
Abstract
In patients with stroke and neurodegenerative diseases, overactivation of poly(ADP-ribose) polymerase-1 (PARP-1) causes harmful effects by inducing apoptosis, necrosis, neuroinflammation, and immune dysregulation. The current study investigated the neuroprotective effect of a novel PARP-1 inhibitor, JPI-289, in an animal model of ischemic stroke. A transient middle cerebral artery occlusion (tMCAO, 2 h) model was used to determine the therapeutic effect and the most effective dose and time window of administration of JPI-289. We also investigated the long-term outcomes of treatment with JPI-289 by diffusion-weighted imaging (DWI) and fluid-attenuated inversion recovery (FLAIR) MRI and by measuring neurological function at 24 h, 7 days, and 28 days after MCAO. The most effective dose and time window of administration of JPI-289 was 10 mg/kg administered 2 h after MCAO with reperfusion. Twenty-four hours after MCAO, infarct volume was reduced by 53% and the number of apoptotic cells was reduced by 56% compared with control. JPI-289 also reduced infarct volume by 16% in the permanent MCAO model. In an MRI-based study, initial infarct volume, as measured using DWI, was similar in the control and JPI-289-treated groups. However, infarct volume and brain swelling were significantly reduced in the group treated with JPI-289 (2 h) at 24 h and 7 days after MCAO. Neurological functions also improved in the group treated with JPI-289 (2 h) until 28 days after MCAO. Inhibition of PARP-1 has neuroprotective effects (reduction of infarct volume and brain swelling) in both tMCAO and pMCAO models of ischemic stroke.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Stroke is the second leading cause of death and a common cause of severe disability in adults [1]. Despite the high mortality and morbidity, treatment options for patients with ischemic stroke remain limited. The only approved medical treatment for the acute stage of ischemic stroke is tissue plasminogen activator (tPA), but it needs to be administered within 4.5 h of having a ischemic stroke and only about 5% of patients are eligible to receive tPA [2]. In addition, tPA may cause reperfusion injury by breaking down the blood–brain barrier (BBB) and causing hemorrhagic transformation of the ischemic brain. Therefore, the development of new and complementary treatment strategies for patients with ischemic stroke is necessary.
Neuroprotective agents have been studied widely for the treatment of stroke, because they are known to reduce reactive oxygen species, inhibit glutamate release, stabilize intracellular Ca2+ levels, prevent mitochondrial collapse, and inhibit neuroinflammation [3]. They are also thought to have roles in preserving the BBB and enhancing the therapeutic time window of tPA [4]. Although, neuroprotective agents have yielded disappointing results in the treatment of ischemic stroke in human trials, careful preclinical assessment and appropriate design/implementation may yield better results [5]. Therefore, clinical trials based on early treatment at prehospital stage or combined with recanalization therapy have been suggested as a new approach to test the effect of neuroprotective agents [5].
Poly(ADP-ribose) polymerase-1 (PARP-1) is a potential neuroprotective target and is a member of a superfamily of cell signaling enzymes present in eukaryotes [6]. PARP-1 is a key regulator of nuclear processes, such as DNA repair, replication, and transcription. However, the excessive activation of PARP-1 in a number of diverse diseases leads to the rapid depletion of intracellular pools of nicotinamide adenine dinucleotide (NAD) and adenosine triphosphate (ATP), resulting in cellular dysfunction and cell death by necrosis and apoptosis [7]. The inhibition of PARP-1 after ischemic stroke is an attractive target for inducing neuroprotection for several reasons. First, following ischemic stroke, PARP-1 is activated in most brain cell types, including neurons and non-neuronal glial cells, and inhibition of PARP-1 might reduce tissue damage by reducing apoptosis and necrosis of all cell types [8]. Second, inhibition of PARP-1 might reduce neuroinflammation, energy depletion, and caspase-independent programmed cell death [9, 10]. Third, inhibition of PARP-1 might regulate the adaptive immune system by the downregulation of pro-inflammatory cytokines, inactivation of T cells and dendritic cells, and differentiation of regulatory T cells [11]. Therefore, multi-targeting PARP-1 inhibitors have been widely studied in ischemic stroke model with positive results [8, 12,13,14,15,16,17,18,19].
We recently identified and developed JPI-289, a novel PARP-1 inhibitor with strong PARP-1 inhibitory activity, and showed that it has beneficial effects on oxygen- and glucose-deprived rat cortical neurons through reduced PARP activity, increased ATP and NAD+ levels, and decreased apoptosis-related molecules such as AIF, cytochrome C, and cleaved caspase-3 [10]. In the present study, we evaluated whether JPI-289 has a role in protecting against brain damage in reperfusion animal models with or without concomitant use of tPA. In addition, we performed diffusion-weighted image (DWI) based randomization pilot study to reduce selection bias.
