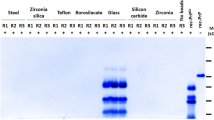
For the detection by Coomassie blue staining, the recombinant seeds with brain homogenate were purified following the protocol developed by Wenborn et al. [72]. For the samples without brain homogenate, 250 µl of the recombinant seeds were digested with 25 µg/ml of PK for 1 h at 42 °C, centrifuged at 19,000g for 1 h at 4 °C and the pellets resuspended in 10 µl of PBS. All the purification products were loaded onto 4–12% NuPAGE Midi gel (Invitrogen Life Technologies), separated and stained with BlueSafe (NZYTech) for 1 h at room temperature.
Detection and characterization of PK-resistant PrPSc in bank vole brain
Brain homogenates (20% w/v) were prepared as previously described [51]. After adding an equal volume of 100 mM of Tris–HCl containing 4% sarkosyl, the homogenates were incubated for 30 min at 37 °C with gentle shaking. PK (Sigma-Aldrich) was added at a final concentration of 200 μg/ml and then the samples were incubated for 1 h at 55 °C with gentle shaking. Protease treatment was stopped with 3 mM of PMSF (Sigma-Aldrich). Aliquots of samples were added with an equal volume of isopropanol/butanol (1:1 v/v) and centrifuged at 20,000g for 5 min. Supernatants were discarded and the pellets were resuspended in denaturing sample buffer and heated for 10 min at 90 °C. Electrophoresis and Western blotting were performed as previously described [51]. The monoclonal antibodies used and their epitope on bank vole PrP were as follow: 9A2 (99–101) and 12B2 (89–93).
Electron microscopy (EM)
Transmission electron microscopy
Misfolded recombinant samples were deposited in freshly glow-discharged carbon-coated gold grids (Electron Microscopy Sciences), washed with de-ionised H20 and stained with freshly prepared, filtered 5% uranyl acetate. The grids were imaged on a JEM-2200FS/CR (JEOL Europe, Croissy-sur-Seine, France) transmission electron microscope, equipped with a field emission gun (FEG) operated at 200 kV.
Cryo-electron microscopy
Misfolded recombinant samples were concentrated up to 100-fold prior to analysis by cryo-electron microscopy. Samples were centrifuged at 19,000g for 1 h at 4 °C. The resultant pellet was resuspended in 10 µl of PBS (Fisher Bioreagents) and stored at − 20 °C until microscopic examination.
Samples were applied directly to a carbon-coated grid, R 2/2 Quantifoil® (Quantifoil), and rapidly plunged into liquid ethane using a Vitrobot Mark III (FEI Inc., Eindhoven, The Netherlands). Sample analysis was performed with a JEM-2200 FS (JEOL) transmission cryo-electron microscope, using an acceleration voltage of 200 kV and defocus ranging from − 1.5 to − 5.0 µm, determined accurately using enhanced power spectra. Images were obtained under low-dose conditions on an UltraScan 4000, 4K × 4K CCD camera (Gatan Inc., Pleasanton, CA, USA).
Samples and transmission experiments
Preparation of in vivo- and in vitro-derived inocula
10% brain homogenates from vole-adapted chronic wasting disease (CWD), CWD-PMCA- and PMCA-misfolded rec-PrPs were diluted 10−1 in sterile PBS prior to intracerebral inoculation into bank voles 109I. The rec-PrPres amount, estimated by Western blot, was comparable in all samples. The inocula for the second passage were prepared as 10% wt/vol homogenates in PBS.
Animal inoculations
Groups of 8-week-old bank voles (Bv109I) were inoculated intracerebrally with 20 µl of homogenate into the left cerebral hemisphere using a sterile disposable 27-gauge hypodermic needle while under ketamine anaesthesia (ketamine 0.1 µg/g). The animals were examined twice a week until neurological clinical signs appeared, after which they were examined daily. Diseased animals were culled at the terminal stage of the disease by exposure to a rising concentration of carbon dioxide but before neurological impairment compromised their welfare, in particular. Survival time was calculated as the interval between inoculation and culling or death. Post-mortem, the brain was removed and divided to be stored at − 80 °C and fixed in formalin.
Neuropathology
Histology, immunohistochemistry and PET-blot analysis were performed on formalin-fixed tissues as previously described [47]. Briefly, brains were trimmed at standard coronal levels, embedded in paraffin wax, cut at 6 μm and stained with haematoxylin and eosin. PrP immunolabeling in immunohistochemistry and PET blot was performed using the 6C2 mAb. The specific brain scoring areas analyzed were: medulla (1), cerebellum (2), superior colliculus (3), hypothalamus (4), thalamus (5), hippocampus (6), septum (7), retrosplenial and adjacent motor cortex (8) and cingulated and adjacent motor cortex (9).
Ethics Statement
Bank voles were obtained from the breeding colony at the Istituto Superiore di Sanità (ISS), Italy. Experiments involving animals followed the “Principles of laboratory animal care” (NIH publication No. 86-23, revised 1985) as well as the guidelines contained in the Italian Legislative Decree 116/92, which transposed the European Directive 86/609/EEC on Laboratory Animal Protection, and then in the Legislative Decree 26/2014, which transposed the European Directive 2010/63/UE on Laboratory Animal Protection. The research protocol was performed under the supervision of the Service for Biotechnology and Animal Welfare of the ISS and was approved by the Italian Ministry of Health (decree number 84/12.B).
Results
In vitro generation of bank vole recombinant misfolded proteins in a brain homogenate environment
Using recombinant PrP from bank voles with polymorphism I109 (rec-PrP I109), complemented with PrP knock-out mouse (Prnp 0/0) brain homogenate as recPMCA substrate, two sets of experiments were performed to produce misfolded rec-PrP I109 by in vitro serial propagation. In the first set, the misfolding of the recombinant protein was induced by adding one among three different pre-formed PrPSc seeds, while in the second, no seed was added to achieve the spontaneous misfolding of PrP.
For the seed-induced misfolding, three different versions of bank vole-adapted PrPSc variants were selected (Fig. 1a, see “Materials and methods” section for details). All of them derive from a chronic wasting disease (CWD) PrPSc isolate, a prion disease-affecting cervids. Such isolate was subsequently passaged to bank voles (Myodes glareolus) and as it adapted to the new host it became the fastest disease-causing prion ever described [24]. The election of bank vole and CWD seeds as a model responds to practical reasons. First, bank vole I109 PrP has shown its ability to misfold spontaneously [69], allowing comparisons between seeded and unseeded misfolded rec-PrPs. Second, it has been considered an almost universal acceptor of prions [68], demonstrating its enhanced ability to adopt different conformations. And third, CWD adapted to bank vole is not just the fastest model of prion disease known, but is also one of the best-characterized ones [24], allowing the discrimination of distinct strains and a proper evaluation of possible conformational variants causing slight phenotypical changes. The three PrPSc seeds, chosen because the slightly different procedures used to generate them could provide additional conformational variability, were used diluted 1:10 in the rec-PrP-containing substrate and 20 serial rounds of recPMCA were performed using 4–6 independent replicates in each round (Fig. 1a). For the spontaneous misfolding experiment, 11 different rec-PrP I109 batches were tested and 20 serial rounds of recPMCA were performed with 4–6 replicates in each round (Fig. 1a). Each replicate was carried individually and independently through the serial rounds of recPMCA.

In vitro propagation experiments. a Rounds (R1-R20) of serial recPMCA using different bank vole (Bv) rec-PrP batches as substrates. Top part of the figure (above dashed line) shows the procedure used to prepare three different seeds (see details in “Materials and methods” section). Bottom part of the figure (below-dashed line) shows unseeded experiments. Unseeded controls of seeded experiments are shown by the same batch number. *Experiment performed until PMCA round 30 with identical results as round 20. Bv rec-PrP batch: A total of 11 different bank vole PrP batches were prepared. Some of these batches were used in several repetitions, being each of the substrates used in these repetitions, different dialysis and preparations of the same protein batch. The nomenclature of the selected samples refers to their migration patterns, being H for High and L for Low (H/L: both forms, H and L might be present as a mixture). b One tube of PMCA round 20 of each PrPres sample (with the exception of round 10 in samples H-unseeded-03 and H-unseeded-04) was selected to show the biochemical analysis of the material generated by recPMCA. Samples were digested with 85 µg/ml of proteinase-K (PK) and analyzed by Western blot using monoclonal antibody Saf83 (1:400) and 12B2 (1:2500). C(rBv): Control, undigested bank vole rec-PrP protein. C(Bv): Control, undigested bank vole whole brain homogenate
The first PK-resistant misfolded recombinant PrPs (rec-PrPres) detectable by WB appeared in different rounds and the misfolding frequency was also quite variable for both sets of experiments (seeded and unseeded) indicating that both the initial misfolding event and its frequency were, as expected, strongly stochastic. Thus, misfolding of the recombinant PrP appeared at rounds ranging from 6 to 17 for seeded samples and from 7 to 19 for the unseeded ones, suggesting that, in both cases, the misfolding of the rec-PrP I109 might be spontaneous in some experiments. However, slight differences should be noted: (i) 4 out of 4 independent seeded experiments gave rise to rec-PrPres, while in the case of the unseeded ones, just 8 out of 16 were positive, (ii) among the seeded replicates, two different electrophoretic migration patterns of rec-PrPres were detected (H for high and L for low), while the unseeded samples showed always the H pattern. Moreover, different protein batches as well as distinct substrate preparations using the same protein batch (separate dialysis), or even the discordant behaviour among the duplicates of each experiment, strongly support the expected existence of a reproducible but highly stochastic process of spontaneous protein misfolding (Fig. 1) [39].