Methods
Animal Preparation and Ischemic Surgery
All animal experimental procedures were performed by the Institutional Animal Care and Use Committee (IACUC) of Hanyang University. Every effort was made to minimize the number of animals used and unnecessary animal suffering. Male Sprague–Dawley (SD) rats, weighing 280 to 320 g, were purchased from Koatech (Pyeongtaek, Korea). Transient middle cerebral artery occlusion (tMCAO, n = 86) was induced by a modification of the intraluminal filament thread method, as described previously [14, 20, 21]. Rats were anesthetized with isoflurane (3% for induction and 2% for surgical procedure) in a mixture of oxygen/nitrous oxide (30/70%). Body temperature was monitored during the surgery by a rectal probe and maintained at 37 ± 0.5 °C with a homeothermic blanket control unit (Harvard Apparatus, UK). Each animal was placed in the supine position, and the left external carotid artery (ECA), internal carotid artery (ICA), and common carotid artery (CCA) were carefully isolated via a midline neck incision. A 20-mm length of 4-0 nylon surgical suture coated with silicon (Xantopren VL plus, Heraeus Kulzer Gmbh & Co. KG, Hanau, Germany) was introduced through the ECA and inserted from the ICA to occlude the origin of the left MCA. For the model of tMCAO, each animal was re-anesthetized, and the monofilament was pulled out to enable reperfusion at 2 h after MCAO. For the model of permanent MCAO (pMCAO), the MCAO was maintained for 24 h before removal of the monofilament (n = 20). Rats displaying neurological deficits were selected for the subsequent studies. The neurologic examination used a neurologic deficit score (NDS) to select for animal models of cerebral ischemia. The NDS comprised of consciousness (0, normal; 1, restless; 2, lethargic; 3, stuporous; 4, death), gait (0, normal; 1, paw adduction; 2, unbalanced walking; 3, circling; 4, unable to stand; 5, no movement), limb tone (0, normal; 1, spastic; 2, flaccid), and pain reflex (0, normal; 2, hypoactive; 4, absent) at 1 h 50 min after MCAO by an investigator who was blind to the experimental groups [22].
Determination of the Most Effective Dose and Time Window
JPI-289 has non-hydroscopic properties and dissolves readily in water [10]. For the dose–response experiment, JPI-289 was dissolved in saline immediately before use and administered at doses of 5 mg/kg (n = 8), 7.5 mg/kg (n = 8), or 10 mg/kg (n = 8). JPI-289 or vehicle (saline; n = 8) was administered by a single intravenous injection into the lateral tail vein 2 h after MCAO. All animals were euthanized on completion of the experiment at 24 h after MCAO.
To determine the optimum therapeutic time window of administration of JPI-289, rats were assigned to five groups. JPI-289 (10 mg/kg) was administered into the tail vein at 2 h (n = 9), 8 h (n = 9), 12 h (n = 9), or 16 h (n = 9) after MCAO. In the vehicle group (n = 9), vehicle was administered at 2 h after MCAO. All animals were euthanized on completion of the experiment at 48 h after MCAO.
For the study of pMCAO, JPI-289 (10 mg/kg, n = 10) or vehicle (n = 10) was administered by tail vein injection 2 h after MCAO. All animals were euthanized on completion of the experiment at 24 h after MCAO.
Infarct volume was evaluated by staining with 2,3,5-triphenyl tetrazolium chloride (TTC), as described previously [23]. Animals were euthanized under anesthesia, and the brains were isolated immediately. The brains were cut into 2-mm coronal slices (+ 4 to − 6 mm from the bregma point) using a brain slicer and then incubated in saline containing 2% TTC at 37 °C for 30 min and fixed in 10% neutral buffered formalin solution. The total infarct volume was measured using ImageJ software (NIH image, version 1.61).
TUNEL Staining
For the assessment of the number of apoptotic cells, rats were assigned to three groups. JPI-289 (10 mg/kg) was administered into the tail vein at either 2 h (n = 3) or 8 h (n = 3) after MCAO. In the vehicle group, vehicle (n = 3) was administered at 2 h after MCAO. At 24 h after MCAO, rats were euthanized before the terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) labeling of apoptotic cells. Brain tissues were embedded in paraffin and cut into 4-μm-thick tissue sections. The sections were stained with a TUNEL kit (In Situ Cell Death Detection Kit, Roche) and colorized with 0.05% 3,3-diaminobenzidine (DAB, Sigma). The number of TUNEL-positive cells was counted from three random fields (400×) within the penumbra for each animal by individuals blinded to the experiment.
Long-Term Effects of JPI-289 Measured Using Diffusion-Weighted Imaging Fluid-Attenuated Inversion Recovery MRI
Using our predetermined dose and therapeutic window of administration (10 mg/kg for JPI-289; administered either 2 or 8 h after MCAO; vehicle administered 2 h after MCAO), we investigated the long-term effects of JPI-289 using DWI in a rat tMCAO model. All magnetic resonance (MR) images were obtained using a 3-T MR machine (Achieva, Philips, Best, The Netherlands) with a four-channel phased array coil (mouse coil, Shanghai Chenguang Medical Technology, Shanghai, China). The temperature of the MRI room was kept at approximately 27 °C.
One hour after MCAO, all rats underwent DWI (TR/TE = 4455/74 ms, slice thickness/gap = 1/1 mm, matrix = 64 × 63, FOV = 50 × 50 mm), and rats with large infarcts (> 100 mm3; infarct volume calculations by Medical Image Processing Analysis and Visualization (MIPAV); National Institutes of Health, Bethesda, MD, USA) were randomized to receive vehicle alone or treatment (10 mg/kg JPI-289, administered either 2 or 8 h after MCAO). A total 15 rats (n = 5/group) underwent fluid-attenuated inversion recovery (FLAIR) MRI (TR/TE/TI = 11,000/77/2800 ms, slice thickness/gap = 1/1 mm; matrix = 128 × 128, FOV = 50 × 50 mm) 24 h, 7 days, or 28 days after MCAO. The infarct volume and hemisphere volume were assessed independently using MIPAV by two experienced radiologists (JYK, YJL) who were blinded to the experimental design. The final infarct volume was calculated from the averages of the two measurements (DWI and FLAIR). Brain swelling was calculated using the following equation: [(affected hemisphere volume / unaffected hemisphere volume) − 1] × 100%.
Behavioral Assessment
To evaluate the long-term neurological outcomes of treatment with JPI-289, we investigated the repair of the neurological deficit and survival following tMCAO in rats (n = 17 each for control, JPI-289 2 h, JPI-289 8 h and n = 10 for sham). All animals underwent neurobehavioral assessment for 3 days before MCAO. Functional activities were evaluated using the NDS, rotarod test, and the modified sticky tape (MST) test. These tests were performed 1 day before surgery (day − 1), then 1, 3, 7, 14, 21, and 28 days after MCAO (dayd 1, 3, 7, 14, 21, 28). NDS was also evaluated just before reperfusion (2 h after MCAO) by an investigator who was blinded to the experimental groups. The distribution of rats to either JPI-289 treatment or control groups was based on pre-reperfusion NDS. NDS was graded on a scale of 0 to 15 (normal score, 0; maximal deficit score, 15) as described previously [22].
An accelerating rotarod was used to measure the motor coordination and balance function of rats. The rats were placed on rungs of the accelerating rotarod, and the time the animals remained on the rotarod was measured. Over 5 min, the speed was slowly increased from 4 to 40 rpm, and the trial ended when the animal fell off the rungs, gripped the device, or spun around for two consecutive revolutions without attempting to walk on the rungs. The animals were trained for 3 days before MCAO. The mean duration (in seconds) on the device was recorded with three trials on the day before surgery (day − 1). Rotarod test data are presented as percentage of duration (three attempts) on the rotarod compared with the internal baseline control (before surgery) [24]. A MST test was performed to evaluate tactile responses and functional asymmetry. A sleeve was created using a 3.0-cm-long piece of green paper tape (Fisher Scientific, Pittsburgh, PA, USA), with a width of 1.0 cm, which was wrapped around the forepaw of the rat so that the tape attached to itself and the claws protrude slightly from the sleeve formed. If attached correctly, the tape sleeve cannot be removed. The typical response is for the rat to vigorously attempt to remove the sleeve by either pulling at the tape with its mouth and/or brushing the tape with its contralateral paw. When the tape was positioned correctly, the rat was then placed in cage and observed for 30 s. Two timers were started: the first should run without interruption and the second should be turned on only while the animal attempts to remove the tape sleeve. The contralateral and ipsilateral limbs were tested separately. The test was repeated three times per testing day, and the best two scores of the day were averaged [25].