Figure 1b shows a representative number (12) of rec-PrPres derived from the initial 20 serial recPMCA experiments.
Biochemical characterization of the different bank vole-misfolded rec-PrPs
H-seeded-02 and L-seeded-02 were selected for further biochemical characterization because of their distinct migration patterns (Supplementary Fig. S1). Both products were selected from round 20 and the rec-PrPres amounts in each seed were normalized by dilution in Prnp 0/0 brain homogenate prior to the propagation or protease-K resistance assays.
First, their ability to self-propagate indefinitely was evaluated. The selected misfolded rec-PrPs were serially propagated for 50 recPMCA rounds at 1:10 dilution in each round, confirming their self-propagating ability. To further determine if the distinct migration patterns observed consistently correspond to different strains with distinguishable propagation capacity, both seeds were serially diluted (from 10−1 to 10−8) in fresh substrate and submitted to a single round of recPMCA. The L-seeded-02 rec-PrPres showed lower propagation ability (the maximum dilution of the seed where rec-PrPres was still detectable was 10−5) than the H-seeded-02, which was able to propagate up to a dilution of 10−8 (Supplementary Fig. S1b). The same seeds diluted 10−2 non-submitted to PMCA were undetectable by Western blot, proving that the signal detected were not due to the remnant seed. Their PK-resistance pattern was also analyzed as it is one of the main characteristics of prions and both samples were subjected to increasing concentrations of PK (from 50 to 2000 µg/ml). Both seeds were resistant to at least 400 µg/ml of PK, although the L-seeded-02 seed showed higher resistance than the H-seeded-02.
In vitro propagation of bank vole-misfolded rec-PrPs by brain PMCA
Classic PMCA, based on the use of a brain homogenate, rather than recombinant PrP, as the substrate (brain-PMCA), has clearly demonstrated its usefulness and infectivity predictive value by its ability to propagate many different prion strains from a variety of mammalian species [15, 14, 65]. With few exceptions [63], once it has been shown that a given putative PrPSc sample can be propagated through brain-PMCA, it is subsequently shown to be infectious in vivo in a homologous host [52]. Therefore, we evaluated the ability of the misfolded rec-PrPs generated previously to propagate in brain-PMCA using two different substrates: one consisting in normal bank vole brain homogenate and the other consisting in brain homogenate from transgenic (Tg) mice expressing BV PrP (TgVole). These animals were selected to validate the results in two different models and for the detection of possible differences in the propagation of the misfolded rec-PrPs due to host-associated factors. All the rec-PrPres used were obtained from round 20 and the amounts normalized by dilution in Prnp 0/0 brain homogenate, so a similar amount of rec-PrPres and an identical seed volume was added in each assay.
All the misfolded recombinant proteins selected as representatives in the first experiment were able to propagate in both brain homogenate-based substrates except H-unseeded-09 in bank vole brain homogenate. Not all the rec-PrPres propagated with the same efficiency (Fig. 2a) nor gave rise to the same misfolded product, as inferred from the different migration patterns of the di-, mono- and unglycosylated PrPs after PK digestion (Fig. 2b). Bank vole and TgVole brain homogenates propagated most of the recombinant seeds similarly, yielding products with similar migration patterns for each seed. Comparing the patterns observed for the original recombinant seeds when propagated in bank vole brain homogenate, High and low patterns are conserved except for the H/L-seeded-03 seed where just the high pattern propagated; and when propagated in TgVole brain homogenate the patterns were maintained except in the case of H-seeded-01 seed that results in both high and low patterns and in the case of H/L-seeded-03 seed where just the low pattern propagated. However, the existence of different migration patterns within duplicates of the same seeds suggests strongly the presence of more than one prion population in the recombinant seeds used. A large amount of conversion was observed with some seeds when the TgVole brain homogenate was used, probably due to the high PrP expression level (4 ×) of this model.

In vitro propagation of misfolded rec-PrPs using brain-based PMCA. a Rounds (R1-R10) of serial PMCA using bank vole or transgenic mice overexpressing bank vole I109 PrP (TgVole) brain homogenates as substrates. The misfolded rec-PrPs: H-seeded-01, H-seeded-02, L-seeded-02, H/L-seeded-03 and H-unseeded-09 from round 20 (Fig. 1a) were used as seeds in replicates of four through ten rounds of serial PMCA. Tubes were considered positive if a classical PrPres pattern was observed. With the exception of H-seeded-02, it was necessary to undergo from 8 to 10 rounds of serial PMCA to observe a classical pattern of Proteinase-K (PK)-resistant bank vole PrP. None of the misfolded proteins were efficiently propagated in any of the mammalian substrates. This may be due to the presence of a strong transmission barrier between the recombinant seeds and the mammalian substrates or due to the presence of a very small amount of seed enciphered a classical prion strain. b Four tubes of round 10 of each PMCA-propagated sample were digested with 85 µg/ml of PK and analyzed by Western blot using monoclonal antibody Saf83 (1:400). At least two biochemical patterns based on migration properties are shown. All unseeded samples remained negative. ↔: Undigested samples. Control, undigested bank vole or TgVole whole brain homogenates
Finally, unseeded reactions with both bank vole brain and TgVole brain homogenates remained negative until the tenth PMCA round, ruling out any cross-contamination during these experiments. This finding contrasts with those obtained with recombinant PrP, where rec-PrPres emerges in almost half of the unseeded experiments. This could be due to the differences between brain-derived PrPC and recombinant PrP, which lacks post-translational modifications such as glycosylations and GPI anchor that have been reported relevant for the conformational diversity of prions [1, 4], although we cannot rule out that it is due to the higher concentration of rec-PrP in the substrate, which is known to favour spontaneous misfolding [68], or to the slightly different PMCA conditions.
Infectivity study in bank voles
To confirm whether misfolded rec-PrPs could be propagated in vivo and could thus be considered bona fide prions, H-seeded-01, H-seeded-02, L-seeded-02, H/L-seeded-03 and H-unseeded-09 recombinant misfolded proteins were inoculated intracerebrally into bank voles bearing the polymorphism I109. As controls, the CWD inocula adapted in vivo and in vitro to bank vole I109 were used. The recombinant seeds were selected as representatives of the possibly different products obtained during in vitro propagation.
All the recombinant inocula were infectious in vivo with incubation times between 133 ± 5 and 172 ± 6 days post-inoculation (dpi) ± SEM and 100% attack rates for most of them in the first passage (Table 1). The analysis of the PK-resistant PrPs from the brains of the affected animals showed that all but two voles accumulated an atypical prion characterized by an electrophoretic migration pattern similar to that observed in voles infected with the human GSS [48], with a predominant 7 kDa PK-resistant band (Fig. 3a). In contrast, two animals inoculated with H/L-seeded-03 and showing abnormally long incubation period (348 and 448 days post-inoculation) accumulated a classical PrPSc type (Fig. 3a). Interestingly, the PrPSc in these two animals was different from PrPSc in voles infected with CWD or scrapie, as both had a lower apparent molecular weight (MW) (Fig. 3a and Supplementary Fig. S2). Furthermore, the two H/L-seeded-03-infected voles were different among them, as judged by both, the glycotype and the differential antibody binding (Supplementary Fig. S2). Voles with atypical PrPSc showed similar neuropathological patterns, mainly characterized by the involvement of white matter areas, such as the alveus of the hippocampus, corpus callosum and white fiber bundles in the striatum. In these areas, disease-associated PrP (PrPd) deposition was moderate-to-intense, in the form of coarse or punctuate deposits (Fig. 3b). Neurodegeneration was mild in grey matter areas such as hippocampus, cerebral cortex and thalamus, characterized by microvacuoles and sparse PrPd deposits in the neuropil (Fig. 3b). Of the two voles with classical PrPSc, only one was available for neuropathological assessment [H/L-seeded-03 (#10 deviant)]. In this, vole neurodegeneration was limited to the mediodorsal thalamic nucleus, where PrPd accumulated mainly in the form of intraneuronal deposits (Fig. 3b). Of note, this neuropathological phenotype was different from that in CWD-infected voles (Fig. 3b).