Embolic Focal Cerebral Ischemia Models with Simultaneous Injection of tPA and JPI-289
An embolic focal cerebral ischemia model was induced in male Wistar rats (n = 27), weighing 180 to 220 g, by a modification of the fibrin-rich clot method, as described previously [26]. To prepare the embolus, the femoral artery of the donor rat was catheterized and blood was transferred directly into 20 cm of PE-50 tubing and kept for 2 h at room temperature. The clot was subsequently refrigerated for 22 h at 4 °C prior to use. The clot was then shifted by gentle shaking for 1 min in a PE10 tube filled with saline. A single 2-cm clot was transferred to a thoracic jugular vein catheter (R-JVC-STD, Braintree Scientific, Braintree, MA, USA) for injection into the MCA. Rats were anesthetized with isoflurane in a mixture of oxygen/nitrous oxide and their body temperature monitored. The left CCA was exposed through a ventral midline incision in the neck. The ECA, ICA, and CCA were carefully isolated and maintained in a Y-shape using a silk suture. A catheter tube filled with the single clot was introduced into the ECA lumen through a small puncture. A 15 to 16-mm length of catheter was gently advanced from the ECA into the lumen of the ICA, and the clot in the catheter was injected into the ICA. The catheter was withdrawn from the right ECA 5 min after the injection, and the ECA was ligated. Rats satisfying the criteria of neurological deficit were selected by NDS and divided into three groups: control (n = 9), tPA (10 mg/kg, n = 9), and tPA plus JPI-289 (20 mg/kg, n = 9). The tPA (Actilyse; Boehringer-Ingelheim, Biberach an der Riss, Germany) and JPI-289 were given as a single intravenous infusion over 30 min into the lateral tail vein 1 h after embolic MCAO. Since continuous infusion of 20 mg/kg JPI-289 was more effective than 10 mg/kg, we used 20 mg/kg for this experiment. All animals were killed on completion of the experiment 24 h after embolic MCAO. Brains were collected and sliced into six serial 2-mm coronal sections, and hemorrhage transformation was assessed by scanning the brain slices with subsequent image analysis [27, 28]. The slices were incubated in phosphate-buffered saline (1× PBS) containing 2 w/v% TTC solution and fixed in formalin, and infarct volumes were determined by scanning the brain sections and subsequent image analysis.
Statistical Analyses
All data are presented as mean ± SEM. Non-parametric data were compared using the Mann–Whitney U test or Kruskal–Wallis test. p values < 0.05 were considered statistically significant. All statistical analyses were performed using SPSS 19.0 software package for Windows (SPSS, Seoul, Korea).
Results
Effect of JPI-289 on Infarct Volume in Transient and Permanent Rat MCAO Models
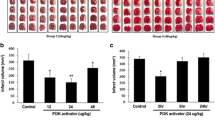
Treatment with JPI-289 significantly reduced the infarct volume by 30% at a dose of 7.5 mg/kg (p < 0.05) and by 53% at a dose of 10 mg/kg (p < 0.05). In the group treated with 5 mg/kg, the infarct volume was reduced by 22% but did not reach statistical significance (p > 0.05, Fig. 1a, b). There were no significant differences in the mean arterial blood pressure, cerebral blood flow, rectal temperature, and arterial blood parameters (pH, pO2, pCO2, and hematocrit) between the vehicle- and JPI-289-treated groups (data not shown).

Optimal therapeutic dose and time window of administration of JPI-289. JPI-289 was injected intravenously at doses of 5, 7.5, and 10 mg/kg 2 h after MCAO; infarct volume was determined by 2,3,5-triphenyl tetrazolium chloride (TTC) staining (n = 8/group) (a). Treatment with 7.5 or 10 mg/kg JPI-289 resulted in significantly reduced infarct volume (b). JPI-289 (10 mg/kg) was injected intravenously at different time points (2, 8, 12, and 16 h after MCAO); then, rats were killed at 48 h, and brains stained with TTC (n = 9/group) (c). Treatment with 10 mg/kg JPI-289 reduced infarct volume up to and including 12 h after MCAO; earlier treatment resulted in greater reductions in infarct volume (d). All data are expressed as mean ± SEM. *p < 0.05 versus vehicle group (Kruskal–Wallis test)
To evaluate the therapeutic time window of JPI-289, an intravenous bolus (10 mg/kg) was introduced at various time points in a rat tMCAO model. JPI-289 reduced the infarct volume by 37% (p < 0.05) compared to the vehicle-treated group when it was administered 2 h after MCAO in the rat tMCAO model, and remained effective after 8 h (22% reduction, p < 0.05) and 12 h (15% reduction, p < 0.05). Sixteen hours after MCAO, the infarct volume was reduced by 11%, but this was not statistically significant (p > 0.05, Fig. 1c, d). The neuroprotective effect of JPI-289 was also confirmed in the rat pMCAO model. JPI-289 (10 mg/kg) reduced the total infarct volume by 16% (p < 0.05) when it was administered 2 h after the onset of MCAO in the rat pMCAO model (Fig. 2).

Effect of JPI-289 on infarct volume in a rat permanent MCAO model. JPI-289 (10 mg/kg) was injected intravenously 2 h after MCAO. a Rats were killed 24 h after MCAO, and infarct volume was determined using TTC staining (n = 10/group). b The group treated with JPI-289 had a reduced infarct volume. All data are expressed as mean ± SEM. *p < 0.05 versus vehicle group (Mann–Whitney U test)
Downregulation of Apoptosis Visualized by TUNEL Staining in a Rat tMCAO Model
TUNEL staining was used to investigate the inhibitory activity of JPI-289 on apoptosis in a rat tMCAO model. TUNEL-positive cells were found in the penumbra region in both control and JPI-289-treated groups. The percentage of TUNEL-positive cells was significantly reduced in the group treated with JPI-289 2 h after MCAO (56% reduction, p < 0.05) compared to the vehicle-treated control group. In the group treated with JPI-289 8 h after occlusion, however, the percentage of TUNEL-positive cells was not significantly reduced (23% reduction, p > 0.05, Fig. 3).

Effect of JPI-289 on apoptosis in the penumbra of rat. JPI-289 (10 mg/kg) was injected intravenously 2 and 8 h after MCAO. Rats were killed after 24 h, and TUNEL staining was used to visualize apoptotic cells (n = 3/group) (a). Treatment with JPI-289 2 h after MCAO reduced apoptosis significantly (b). All data are expressed as mean ± SEM. *p < 0.05 versus vehicle group (Kruskal–Wallis test)
Long-Term Efficacy of JPI-289 Measured by Diffusion-Weighted Imaging and Fluid-Attenuated Inversion Recovery MRI
Images representative of serial MRIs after MCAO are shown in Fig. 4a. Rats with an infarct volume greater than 100 mm2, as determined by DWI, were randomized to receive vehicle alone, or treatment (JPI-289 10 mg/kg administered at either 2 or 8 h after MCAO). At the start of the experiment, the mean infarct volumes of the control group, the group receiving JPI-289 2 h after MCAO, and the group receiving JPI-289 8 h after MCAO were 197.2 ± 61.0, 187.7 ± 62.9, and 193.5 ± 52.9 mm3, respectively, with no significant difference between groups (p = 0.917, Fig. 4b). FLAIR MRI at 24 h and 7 days after MCAO, however, showed that the infarct volume was significantly reduced in the group that received JPI-289 2 h after MCAO, compared with the control group (Fig. 4b). The mean infarct volume in the control group was 328.0 ± 41.9 and 236.9 ± 38.6 mm3 at 24 h and 7 days after MCAO, respectively, whereas that of the group treated with JPI-289 2 h after MCAO was 221.9 ± 77.7 mm3 (p = 0.016) and 160.0 ± 56.5 mm3 (p = 0.028) at 24 h and 7 days after MCAO, respectively. The mean infarct volume in the group treated with JPI-289 8 h after MCAO was smaller than that of the control group (281.2 ± 60.0 and 206.6 ± 43.8 mm3, at 24 h and 7 days after MCAO, respectively), but this did not reach statistical significance. On day 28 after MCAO, the JPI-289-treated groups continued to have smaller mean infarct volumes than the control group, but this difference was not statistically significant (Fig. 4b).

Efficacy of JPI-289 measured by diffusion-weighted imaging and fluid-attenuated inversion recovery (FLAIR) MRI. Images are representative of serial MRIs after MCAO (a). Although initial infarct size, measured by diffusion MRI, was similar in all the groups, fluid-attenuated inversion recovery (FLAIR) MRI reveals a significant reduction of infarct volume after 24 h and 7 days in the group that received JPI-289 2 h after MCAO (n = 5/group) (b). Brain swelling measured by MRI was significantly reduced 7 days following MCAO in the group treated with JPI-289 after 2 h (c). All data are expressed as mean ± SEM. *p < 0.05 versus control group (Mann–Whitney U test)
Brain swelling was also reduced in the JPI-289-treated groups, with significant reduction in brain swelling 7 days after MCAO in the group receiving JPI-289 2 h after MCAO (p = 0.047). Brain swelling was reduced on all other days after MCAO, in the JPI-289-treated groups compared to the control group, but this was not statistically significant (Fig. 4c).