Analyses of brains from misfolded rec-PrPs inoculated bank voles I109. a Biochemical analysis of Proteinase-K (PK)-resistant PrPSc in brain homogenates from bank vole I109 inoculated with the misfolded rec-PrPs: H-seeded-01, H-seeded-02, L-seeded-02, H/L-seeded-03 and H-unseeded-09. Representative vole brain homogenates were digested with 200 µg/ml of PK. All the H-seeded-01, H-seeded-02, L-seeded-02 and most of the H/L-seeded-03 inoculated vole brains accumulated an atypical prion characterized by an electrophoretic migration pattern similar to that observed in voles infected with the human GSS [50], with a predominant 7 kDa PK-resistant band. Two animals inoculated with H/L-seeded-03 accumulated a classical PrPSc type also different from PrPSc in voles infected with CWD or scrapie. 9A2 monoclonal antibody (1:400). MW: Molecular weight. b Brain deposition of PrPd was assessed by immunohistochemistry using the monoclonal antibody 6C2 (1:200). For the group of voles inoculated with vole-CWD and vole-CWD-PMCA a single representative vole is shown. Similarly, for the groups of voles inoculated with seeded recombinant inocula (H-seeded-01, H-seeded -02, L-seeded-02, H/L-seeded-03) a single representative vole is shown. Voles with atypical PrPSc (H-seeded-01, H-seeded-02, L-seeded-02, H/L-seeded-03 and H-unseeded-09) showed similar PrPd deposition patterns, mainly characterized by the involvement of white matter areas (left panels), and much less or not at all in grey matter areas (right panels). By contrast, CWD-infected voles showed diffuse PrPd deposition in the neuropil of several brain areas but not in the hippocampus. One vole with classical PrPSc [H/L-seeded-03 (#10 deviant)] showed a neurodegeneration limited to the mediodorsal thalamic nucleus, where PrPd accumulated mainly in the form of intraneuronal deposits. Bars, 50 µm. c Brain lesion profiles (determined by quantitative assessment of neuronal vacuolation in narrowly defined areas of the brain) from all groups of voles inoculated with misfolded rec-PrPs (H-seeded-01, H-seeded-02, L-seeded-02, H/L-seeded-03 and H-unseeded-09) showed similar patterns, which were different from those caused by inoculation with in vivo-adapted CWD or in vitro-propagated CWD. The two voles showing classical PrPSc in the H/L-seeded-03 group were excluded from this analysis
To attain a more objective comparison of distribution and severity of spongiform degeneration among groups of voles inoculated with seeded and unseeded misfolded rec-PrPs or in vitro-propagated CWD, brain lesion profiles were determined by quantitative assessment of neuronal vacuolation in narrowly defined areas of the brain. All groups of voles inoculated with misfolded rec-PrPs showed similar lesion profiles, which were different from those caused by inoculation with CWD or in vitro-passaged CWD (Fig. 3c; the two voles showing classical PrPSc in the H/L-seeded-03 group have been excluded from this analysis).
Overall, these findings prove that bank vole-misfolded rec-PrPs are infectious and show that at least two different strains (classical and atypical), distinct from CWD, were recovered from the various misfolded rec-PrPs, irrespective of their spontaneous or seeded origin. Furthermore, one misfolded rec-PrP, H/L-seeded-03, putatively encoded for a mixture of recombinant prion strains.
Selection of different misfolded recombinant proteins using different cofactors
To determine whether different strains could be selected using different propagation environments, by adding components already proven to interact with mammalian prions, inoculum H-seeded-02 (100% attack rate and an atypical electrophoretic pattern) was subjected to four different substrates devoid of brain homogenate: (1) without any cofactor, (2) with dextran sulfate [7], (3) with RNA [20] or (4) with plasmid DNA [46]. Thus, the only components of these substrates were rec-PrP, conversion buffer and the specific aforementioned compound. These compounds (putative cofactors) were chosen based on previous data showing that each of them could alter the in vitro propagation of some prions. H-seeded-02 was used as a seed for the four distinct substrates and all were submitted to 25 (1:10 dilution) serial rounds of recPMCA. Figure 4 shows the distinct migration pattern of one out of four resultant misfolded rec-PrPs (the one complemented with dextran sulfate) further supporting the theory of a mixture of strains in the original seed.

In vitro propagation of the misfolded rec-PrP H-seeded-02 in different substrates. The misfolded rec-PrP H-seeded-02 acting as seed was subjected serially to recPMCA using four different substrates: no cofactors, dextran, RNA and plasmid. The seed was diluted between 1:3 to 1:10 in each round of recPMCA until round 25 where no original seed was left. A representative tube of round 25 of each sample was digested with 85 µg/ml of Proteinase-K (PK) and analyzed by Western blot using monoclonal antibody Saf83 (1:400). All samples showed a PK-resistant band above 15 kDa. While the size of the band in the samples H-seeded-No cofactor, H-seeded-RNA and H-seeded-Plasmid was similar to the one observed in the seed H-seeded-02, the size of the band in the sample containing the misfolded protein propagated using dextran sulfate was lower and was named as L-seeded-Dextran. Undigested samples containing bank vole rec-PrP and brain were used as controls
In vitro propagation of cofactor-selected misfolded rec-PrPs by brain-PMCA
All four misfolded rec-PrP obtained after serial recPMCA rounds in the selective substrates (H-seeded-No cofactor, L-seeded-Dextran, H-seeded-RNA and H-seeded-Plasmid) were subjected to brain-PMCA to evaluate their infectivity in vitro in substrates based on TgVole and bank vole I109 brain homogenates.
The propagation ability of the cofactor-selected prion populations using brain-PMCA was significantly superior to the propagation ability shown by the original seeds (putatively strain mixtures). While those in the Prnp 0/0 environment required from 4 to 10 serial rounds to develop a detectable amount of glycosylated mammalian-misfolded PrP (Fig. 2), the cofactor-selected ones required 1–4 rounds only (Fig. 5). This suggests further the existence of a mixed prion population in the original H-seeded-02 recombinant seed, which contains much lower amounts of the strains able to propagate in brain-PMCA than the products selected through the different cofactor-containing substrates. However, we cannot exclude the presence of inhibitors in the Prnp 0/0 brain homogenate that makes the propagation of the seeds less efficient.

In vitro propagation of cofactor-selected misfolded rec-PrPs using brain-based PMCA. a Rounds (R1–R5) of serial PMCA using bank vole or transgenic mice overexpressing bank vole I109 PrP (TgVole) brain homogenates as substrates. The misfolded rec-PrPs: H-seeded-No cofactor, L-seeded-Dextran, H-seeded-RNA and H-seeded-Plasmid were used as seeds in replicates of four through five rounds of serial PMCA. Tubes were considered positive if a classical PrPres pattern was observed. All the seeds were efficiently propagated over a mammalian substrate, with seeds L-seeded-Dextran and H-seeded-RNA being particularly efficient. b Four tubes of round 5 of each PMCA-propagated sample were digested with 85 µg/ml of Proteinase-K (PK) and analyzed by Western blot using monoclonal antibody Saf83 (1:400). TgVole-based substrate was slightly more efficient than the bank vole-based substrate. At least three biochemical patterns, based on their migration properties, are shown; the lowest in the H-seeded-No cofactor sample, the intermediate-low in the L-seeded-Dextran sample and the highest in the H-seeded-Plasmid sample. All unseeded samples remained negative. Control substrate: undigested bank vole or TgVole whole brain homogenates
Similar to the different electrophoretic migration patterns observed in the brain-PMCA products derived from the seeds complemented with Prnp 0/0 brain homogenate, electrophoresis of the selected seeds also resulted in different patterns, suggesting the existence and selection of different misfolded rec-PrP populations (Fig. 5).
Infectivity of the cofactor-selected misfolded rec-PrPs in bank voles
All the selected prions (H-seeded-No cofactor, L-seeded-Dextran, H-seeded-RNA and H-seeded-Plasmid) were infectious by this method and showed variations in attack rates, between 78–100%, and in incubation times (Table 2), thus suggesting strain differences. Recombinant prions propagated with RNA as a cofactor (H-seeded-RNA) had the shortest incubation time with the least variability and a 100% attack rate. In turn, recombinant prions propagated in the absence of any cofactors (H-seeded-No cofactor) showed the longest incubation times and a 78% attack rate. In contrast to rec-PrP inocula derived from KO brain-supplemented PMCA, cofactor-selected misfolded rec-PrPs mainly propagated classical PrPSc after transmission in voles (Table 2). Indeed, H-seeded-No cofactor, L-seeded-Dextran and H-seeded-RNA-induced classical PrPSc in all recipient voles, which was mainly diglycosylated and with MW lower than vole-CWD (Fig. 6a). In contrast, H-seeded-Plasmid retained the ability to induce atypical PrPSc as the original H-seeded-02 recombinant seed, albeit not completely as it also induced classical PrPSc in a single vole. This vole had the longest incubation time (Table 2) and propagated a classical PrPSc whose PK-resistant core had an unusually high MW (Fig. 6a). Of note, in many voles, classical PrPSc was accompanied by variably low levels of atypical PrPSc (Fig. 6a) thus suggesting that both, atypical and classical strains might have been encoded in the various cofactor-selected rec-PrPs. It is also noteworthy that PrPSc features in voles with classical PrPSc correlated with the in vitro propagation in brain-PMCA with respect to the presence of different electrophoretic migration patterns (Figs. 5b and 6a). Cofactor-selected misfolded PrPs caused variable patterns of brain regional distribution of spongiform neurodegeneration, which were strongly dependent on the molecule used as cofactor during PMCA (Fig. 6b). Interestingly, neurodegenerative changes and PrPd deposition caused by misfolded PrP propagated using plasmid as a cofactor, were similar to those associated to the original H-seeded-02 (Figs. 6b and 3b, c), suggesting that the strain was preserved during PMCA propagation in the presence of plasmid. The only vole with classical PrPSc in this group also showed a deviant pathological phenotype, characterized by diffuse PrPd deposition in the neuropil of several subcortical areas (Fig. 6b). Voles infected with H-seeded-No cofactor, L-seeded-Dextran and H-seeded-RNA showed neuropathological patterns different from the original seeds, vole-CWD and H-seeded-02, and also different among them (Fig. 6b).

Analyses of brains from cofactor-selected misfolded rec-PrPs inoculated bank voles I109. a Biochemical analysis of Proteinase K (PK)-resistant PrPSc in brain homogenates from bank vole I109 inoculated with the cofactor-selected misfolded rec-PrPs: H-seeded-No cofactor, L-seeded-Dextran, H-seeded-RNA and H-seeded-Plasmid. Representative vole brain homogenates were digested with 200 µg/ml of PK. At least three distinct migration patterns were observed. While samples H-seeded-No cofactor, L-seeded-Dextran and H-seeded-RNA, all showed a classical pattern with apparent MW lower than scrapie and CWD controls, the H-seeded-Plasmid showed two types of patterns: an atypical pattern such as the brain-complemented original seed and a well-distinguishable classical pattern, characterized by a MW clearly higher than scrapie and CWD controls. Of note, minimal and variable amount of 7 kDa PK-resistant fragment was detected also in voles infected with recombinant prions and showing classical PrPSc, but not in voles infected with scrapie or CWD. 9A2 monoclonal antibody (1:400). MW: Molecular weight. b Neuropathological study of brains from bank voles I109 inoculated with the cofactor-selected misfolded rec-PrPs: H-seeded-No cofactor, L-seeded-Dextran, H-seeded-RNA and H-seeded-Plasmid. In the left column are shown the lesion profiles from the groups of voles indicated. In the right columns, it is shown the deposition of PrPd in two brain areas, the thalamus and the hippocampus, as assessed by immunohistochemistry using the monoclonal antibody 6C2 (1:300). The single vole with classical PrPSc in this group (H-seeded-Plasmid #4) had a distinctive neuropathological pattern, with intense neuropil-associated and peri-neuronal PrPd deposition mainly restricted to subcortical areas. All other groups were different among them in terms of lesion profiles and PrPd deposition patterns: in H-seeded-No cofactor spongiform degeneration was characterized by rather large vacuoles in thalamus, hypothalamus and mesencephalon and PrPd deposition was mainly intraneuronal and intraglial; in H-seeded-RNA the hypothalamus was spared and neuropathology and PrPd deposition were also evident in hippocampus and cerebral cortex; L-seeded-Dextran was characterized by large PrPd plaques accompanied by neuropil-associated PrPd. Bars, 50 µm
Overall, these results show that misfolded rec-PrP populations propagated in the absence of brain homogenate-complemented substrate, and even in the absence of any cofactor, are infectious. Furthermore, these misfolded rec-PrPs were more prone to encode classical prion strains than brain homogenate-complemented misfolded rec-PrPs, although the co-presence of classical and atypical PrPSc in several voles, as well as the results obtained with H-seeded-Plasmid, suggest that variable amounts of atypical prions were also encoded by cofactor-selected misfolded PrPs. Finally, the different phenotypic patterns observed in voles suggest that diverse classical strains could have been selected during in vitro propagation under different cofactor conditions.
Second passage experiments to confirm the existence of different prion strains
To confirm that the strong phenotypic differences observed in primary passages corresponded to different strains, second passages using H-seeded-01, H-seeded-02, H-seeded-No cofactor and L-seeded-Dextran inoculated bank vole brain homogenates were performed. Second passages infected voles with very short incubation times, comprised between 34 and 103 dpi (Table 3). The strongly reduced incubation times of the classical prion strains between the first and second passage suggest the existence of a transmission barrier (H-seeded-No cofactor: 424 vs. 82 dpi, respectively, and L-seeded-Dextran: 239 and 61 dpi, respectively) (Tables 2 and 3). The use of a recombinant material (devoid of GPI anchoring and glycosylation) likely requires some adaptation to a mammalian host. However, it is difficult to rule out that the longer incubation periods of the first passage could be due to low titers of the seeds after a necessary 1:10 dilution.
In keeping with the different incubation times, different phenotypes of disease were observed in voles, which allowed defining four different prion strains (Supplementary Fig. S3). As previously observed, although it is known that PMCA-adapted strains may suffer a prolongation in the incubation times with respect to the original brain-derived strains [24], in a second passage, CWD was recovered from the original strain subjected to brain-PMCA, but none of the recombinant strains resembled the CWD seed. The emergence of at least three different strains was confirmed: an atypical prion strain with a shortened incubation time of ~ 100 dpi in a second passage, which was recovered from both brain homogenate-complemented inocula, H-seeded-01 and H-seeded-02, and two classical prion strains (Classical A and Classical B), encoded by cofactor-selected recombinant seeds, characterized by slightly different incubation times (Table 3) and clearly different neuropathological patterns (Supplementary Fig. S3).
Selection vs. de novo generation
To determine if the four strains obtained from the substrates complemented with different cofactors were really a result of selection or due to de novo generation, we examined the spontaneous misfolding ability of the rec-PrP I109 in the selective substrates. Unseeded recPMCA serial rounds with the four substrates yielded misfolded rec-PrPs spontaneously (H-unseeded-No cofactor, L-unseeded-Dextran, H-unseeded-RNA and H-unseeded-Plasmid) with similar electrophoretic migration patterns to those observed during the previous selective study (Fig. 7b vs. 4 and Fig. 7c). These data appear to confirm the association between strain and cofactor, regardless of whether they were selected from the brain-complemented seed (Fig. 4) or spontaneously misfolded in the cofactor-complemented substrates (Fig. 7b). The number of recPMCA rounds needed to obtain each de novo strain varied between three rounds for the RNA-complemented protein to 40 with no cofactor (Fig. 7a). Such a dispersion in the generation round number suggests that spontaneous misfolding in the cofactor-complemented substrates was hindered with respect to the previous study in which the same substrates were seeded with a brain homogenate-complemented recombinant seed (H-seeded-02).

Unseeded in vitro propagation using different cofactors. a Rounds (R1-R47) of serial recPMCA using bank vole rec-PrP complemented with different cofactors (No cofactor, dextran sulfate, RNA or plasmid) as substrates. Red highlighted squares indicate selected samples for further studies and the coloured circles indicates a biochemically different rec-PrPres pattern [red for high (H) and blue for low (L)]. All the cofactors were helpful in triggering spontaneous misfolding. RNA was significantly more efficient than dextran or plasmid. The substrate without any cofactor needed 40 rounds to visualize the first misfolded protein. b One tube of round 47 of each PrPres sample was selected to show the biochemical analysis of the material generated after serial recPMCA. Samples were digested with 85 µg/ml of Proteinase-K (PK) and analyzed by Western blot using monoclonal antibody Saf83 (1:400). All samples showed a PK-resistant band greater than 15 kDa. Similarly to that observed in Fig. 4, while the size of the bands in the samples H-unseeded-No cofactor, H-unseeded-RNA and H-unseeded-Plasmid were similar to each other, the size of the band in the sample containing the misfolded protein propagated using dextran sulfate (L-unseeded-Dextran) was lower. Undigested samples containing bank vole rec-PrP and brain were used as controls. c Misfolded proteins from the two groups (selected or seeded vs. de novo generated or unseeded): H-seeded-No cofactor, L-seeded-Dextran, H-seeded-RNA, H-seeded-Plasmid, H-unseeded-No cofactor, L-unseeded-Dextran, H-unseeded-RNA and H-unseeded-Plasmid were digested with 25 µg/ml PK and purified by centrifugation. H-seeded-02 was purified using a specific procedure (see “Materials and methods”) Samples were visualised by Coomassie blue staining on SDS-PAGE
The propagation study of the de novo-generated seeds by brain-PMCA showed close agreement with the results of the strains putatively selected from the Prnp 0/0 brain homogenate-complemented seed. This is clear in the number of brain-PMCA rounds needed to misfold the PrPs from bank vole and TgVole brain homogenates, when seeded with either selected or spontaneously generated recombinant prions in substrates containing dextran sulfate, RNA and plasmid DNA (Supplementary Fig. S4 vs. Fig. 5a) but not for the non-cofactor-complemented seeds. Additionally, the electrophoretic migration patterns of these brain-PMCA products are also similar (Supplementary Fig. S4b vs. Fig. 5b).
Electron microscopy of bank vole-misfolded rec-PrPs
The misfolded rec-PrPs prepared using different substrates were concentrated 100 times and subjected to cryo-electron microscopy (cryo-EM). All samples contained rod-like structures with a length of 100–200 nm (Supplementary Fig. S5a) with an overall similar appearance as GPI-anchorless PrPSc samples purified from RML-inoculated GPI-anchorless transgenic mouse brains [64]. As with these samples, the rods consisted of laterally aggregated ~ 10 nm fibrils. No obvious differences between different misfolded rec-PrPs were discernible. The samples were also subjected to transmission electron microscopy. Supplementary Fig. S5b shows a typical image from a misfolded L-seeded-Dextran containing the same short rod-like structures seen by cryo-EM, again, very similar to those seen in GPI-anchorless PrPSc and wild-type PrPSc samples isolated from prion-infected brains [61]. Frequently, the individual ~ 10 nm fibrils making up the rods by lateral association were discernible. Similar fibrils were detected in brain-derived prion preparations [57].
Discussion
In this study, we obtained several recombinant misfolded PrPs endowed with in vivo infectivity. Importantly, in vitro and in vivo experiments clearly showed that these recombinant prions encoded for different biological properties and thus can be considered different prion strains, or mixtures thereof, whose emergence mainly depended on the molecules used as replication cofactors. A graphical summary of the experiments is reported in Fig. S7. Since complete titration experiments were not performed with the recombinant seeds generated in vitro, we cannot confirm that their specific infectivity is comparable to brain-derived mammalian strains, although their infectious nature in vivo is beyond doubts. The arousal of such a variety of conformations or strains may have been favoured by the choice of the bank vole as model system for this study. Since its PrP is known as a universal acceptor of prions [68], there is a possibility that the variety of conformations derive from the ability of its PrP to adopt different structural variations around the general architectural theme of PrPSc [64]. Nonetheless, the existence of different prion strains in many distinct mammalian species suggests that the same phenomenon, perhaps to a lesser extent, may occur also with other PrPs. Precisely, bank vole PrP I109 was chosen for this study because its enhanced misfolding ability could facilitate the observation of easily distinguishable conformational variants or strains.