Effect of JPI-289 on Long-Term Behavior in tMCAO Model
We started with 17 rats in each group; however, three rats in the control group, three rats in the group treated with JPI-289 2 h after MCAO, and two rats in the group treated with JPI-289 8 h after MCAO were expired before 3 days after MCAO. This left 14 rats in the control group, 14 rats in the group treated with JPI-289 2 h after MCAO, and 15 rats in the group treated with JPI-289 8 h after MCAO for further analysis. The results of behavior tests (NDS, rotarod, and MST test) after MCAO showed no differences between each group for the first 3 days after MCAO. From day 7, however, rats treated with JPI-289 2 h after MCAO showed significant improvement in motor and sensory function, as measured by NDS and the MST test, than the control group. Rats treated with JPI-289 8 h after MCAO also showed beneficial effects 14 days after tMCAO. However, there was no significant difference between the two JPI-289-treated groups at any time point (Fig. 5).

The effect of JPI-289 on long-term behavior function in rats. Behavioral function tests (neurological deficiency scale (NDS) (a), rotarod (b), and modified sticky-tape test (c)) before and after MCAO show the beneficial effects of early treatment with JPI-289 on the motor and sensory functions of rats (n = 10 for sham, n = 14 for control, n = 14 for JPI-289 2 h, n = 15 for JPI-289 8 h). All data are expressed as mean ± SEM. *p < 0.05 versus control group (Mann–Whitney U test)
Effect of JPI-289 on Infarct Volume and Hemorrhage Transformations in a Rat Embolic Focal Cerebral Ischemia Model
Average infarct volumes relative to the vehicle control group were reduced by 49% (p < 0.01) and 58% (p < 0.01) in the group treated with tPA only and the group treated with JPI-289 and tPA, respectively, at 24 h after embolic stroke onset (Fig. 6a, b). Gross hemorrhage in the ipsilateral lesion was detected in five of nine rats in the control group, seven of nine in the tPA group, and four of nine in the JPI-289 plus tPA group (Fig. 6a). Treatment with tPA at 1 h after ischemia increased the area with hemorrhage measured at 24 h after embolic MCAO relative to the control group, whereas JPI-289 plus tPA reduced the hemorrhage area significantly compared with the tPA group (Fig. 6c).

Effect of JPI-289 on infarct volumes and hemorrhage transformation in a rat embolic focal cerebral ischemia model. tPA (10 mg/kg) or tPA plus JPI-289 (20 mg/kg) was injected by intravenous infusion over 30 min at 1 h after embolic MCAO, and the rats were killed at 24 h (n = 9/group). Hemorrhage transformation was determined by scanning brain tissue slices, and infarct volumes by TTC staining (a). Infarct volumes relative to the control group were reduced in both the tPA only and the combined JPI-289 and tPA group (b). Hemorrhage area was significantly lower in the JPI-289 and tPA group than in the tPA only group (c). All data are expressed as means ± SEMs. *p < 0.05 versus vehicle group (Kruskal–Wallis test)
Discussion
The present study demonstrates that early inhibition of PARP-1 by JPI-289 reduces infarct volume in rat tMCAO and pMCAO models with reduced apoptosis. In addition, the reduction of infarct volume persists until 28 days after MCAO, and the behavior of rats improved following treatment with JPI-289 until 28 days after MCAO. Furthermore, JPI-289 reduced tPA-related hemorrhagic transformation in embolic stroke model. These results suggest that JPI-289 might be neuroprotective during the acute stage of ischemic stroke.
PARP-1 plays a significant role in the acute stage of ischemic stroke. It is a nuclear enzyme that participates in DNA repair, cell cycle control, and the genomic stability response to hypoxia. During mild DNA damage, polyADP-ribosylation facilitates DNA repair, which leads to cell survival. However, excessive PARP-1 activation contributes to an energy-consuming cellular process, which leads to cellular NAD and ATP depletion, mitochondrial dysfunction, overall cellular dysfunction, and ultimately neuronal cell death [7, 29]. By downregulating PARP-1, neuronal injury during ischemia might be prevented and, for this reason, PARP-1 inhibitors have been studied extensively as attractive therapeutic targets for stroke drug development. 3-Aminobenzamide (3-AB), a classical PARP inhibitor, reduces neuronal cell death in in vivo models of ischemic stroke [14]. In addition, other PARP inhibitors, including PJ-34 (N-(6-oxo-5,6-dihydro-phenanthridin-2-yl)-N,N-dimethylacetamide), MP-124 (an isoquinoline derivative), and thienyl-isoquinolone (TIQ-A) have more potent and selective inhibitory effects on PARP-1 activity than 3-AB in animal models of ischemic stroke [12, 13, 30]. Olaparib, which was initially approved for cancer therapy by the US Food and Drug Administration, also reduces cerebral ischemia and BBB damage successfully [19].