Recombinant prions grown in Prnp0/0 brain homogenate-supplemented PMCA reactions
Although several different recombinant prions have been generated spontaneously or using seeds obtained from field isolates [16, 20, 36, 37, 41, 66], the generation of recombinant prions in a controlled manner reproducing the biochemical and biological properties of those naturally occurring prion strains had not been accomplished to date. The fact that both non-infectious- and infectious-misfolded recombinant proteins could be generated applying similar conditions directed research on the molecular basis of infectivity and, as a consequence, many studies emerged suggesting different molecules (cofactors) as responsible for the infectivity or absence of infectivity of certain misfolded PrPs [19, 21, 56, 66]. In the present study, all the cofactors that could play some role in vivo were putatively provided by the Prnp 0/0 brain homogenate environment. By mimicking as closely as possible the natural scenario in which prions are propagated, we expected to reproduce the CWD-derived strain used for seeding [24] and, in the case of unseeded reactions, to obtain the same strain that causes spontaneous disease in vivo [69]. However, we could not faithfully reproduce an in vivo strain on rec-PrP. This could be due to the differences between brain-derived PrPC and recombinant PrP, which lacks post-translational modifications such as glycosylations and GPI-anchor that have been reported relevant for the conformational diversity of prions [1, 4]. Still, we confirmed that obtaining infectious recombinant prions in vitro is possible (although unrelated to the original seeds) and our study shows that they probably arise from a mixture of conformations with distinct properties.
The generation of recombinant infectious prions seems largely independent from seeding with brain-derived prions, since it also occurred spontaneously in unseeded reactions and an atypical strain was recovered in vivo from both, seeded and unseeded reactions. The spontaneous formation of atypical strains observed here is reminiscent of naturally or experimentally occurring spontaneous atypical strains such as Nor98 in sheep [6], variably protease-sensitive prionopathy (VPSPr) [74] or GSS in humans, and, perhaps not surprisingly, the spontaneous prion strain in transgenic mice overexpressing the same bank vole PrP109I prion protein here used [67]. Importantly, the same recombinant prions leading to the atypical strain after transmission in bank voles were shown to reproduce different classical PrPSc types once propagated in vitro by brain-PMCA, thus strongly suggesting that the recombinant inocula contained a mixture of classical and atypical strains. The fact that two animals inoculated with H/L-seeded-03 survived longer (348 and 448dpi) showing a classical electrophoretic pattern on Western blots, similar to those observed by brain-PMCA, further supports the existence of a strain mixture in H/L-seeded-03 recombinant inoculum. This may be explained by a low titre of the fastest propagating atypical strain—supported by the survival of two animals from the same group—and a slower propagating classical strain, which would have affected only those animals which lived longer. Thus, as there was little or no selection pressure due to the presence of complete brain homogenate in these recPMCA reactions, it can be expected that any mixtures generated would contain different proportions of conformational variants of PrPSc, in the form of quasi-species, each of them with individual attributes with respect to infectivity and in vitro propagation propensities (Fig. S6).
Recombinant prions grown in single cofactor-supplemented PMCA reactions
Although previous reports of some cofactors favouring spontaneous prion formation exist [22, 66], such as certain polyanions and lipids, we focused on three different molecules with a similar polyanionic nature to assure putatively similar selection mechanisms. Propagation of H-seeded-02, which presented an atypical pattern in vivo, in substrates containing specific cofactors gave rise to prions that showed a classical pattern except for the one propagated with plasmid DNA. This suggests that specific cofactors could drive the selection of strains that were minority in the initial mixture and which manifested just in those cases where the fastest atypical strain was outcompeted for unknown reasons. Indeed, brain-PMCA studies with the selected strains predicted a classical strain behaviour and distinctive electrophoretic migration patterns. This behaviour was confirmed in vivo with the exception of the seed propagated with plasmid DNA, which resulted in a mixture of atypical and classical patterns after propagation in vivo. Thus, the substrate complemented with plasmid seems not to specifically favour classical strains such as other cofactors but instead propagated a mixture similar to that generated in the presence of Prnp 0/0 brain homogenate. The rest of the cofactor-selected seeds mainly encoded for classical strains, although they could have also contained minimal quantities of the atypical strain. These findings imply that the infectious properties of the recombinant strains generated in vitro were variable and highly dependent on the cofactor, highlighting the capacity of this recombinant PrP to misfold into a number of different structures. Actually, that the presence of cofactors can shift prion strain replication preference has already been demonstrated using other components [23]. Thus, it is highly probable that by choosing other polyanionic cofactors, many other strains could arise with different properties.
The similarities between the biochemical patterns of seeded versus spontaneously generated misfolded proteins in the presence of specific cofactors (Figs. 4 and 7b) further support the notion that the cofactors played a role in the predominance of certain strains. This resemblance was further confirmed through brain-PMCA where similar patterns were observed also when using seeds either selected (seeded) or spontaneously generated (unseeded) in dextran sulfate, RNA and plasmid DNA-containing substrates.
Recombinant prions grown in PMCA reactions without cofactors
An important finding of this study was that the propagation of H-seeded-02 recombinant prion in recPMCA without cofactors led to the replication of an infectious recombinant prion. This finding, in agreement with previous studies [36], suggests that cofactors are not an essential component of the infectious particle. Oddly, despite the similarity of the electrophoretic patterns between seeded and unseeded recombinant prion populations propagated without any cofactor, a completely different behaviour was shown when infectivity was evaluated in vitro. Indeed, in contrast to its seeded counterpart, the spontaneous recombinant prions generated by recPMCA in absence of cofactors were unable to propagate in brain-PMCA reactions. This suggests that spontaneously misfolded PrPs in the absence of any cofactor could have low or no infectivity. Although this hypothesis needs confirmation by bioassay in vole, it seems supported by the finding that H-seeded-02 grown in absence of any cofactor was the least infectious in our experiments, having the longest incubation time and the lowest attack rate (see Table 2). Therefore, it appears that the absence of cofactors probably imposes fewer restrictions on the misfolding process, allowing higher variability and putatively higher propensity for misfolding in non-infectious conformations.
Relevance for the pathogenesis of prion diseases
All these results point out that complementation with brain homogenate allows the spontaneous generation of a high number of infectious recombinant prion strains named quasi-species, where the most abundant and fastest would show an atypical pattern in vivo [17, 71]. Each of the strains from the mixture might be selected by the use of specific cofactors which may favour the propagation of one particular strain or a small group of similar strains (Supplementary Fig. S6). As an infectious strain was propagated in the absence of any cofactor, we can conclude that no essential cofactor exists; rather, there exist many strains with diverse ways to propagate, at least in vitro. Previous reports showing the necessity of cofactors for infectious recombinant prion generation may probably be a consequence of the different experimental models used, in which distinct recombinant prion strains were handled [59].
The idea that cofactors play a pivotal role in driving conformational selection of prions has several implications for understanding the pathogenesis of prion diseases and may be also important in other neurodegenerative diseases associated with protein misfolding [26]. If the replication of a given strain heavily relies on specific cofactors, this may contribute to explain some of the pathological features which typify strains, such as the differential tissue distribution (i.e., presence or absence in lymphoid tissues), cellular localization (intracellular, membrane-attached or extracellular), involvement of specific cell types (neurons or glia) or the regional distribution of aggregates in the brain. Our results support this idea since prions which preferentially propagated in Prnp 0/0 brain homogenate showed very specific preferential targeting in vivo too, characterized by PrPSc accumulation in some white matter areas of the brain (Fig. S7). In sharp contrast, recombinant prions propagated in substrates containing specific cofactors preferentially accumulated in different grey matter brain compartments (Fig. S7). These classical strains, which needed different cofactors to sustain their propagation in vitro, may be dependent on other cofactors in vivo, driving them to distinct brain (grey matter) and cellular compartments (intraneuronal, intraglial or neuron associated). These data suggest that the atypical strain propagated in Prnp 0/0 brain homogenate environment needs some brain-derived cofactor, which is absent in no-cofactor substrate and is not well mimicked by dextran sulfate or RNA and only partially by plasmid DNA (Fig. S7). This phenomenon would also explain why cell culture models for prion infection have so strict strain preferences [40]. In fact, selective trafficking of prion strains through specific compartments in cell culture has been recently proposed to control access to cofactors or environments required for the replication of certain strains [28]. The fact that the type of strains or conformations is strongly dependent on the available cofactors could be also relevant for interspecies transmission of prions, since some cofactor or its relative abundance might be different in distinct species. This was observed in vivo on transmission experiments to transgenic mice expressing bank vole PrP, which were unable to replicate completely the original strain coming from bank vole brain homogenates, putatively due to non-PrP species-specific factors that could drive strain selection during interespecies transmissions [27].
Conclusions
Despite this advance in being able to generate a diversity of infectious prion strains and the capacity to propagate them selectively using different cofactors, there are still questions to address: (i) why do the polyanionic compounds specifically select distinct strains?, (ii) why do recombinant strains with almost identical biochemical patterns (see RNA vs. plasmid) show strikingly different patterns after in vivo propagation?, or (iii) is it possible to modify the behaviour of the different strains by switching cofactors once the strains have been selected or spontaneously generated in the presence of another cofactor?
The search for a protein X [73] or cofactors involved as propagation helpers in vivo has been long considered the Holy Grail necessary to understand the molecular basis of prion strain propagation. The ability to handle prions in vitro [14] has enabled us to investigate the long searched cofactor thought to be responsible for the generation of bona fide prions in vitro [23, 66, 59]. However, the results of the present study indicate that cofactors are not necessary for prion propagation but do influence its outcome significantly. Thus, despite not being part of the infectious particle, cofactors appear to be involved in the propagation and/or maintenance of certain strains, probably restricting variability by favouring the generation of some of them. The idea that strain properties can be driven by changing or limiting cofactors present during the misfolding process may have several implications for understanding their ability to differentially propagate in distinct tissues and hosts, which may be also relevant for other neurodegenerative disorders for which conformational variants have been described. The use of recombinant proteins in the prion field and the possibility to propagate, handle and select a diversity of infectious strains takes us a step closer to deciphering one of the most intriguing features of prions, the strain phenomenon.
References
Aguilar-Calvo P, Xiao X, Bett C, Erana H, Soldau K, Castilla J, Nilsson KP, Surewicz WK, Sigurdson CJ (2017) Post-translational modifications in PrP expand the conformational diversity of prions in vivo. Sci Rep 7:43295. https://doi.org/10.1038/srep43295
Aguzzi A (2006) Prion diseases of humans and farm animals: epidemiology, genetics, and pathogenesis. J Neurochem 97:1726–1739. https://doi.org/10.1111/j.1471-4159.2006.03909.x
Atarashi R, Moore RA, Sim VL, Hughson AG, Dorward DW, Onwubiko HA, Priola SA, Caughey B (2007) Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat Methods 4:645–650. https://doi.org/10.1038/nmeth1066
Baron GS, Hughson AG, Raymond GJ, Offerdahl DK, Barton KA, Raymond LD, Dorward DW, Caughey B (2011) Effect of glycans and the glycophosphatidylinositol anchor on strain dependent conformations of scrapie prion protein: improved purifications and infrared spectra. Biochemistry 50:4479–4490. https://doi.org/10.1021/bi2003907
Barria MA, Mukherjee A, Gonzalez-Romero D, Morales R, Soto C (2009) De novo generation of infectious prions in vitro produces a new disease phenotype. PLoS Pathog 5:e1000421. https://doi.org/10.1371/journal.ppat.1000421
Benestad SL, Sarradin P, Thu B, Schonheit J, Tranulis MA, Bratberg B (2003) Cases of scrapie with unusual features in Norway and designation of a new type, Nor98. Vet Rec 153:202–208
Beringue V, Adjou KT, Lamoury F, Maignien T, Deslys JP, Race R, Dormont D (2000) Opposite effects of dextran sulfate 500, the polyene antibiotic MS-8209, and Congo red on accumulation of the protease-resistant isoform of PrP in the spleens of mice inoculated intraperitoneally with the scrapie agent. J Virol 74:5432–5440
Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Caughey B (1995) Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature 375:698–700. https://doi.org/10.1038/375698a0
Bidhendi EE, Bergh J, Zetterstrom P, Andersen PM, Marklund SL, Brannstrom T (2016) Two superoxide dismutase prion strains transmit amyotrophic lateral sclerosis-like disease. J Clin Invest 126:2249–2253. https://doi.org/10.1172/JCI84360
Bruce ME (1993) Scrapie strain variation and mutation. Br Med Bull 49:822–838
Bruce ME (2003) TSE strain variation. Br Med Bull 66:99–108
Castilla J, Gonzalez-Romero D, Saa P, Morales R, De Castro J, Soto C (2008) Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell 134:757–768. https://doi.org/10.1016/j.cell.2008.07.030
Castilla J, Gutierrez-Adan A, Brun A, Doyle D, Pintado B, Ramirez MA, Salguero FJ, Parra B, Segundo FD, Sanchez-Vizcaino JM, Rogers M, Torres JM (2004) Subclinical bovine spongiform encephalopathy infection in transgenic mice expressing porcine prion protein. J Neurosci 24:5063–5069. https://doi.org/10.1523/JNEUROSCI.5400-03.200424/21/5063
Castilla J, Morales R, Saa P, Barria M, Gambetti P, Soto C (2008) Cell-free propagation of prion strains. EMBO J 27:2557–2566. https://doi.org/10.1038/emboj.2008.181
Castilla J, Saa P, Hetz C, Soto C (2005) In vitro generation of infectious scrapie prions. Cell 121:195–206. https://doi.org/10.1016/j.cell.2005.02.011
Choi JK, Cali I, Surewicz K, Kong Q, Gambetti P, Surewicz WK (2016) Amyloid fibrils from the N-terminal prion protein fragment are infectious. Proc Natl Acad Sci USA 113:13851–13856. https://doi.org/10.1073/pnas.1610716113
Collinge J (2010) Medicine. Prion strain mutation and selection. Science 328:1111–1112. https://doi.org/10.1126/science.1190815
Collinge J, Clarke AR (2007) A general model of prion strains and their pathogenicity. Science 318:930–936. https://doi.org/10.1126/science.1138718
Deleault NR, Geoghegan JC, Nishina K, Kascsak R, Williamson RA, Supattapone S (2005) Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J Biol Chem 280:26873–26879. https://doi.org/10.1074/jbc.M503973200
Deleault NR, Harris BT, Rees JR, Supattapone S (2007) Formation of native prions from minimal components in vitro. Proc Natl Acad Sci USA 104:9741–9746. https://doi.org/10.1073/pnas.0702662104
Deleault NR, Lucassen RW, Supattapone S (2003) RNA molecules stimulate prion protein conversion. Nature 425:717–720. https://doi.org/10.1038/nature01979
Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, Supattapone S (2012) Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc Natl Acad Sci USA 109:8546–8551. https://doi.org/10.1073/pnas.1204498109
Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, Rees JR, Supattapone S (2012) Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci USA 109:E1938–E1946. https://doi.org/10.1073/pnas.1206999109
Di Bari MA, Nonno R, Castilla J, D’Agostino C, Pirisinu L, Riccardi G, Conte M, Richt J, Kunkle R, Langeveld J, Vaccari G, Agrimi U (2013) Chronic wasting disease in bank voles: characterisation of the shortest incubation time model for prion diseases. PLoS Pathog 9:e1003219. https://doi.org/10.1371/journal.ppat.1003219
Elezgarai SR, Fernández-Borges N, Erana H, Sevillano A, Moreno J, Harrathi C, Saá P, Gil D, Kong Q, Requena JR, Andreoletti O, Castilla J (2017) Generation of a new infectious recombinant prion: a model to understand Gerstmann–Sträussler–Scheinker syndrome. Sci Rep. https://doi.org/10.1038/s41598-017-09489-3
Erana H, Venegas V, Moreno J, Castilla J (2017) Prion-like disorders and Transmissible Spongiform Encephalopathies: an overview of the mechanistic features that are shared by the various disease-related misfolded proteins. Biochem Biophys Res Commun 483:1125–1136. https://doi.org/10.1016/j.bbrc.2016.08.166
Espinosa JC, Nonno R, Di Bari M, Aguilar-Calvo P, Pirisinu L, Fernandez-Borges N, Vanni I, Vaccari G, Marin-Moreno A, Frassanito P, Lorenzo P, Agrimi U, Torres JM (2016) PrPC governs susceptibility to prion strains in bank vole, while other host factors modulate strain features. J Virol 90:10660–10669. https://doi.org/10.1128/JVI.01592-16
Fehlinger A, Wolf H, Hossinger A, Duernberger Y, Pleschka C, Riemschoss K, Liu S, Bester R, Paulsen L, Priola SA, Groschup MH, Schatzl HM, Vorberg IM (2017) Prion strains depend on different endocytic routes for productive infection. Sci Rep 7:6923. https://doi.org/10.1038/s41598-017-07260-2
Fernandez-Borges N, de Castro J, Castilla J (2009) In vitro studies of the transmission barrier. Prion 3:220–223
Fernández-Borges N, Erana H, Elezgarai SR, Harrathi C, Venegas V, Castilla J (2017) A quick method to evaluate the effect of the amino acid sequence in the misfolding proneness of the prion protein. In: Lawson VA (ed) Prions: methods and protocols. Springer, Berlin
Ghaemmaghami S, Ahn M, Lessard P, Giles K, Legname G, DeArmond SJ, Prusiner SB (2009) Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog 5:e1000673. https://doi.org/10.1371/journal.ppat.1000673
Gonzalez-Montalban N, Lee YJ, Makarava N, Savtchenko R, Baskakov IV (2013) Changes in prion replication environment cause prion strain mutation. FASEB J 27:3702–3710. https://doi.org/10.1096/fj.13-230466
Heilbronner G, Eisele YS, Langer F, Kaeser SA, Novotny R, Nagarathinam A, Aslund A, Hammarstrom P, Nilsson KP, Jucker M (2013) Seeded strain-like transmission of beta-amyloid morphotypes in APP transgenic mice. EMBO Rep 14:1017–1022. https://doi.org/10.1038/embor.2013.137
Jackson WS, Borkowski AW, Faas H, Steele AD, King OD, Watson N, Jasanoff A, Lindquist S (2009) Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron 63:438–450. https://doi.org/10.1016/j.neuron.2009.07.026
Jones EM, Surewicz WK (2005) Fibril conformation as the basis of species- and strain-dependent seeding specificity of mammalian prion amyloids. Cell 121:63–72. https://doi.org/10.1016/j.cell.2005.01.034
Kim JI, Cali I, Surewicz K, Kong Q, Raymond GJ, Atarashi R, Race B, Qing L, Gambetti P, Caughey B, Surewicz WK (2010) Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem 285:14083–14087. https://doi.org/10.1074/jbc.C110.113464