We recently developed a novel PARP-1 inhibitor, JPI-289, which has non-hydroscopic properties and dissolves readily in water [10]. This compound exhibits potent PARP-1 inhibition in in vitro models, with possible neuroprotective function. In the present study, we confirmed the early neuroprotective function of JPI-289 when rats were treated after tMCAO, a model of ischemic stroke. JPI-289 administered 2 h after MCAO reduced infarct volume by 53% 24 h after MCAO. Our studies suggest that, in comparison to PJ-34 and MP-124 (which were given at either 1 or 1.5 h after tMCAO [12, 13]), JPI-289 is more effective in reducing infarct volume. The saturated piperidine ring and morpholine in the JPI-289 are predicted to generate a stronger hydrophobic interaction and hydrogen bonds and lead to greater solubility and PARP-1 inhibitory activity than PJ-34 or MP-124 [10, 31, 32].
In addition to a role in neuroprotection role, PARP-1 inhibitors are known to play a role in the reduction of inflammatory responses and the modulation of adaptive immunity [11, 33]. PARP-1 sustains the expression of cytokines, chemokines, and other inflammatory mediators, such as tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), IL-6, interferon-γ (IFN-γ), inducible nitric oxide synthase (iNOS), nuclear factor-κB (NF-κB), and high mobility group box 1 protein (HMGB1). In addition, PARP-1 increases the expression of several adhesion molecules (intercellular adhesion molecule 1, vascular cell adhesion molecule, P-selectin, E-selectin, caludin-5, zonula occludens-1, and vascular endothelial cadherin), chemoattractant chemokines (IL-8, macrophage inflammatory proteins 1 and 2, monocyte chemoattractant protein 1), and matrix metalloproteinase 9, which are all involved in reperfusion injury and hemorrhagic transformation after ischemic stroke [18, 33]. Thus, PARP-1 inhibition has been studied for its potential to stabilize the BBB after ischemic stroke and has been shown to prevent BBB component degradation and neuroinflammation [16, 18]. In the current study, we found that treatment with JPI-289 reduced brain swelling in tMCAO model and decreased hemorrhagic complication of tPA in embolic stroke model. Although the detailed molecular mechanism was not evaluated in this study, we believe that JPI-289 stabilizes the BBB and reduces neuroinflammation. Another beneficial function of PARP-1 inhibitors is the regulation of the immune system either by modulating the ability of dendritic cells to stimulate T cells or by directly affecting the differentiation and function of T and B cells [33]. As an increase in the number of regulatory T cells exerts neuroprotective effects in acute experimental models of stroke [34], the immune modulatory functions of JPI-289 need to be investigated in future studies.
Many previous experimental studies evaluating neuroprotective agents in acute ischemic stroke have low-quality study design and overestimate efficacy [35], so STAIR (Stroke Therapy Academic Industry Roundtable) made the following recommendations: (1) there should be an adequate dose–response curve; (2) the time window should be defined in a well-characterized model; (3) studies should be blinded, physiologically controlled, and reproducible; (4) histological and functional outcomes should be assessed both acutely and over the long term; (5) studies should be carried out initially in rodents, and then in species with gyrencephalic brains; and (6) consider permanent occlusion and then transient in most cases [36]. To adhere to these recommendations, our study used DWI to randomize the experimental groups, and long-term outcomes were assessed, together with behavioral testing and FLAIR MRI, until 28 days after MCAO. As we reported previously, 1 h after MCAO and the baseline, infarct volume was similar in all groups [21]. We also performed FLAIR MRI at 24 h, 7 days, and 28 days after MCAO to accurately confirm infarct volume and brain swelling and to show the trends in infarct volume and brain swelling. Behavior tests were also performed to the same schedule to demonstrate the long-term beneficial effects of JPI-289 when used to treat the effects of acute ischemic stroke. Although a sample size calculation was not performed in this study, contrary to the STAIR recommendations, we believe that this study was well regulated and minimized biases. Further investigations in gyrencephalic species are needed to consolidate our findings.