Legname G, Baskakov IV, Nguyen HO, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB (2004) Synthetic mammalian prions. Science 305:673–676. https://doi.org/10.1126/science.1100195305/5684/673
Li J, Browning S, Mahal SP, Oelschlegel AM (2010) Weissmann C Darwinian evolution of prions in cell culture. Science 327:869–872. https://doi.org/10.1126/science.1183218
Maggioni GM, Mazzotti M (2015) Modelling the stochastic behaviour of primary nucleation. Faraday Discuss 179:359–382. https://doi.org/10.1039/c4fd00255e
Mahal SP, Baker CA, Demczyk CA, Smith EW, Julius C, Weissmann C (2007) Prion strain discrimination in cell culture: the cell panel assay. Proc Natl Acad Sci USA 104:20908–20913. https://doi.org/10.1073/pnas.0710054104
Makarava N, Kovacs GG, Bocharova O, Savtchenko R, Alexeeva I, Budka H, Rohwer RG, Baskakov IV (2010) Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol 119:177–187. https://doi.org/10.1007/s00401-009-0633-x
Makarava N, Savtchenko R, Alexeeva I, Rohwer RG, Baskakov IV (2016) New molecular insight into mechanism of evolution of mammalian synthetic prions. Am J Pathol 186:1006–1014. https://doi.org/10.1016/j.ajpath.2015.11.013
Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J (1994) 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol 8:121–127. https://doi.org/10.1007/BF02780662
Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M (2006) Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 313:1781–1784. https://doi.org/10.1126/science.1131864
Morales R, Abid K, Soto C (2007) The prion strain phenomenon: molecular basis and unprecedented features. Biochim Biophys Acta 1772:681–691. https://doi.org/10.1016/j.bbadis.2006.12.006
Nandi PK, Leclerc E (1999) Polymerization of murine recombinant prion protein in nucleic acid solution. Arch Virol 144:1751–1763
Nonno R, Di Bari MA, Cardone F, Vaccari G, Fazzi P, Dell’Omo G, Cartoni C, Ingrosso L, Boyle A, Galeno R, Sbriccoli M, Lipp HP, Bruce M, Pocchiari M, Agrimi U (2006) Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog 2:e12. https://doi.org/10.1371/journal.ppat.0020012
Parchi P, Chen SG, Brown P, Zou W, Capellari S, Budka H, Hainfellner J, Reyes PF, Golden GT, Hauw JJ, Gajdusek DC, Gambetti P (1998) Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Straussler-Scheinker disease. Proc Natl Acad Sci USA 95:8322–8327
Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van den Haute C, Melki R, Baekelandt V (2015) alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522:340–344. https://doi.org/10.1038/nature14547
Pirisinu L, Di Bari MA, D’Agostino C, Marcon S, Riccardi G, Poleggi A, Cohen ML, Appleby BS, Gambetti P, Ghetti B, Agrimi U, Nonno R (2016) Gerstmann-Straussler-Scheinker disease subtypes efficiently transmit in bank voles as genuine prion diseases. Sci Rep 6:20443. https://doi.org/10.1038/srep20443
Pirisinu L, Marcon S, Di Bari MA, D’Agostino C, Agrimi U, Nonno R (2013) Biochemical characterization of prion strains in bank voles. Pathogens 2:446–456. https://doi.org/10.3390/pathogens2030446
Saa P, Castilla J, Soto C (2006) Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J Biol Chem 281:35245–35252. https://doi.org/10.1074/jbc.M603964200
Saborio GP, Permanne B, Soto C (2001) Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411:810–813. https://doi.org/10.1038/35081095
Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB (1998) Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med 4:1157–1165. https://doi.org/10.1038/2654
Schmitz M, Dittmar K, Llorens F, Gelpi E, Ferrer I, Schulz-Schaeffer WJ, Zerr I (2016) Hereditary human prion diseases: an update. Mol Neurobiol. https://doi.org/10.1007/s12035-016-9918-y
Shaked GM, Meiner Z, Avraham I, Taraboulos A, Gabizon R (2001) Reconstitution of prion infectivity from solubilized protease-resistant PrP and nonprotein components of prion rods. J Biol Chem 276:14324–14328. https://doi.org/10.1074/jbc.M007815200M007815200
Sim VL, Caughey B (2009) Ultrastructures and strain comparison of under-glycosylated scrapie prion fibrils. Neurobiol Aging 30:2031–2042. https://doi.org/10.1016/j.neurobiolaging.2008.02.016
Stohr J, Condello C, Watts JC, Bloch L, Oehler A, Nick M, DeArmond SJ, Giles K, DeGrado WF, Prusiner SB (2014) Distinct synthetic Abeta prion strains producing different amyloid deposits in bigenic mice. Proc Natl Acad Sci USA 111:10329–10334. https://doi.org/10.1073/pnas.1408968111
Supattapone S (2014) Elucidating the role of cofactors in mammalian prion propagation. Prion 8:100–105
Taniguchi-Watanabe S, Arai T, Kametani F, Nonaka T, Masuda-Suzukake M, Tarutani A, Murayama S, Saito Y, Arima K, Yoshida M, Akiyama H, Robinson A, Mann DM, Iwatsubo T, Hasegawa M (2016) Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol 131:267–280. https://doi.org/10.1007/s00401-015-1503-3
Terry C, Wenborn A, Gros N, Sells J, Joiner S, Hosszu LL, Tattum MH, Panico S, Clare DK, Collinge J, Saibil HR, Wadsworth JD (2016) Ex vivo mammalian prions are formed of paired double helical prion protein fibrils. Open Biol 6:160035. https://doi.org/10.1098/rsob.160035
Vanik DL, Surewicz KA, Surewicz WK (2004) Molecular basis of barriers for interspecies transmissibility of mammalian prions. Mol Cell 14:139–145
Vanni I, Migliore S, Cosseddu GM, Di Bari MA, Pirisinu L, D’Agostino C, Riccardi G, Agrimi U, Nonno R (2016) Isolation of a defective prion mutant from natural scrapie. PLoS Pathog 12:e1006016. https://doi.org/10.1371/journal.ppat.1006016
Vazquez-Fernandez E, Vos MR, Afanasyev P, Cebey L, Sevillano AM, Vidal E, Rosa I, Renault L, Ramos A, Peters PJ, Fernandez JJ, van Heel M, Young HS, Requena JR, Wille H (2016) The structural architecture of an infectious mammalian prion using electron cryomicroscopy. PLoS Pathog 12:e1005835. https://doi.org/10.1371/journal.ppat.1005835
Vidal E, Fernandez-Borges N, Pintado B, Erana H, Ordonez M, Marquez M, Chianini F, Fondevila D, Sanchez-Martin MA, Andreoletti O, Dagleish MP, Pumarola M, Castilla J (2015) Transgenic mouse bioassay: evidence that rabbits are susceptible to a variety of prion isolates. PLoS Pathog 11:e1004977. https://doi.org/10.1371/journal.ppat.1004977
Wang F, Wang X, Yuan CG, Ma J (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327:1132–1135. https://doi.org/10.1126/science.1183748
Watts JC, Giles K, Bourkas ME, Patel S, Oehler A, Gavidia M, Bhardwaj S, Lee J, Prusiner SB (2016) Towards authentic transgenic mouse models of heritable PrP prion diseases. Acta Neuropathol. https://doi.org/10.1007/s00401-016-1585-6
Watts JC, Giles K, Patel S, Oehler A, DeArmond SJ, Prusiner SB (2014) Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog 10:e1003990. https://doi.org/10.1371/journal.ppat.1003990
Watts JC, Giles K, Stohr J, Oehler A, Bhardwaj S, Grillo SK, Patel S, DeArmond SJ, Prusiner SB (2012) Spontaneous generation of rapidly transmissible prions in transgenic mice expressing wild-type bank vole prion protein. Proc Natl Acad Sci USA 109:3498–3503. https://doi.org/10.1073/pnas.1121556109
Weissmann C, Flechsig E (2003) PrP knock-out and PrP transgenic mice in prion research. Br Med Bull 66:43–60
Weissmann C, Li J, Mahal SP, Browning S (2011) Prions on the move. EMBO Rep 12:1109–1117. https://doi.org/10.1038/embor.2011.192
Wenborn A, Terry C, Gros N, Joiner S, D’Castro L, Panico S, Sells J, Cronier S, Linehan JM, Brandner S, Saibil HR, Collinge J, Wadsworth JD (2015) A novel and rapid method for obtaining high titre intact prion strains from mammalian brain. Sci Rep 5:10062. https://doi.org/10.1038/srep10062
Yehiely F, Bamborough P, Da Costa M, Perry BJ, Thinakaran G, Cohen FE, Carlson GA, Prusiner SB (1997) Identification of candidate proteins binding to prion protein. Neurobiol Dis 3:339–355. https://doi.org/10.1006/nbdi.1997.0130
Zou WQ, Puoti G, Xiao X, Yuan J, Qing L, Cali I, Shimoji M, Langeveld JP, Castellani R, Notari S, Crain B, Schmidt RE, Geschwind M, Dearmond SJ, Cairns NJ, Dickson D, Honig L, Torres JM, Mastrianni J, Capellari S, Giaccone G, Belay ED, Schonberger LB, Cohen M, Perry G, Kong Q, Parchi P, Tagliavini F, Gambetti P (2010) Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol 68:162–172. https://doi.org/10.1002/ana.22094
Acknowledgments
This work was supported financially by Spanish Government Grants AGL2015-65046-C2-1-R, PCIN‐2013‐065 and BFU2013-48436-C2-1-P, and a Basque Government Grant 2014111157. The authors would like to thank the following for their support: IKERBasque foundation, vivarium and maintenance from CIC bioGUNE and Patricia Piñeiro and Maite Pérez for technical support. Oxford Protein Production Facility UK (OPPF) for the plasmid pOPIN E, Dr. Ester Vázquez Fernández for useful advice in the initial steps. Dr. Mark P. Dagleish (Moredun Research Institute) for useful discussion and advice.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
401_2017_1782_MOESM1_ESM.pdf
Supplementary material 1 (PDF 2304 kb) Fig. S1 Biochemical characterization of two distinct misfolded rec-PrPs. a Western blot representing two duplicates of L-seeded-02 and H-seeded-02, two misfolded rec-PrPs generated after serial PMCA propagation (see Fig. 1). Two distinct migration patterns were observed (Low and High) after digestion with 85 µg/ml of Proteinase-K (PK). b Western blot of amplified L-seeded-02 and H-seeded-02 seeds subjected to a single round of 24-h recPMCA. Each misfolded rec-PrP was subjected to serial dilutions (from 10−1 to 10−8) previous to in vitro propagation. The L-seeded-02 seed was able to amplify until a dilution of 10−5 while the H-seeded-02 seed amplified until dilution of 10−8. All the samples were digested with 85 µg/ml of PK. Despite all the samples were run at the same time, the blot was cropped as indicated by the vertical dotted line to avoid displaying unrelated samples. c PK-resistance assay of the two distinct misfolded rec-PrPs. Representative Western blot of the resistance of L-seeded-02 and H-seeded-02 misfolded rec-PrPs to increasing concentrations of PK. The samples were treated for 1 h at 42 °C with 50, 100, 200, 300, 400, 600, 800, 1600 and 2000 μg/ml of PK. Both misfolded proteins showed a high resistance to PK digestion (up to 400 μg/ml) with L-seeded-02 showing higher resistance (at least 2,000 μg/ml). Membranes were developed with SAF83 monoclonal antibody (1:400). Mw: Molecular weight. Fig. S2 Biochemical analysis of Proteinase K (PK)-resistant PrPSc in brain homogenates from bank vole I109 inoculated with different misfolded rec-PrPs. a The same Western blot with mAb 9A2 (1:400) shown in Fig. 3a is here compared with a replica blot revealed with mAb 12B2 (1:500), as indicated. With both antibodies, the two voles infected with H/L-seeded-03 and having classical PrPSc show clearly different glycosylation patterns. Note that, while the brain-derived classical prion strains, CWD and scrapie, show similar signal intensities with the two antibodies, PrPSc from voles infected with recombinant prions (H/L-seeded-03) seems weaker with 12B2 than with 9A2. As 9A2 and 12B2 maps, two nearby epitopes in the region cleaved by PK (amino acids 99-101 and 89-93, respectively) the relative presence of the 12B2 epitope is a measure of the N-terminal cleavage by PK and thus, indirectly, of conformational differences among PrPSc aggregates. b Measurements of the signal ratio between 9A2 and 12B2 replica blots where a high signal ratio indicates a less N-terminal PK-cleavage site. Note that, the ratio is higher in PrPSc from voles infected with recombinant prions (H/L-seeded-03) than in CWD or scrapie, in keeping with their lower apparent molecular weight, suggesting that N-terminal cleavage by PK is less N-terminal in these samples. PrPSc from the two animals showing a classical PrPres pattern are thus different between them (glycotype) and also from PrPSc in voles infected with CWD or scrapie (differential antibody binding). Fig. S3 Molecular and pathological and phenotypes in vole-adapted prion strains derived from recombinant prions. a Biochemical analysis of PK-resistant PrPSc in brain homogenates from bank vole I109 after second passage of PMCA-derived vole CWD and misfolded rec-PrPs (H-seeded-01, H-seeded-02, L-seeded-Dextran, H-seeded-No cofactor). Representative vole brain homogenates were digested with 200 µg/ml of PK and analyzed by Western blot with monoclonal antibodies 9A2 (1:400) and 12B2 (1:500). Overall, all inocula preserved the original PrPSc type, H-seeded-01 and H-seeded-02 propagating atypical 7 kDa protease-resistant PrPSc, and cofactor-selected recombinant prions propagating classical PrPSc. b As 9A2 and 12B2 maps two nearby epitopes in the region cleaved by PK (amino acids 99-101 and 89-93, respectively), the relative presence of the 12B2 epitope is a measure of the N-terminal cleavage by PK and thus, of conformational differences among PrPSc aggregates. This can be measured by the signal ratio between 9A2 and 12B2 replica blots, where a high signal ratio indicates a less N-terminal PK-cleavage site and correlates with a lower apparent MW. Note that, the ratio is higher in PrPSc from voles infected with recombinant prions than in CWD, suggesting that cleavage by PK is less N-terminal in these samples. Furthermore, a different N-terminal cleavage can be observed even between H-seeded-No cofactor and L-seeded-Dextran. c Patterns of neurodegeneration assessed by lesion profiles (left column) and PET-blot analysis of deposition of PK-resistant PrPSc with monoclonal antibody 6C2 (1:300). H-seeded-01 and H-seeded-02 showed overlapping distribution of spongiform degeneration and PrPSc deposition, characterized by strong PrPSc deposition in white matter tracts, such as the alveus of the hippocampus, corpus callosum and fiber bundles in the striatum and blind involvement of subcortical areas. In contrast, subcortical involvement and grey matter PrPSc deposition were the hallmarks of H-seeded-No cofactor, which closely mirrored the phenotype observed after primary passage of the same inoculum (compare with Fig. 6), despite the dramatic reduction of the incubation times (424 dpi vs. 82 dpi). L-seeded-Dextran showed both, cortical and subcortical involvement. Of note, along with PrPSc deposition in grey matter areas, also white matter deposition was observed in L-seeded-Dextran. Fig. S4 In vitro propagation of unseeded misfolded rec-PrPs (generated in cofactor-containing substrate) using brain-based PMCA. a Rounds (R1-R5) of serial PMCA using bank vole or transgenic mice overexpressing bank vole I109 PrP (TgVole) brain homogenates as substrates. The misfolded rec-PrPs: H-unseeded-No cofactor, L-unseeded-Dextran, H-unseeded-RNA and H-unseeded-Plasmid were used as seeds in replicates of four through five rounds of serial PMCA. Tubes were considered positives if a classical PrPres pattern was observed on Western blot. With the exception of H-unseeded-No cofactor, the rest of the samples were efficiently propagated over a mammalian substrate, with L-unseeded-Dextran and H-unseeded-RNA being particularly efficient seeds. These results correlate strongly with those observed in Fig. 5a. b Four tubes of round 5 of each PMCA-propagated sample were digested with 85 µg/ml of Proteinase-K (PK) and analyzed by Western blot using monoclonal antibody Saf83 (1:400). Both TgVole-based and bank vole-based substrates were similarly efficient. At least two biochemical patterns based on migration properties are shown; a low migration of the L-unseeded-Dextran and a high migration of the H-unseeded-Plasmid. All unseeded samples remained negatives. Control substrates: undigested bank vole or TgVole whole brain homogenates. Mw: Molecular weight. Fig. S5 Electronic microscopy analysis of the cofactor-selected misfolded rec-PrPs. a Cryo-EM images of the cofactor-selected misfolded rec-PrPs. The sample was concentrated 100 times by sedimentation without Proteinase-K (PK)-digestion. Rod-like structures, of 100 to 200 nm in length, are conspicuous. The rods are made up of 10 nm wide fibrils laterally associated or bundled, and are very similar to those seen in preparations of GPI-anchorless PrPSc isolated from brain [64]. Scale bars: 200 nm. b Negative stain TEM images of the dextran-selected misfolded rec-PrP (L-seeded-Dextran) and mouse GPI-anchorless PrPSc. Samples were deposited on freshly glow-discharged carbon-coated gold grids and stained with 5% uranyl acetate. As with cryo-EM images, rods visible in the misfolded rec-PrP samples are very similar to those seen in GPI-anchorless PrPSc samples obtained from brain. Scale bars: 100 and 500 nm. Fig. S6 Modelling the generation and selection of different misfolded rec-PrPs Cartoon showing the putative generation of three different misfolded rec-PrPs (purple, orange and blue figures) after serial rounds of unseeded recPMCA. Different misfolding ratios are obtained as consequence as the presence of brain homogenate (BH) components. A filter representing a specific component (putative cofactor) is drawn as sieve to filter (preferential propagation of) just certain misfolded rec-PrPs. The serial rounds of PMCA select positively the recombinant prion strain that is favoured at expenses of the rest of the strains that could even disappear after a larger number of rounds. Fig. S7 Schematic representation of in vitro propagated recombinant prions and histopathological findings associated to their inoculation in bank vole. An overview of the procedures performed and the results generated along this work. The scheme has been divided in two parts: in vitro and in vivo (bioassay) studies. The in vitro part shows how different types of substrates (Prnp 0/0 brain homogenate and specific cofactors) generated different misfolded rec-PrPs. The bioassay part shows the prion strains resulting in vivo and the major histopathological findings observed after their inoculation in bank vole.
Rights and permissions
About this article

Cite this article
Fernández-Borges, N., Di Bari, M.A., Eraña, H. et al. Cofactors influence the biological properties of infectious recombinant prions. Acta Neuropathol 135, 179–199 (2018). https://doi.org/10.1007/s00401-017-1782-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-017-1782-y