There are several potential limitations to this study. First, this is a pilot study with a small sample size, and we did not calculate sample size. Although the STAIR guidelines recommend a sample size calculation before undertaking an animal study, it was not possible for the current study because the differences in infarct size and estimated efficacy of JPI-289 were not known. Second, the experiments described in the current study were only performed in male rats. As responses to PARP-1 inhibitors have been shown to differ according to sex [37, 38], future studies of PARP-1 inhibitors in animal stroke models should use female animals. Third, the molecular mechanisms of JPI-289 action were not examined in the rat brain. Although reduced inflammation and apoptosis were demonstrated in a previous in vitro study, additional experiments are needed to delineate the molecular mechanisms of JPI-289 action on ischemic stroke.
We have demonstrated the positive effects of early PARP-1 inhibition in a rat model of acute ischemic stroke, including decreases in infarct volume, brain swelling, apoptosis, and hemorrhagic transformation. We also found that long-term follow-up with DWI and FLAIR imaging was both feasible and applicable to animal studies, as was the blinding of investigators to experimental design. These findings demonstrate that JPI-289 is a promising therapy for the treatment of ischemic stroke. Specifically, administration of JPI-289 together with tPA could exert more protective function against ischemic damage.
References
Murray CJ, Vos T, Lozano R, Naghavi M, Flaxman AD, Michaud C, Ezzati M, Shibuya K et al (2012) Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380(9859):2197–2223. https://doi.org/10.1016/S0140-6736(12)61689-4
Wardlaw JM, Murray V, Berge E, del Zoppo G, Sandercock P, Lindley RL, Cohen G (2012) Recombinant tissue plasminogen activator for acute ischaemic stroke: an updated systematic review and meta-analysis. Lancet 379(9834):2364–2372. https://doi.org/10.1016/S0140-6736(12)60738-7
Reis C, Akyol O, Ho WM, Araujo C, Huang L, Applegate Ii R, Zhang JH (2017) Phase I and phase II therapies for acute ischemic stroke: an update on currently studied drugs in clinical research. Biomed Res Int 2017(4863079):1–14. https://doi.org/10.1155/2017/4863079
Pena ID, Borlongan C, Shen G, Davis W (2017) Strategies to extend thrombolytic time window for ischemic stroke treatment: an unmet clinical need. J Stroke 19(1):50–60. https://doi.org/10.5853/jos.2016.01515
Savitz SI, Baron JC, Yenari MA, Sanossian N, Fisher M (2017) Reconsidering neuroprotection in the reperfusion era. Stroke 48(12):3413–3419. https://doi.org/10.1161/STROKEAHA.117.017283
Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG (2010) PARP inhibition: PARP1 and beyond. Nat Rev Cancer 10(4):293–301. https://doi.org/10.1038/nrc2812
Strosznajder RP, Czubowicz K, Jesko H, Strosznajder JB (2010) Poly(ADP-ribose) metabolism in brain and its role in ischemia pathology. Mol Neurobiol 41(2-3):187–196. https://doi.org/10.1007/s12035-010-8124-6
Moroni F, Chiarugi A (2009) Post-ischemic brain damage: targeting PARP-1 within the ischemic neurovascular units as a realistic avenue to stroke treatment. FEBS J 276(1):36–45. https://doi.org/10.1111/j.1742-4658.2008.06768.x
Fatokun AA, Dawson VL, Dawson TM (2014) Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol 171(8):2000–2016. https://doi.org/10.1111/bph.12416
Kim Y, Kim YS, Noh MY, Lee H, Joe B, Kim HY, Kim J, Kim SH et al (2017) Neuroprotective effects of a novel poly(ADP-ribose) polymerase-1 inhibitor, JPI-289, in hypoxic rat cortical neurons. Clin Exp Pharmacol Physiol 44(6):671–679. https://doi.org/10.1111/1440-1681.12757
Laudisi F, Sambucci M, Pioli C (2011) Poly (ADP-ribose) polymerase-1 (PARP-1) as immune regulator. Endocr Metab Immune Disord Drug Targets 11(4):326–333. https://doi.org/10.2174/187153011797881184
Egi Y, Matsuura S, Maruyama T, Fujio M, Yuki S, Akira T (2011) Neuroprotective effects of a novel water-soluble poly(ADP-ribose) polymerase-1 inhibitor, MP-124, in in vitro and in vivo models of cerebral ischemia. Brain Res 1389:169–176. https://doi.org/10.1016/j.brainres.2011.03.031
Haddad M, Rhinn H, Bloquel C, Coqueran B, Szabo C, Plotkine M, Scherman D, Margaill I (2006) Anti-inflammatory effects of PJ34, a poly(ADP-ribose) polymerase inhibitor, in transient focal cerebral ischemia in mice. Br J Pharmacol 149(1):23–30. https://doi.org/10.1038/sj.bjp.0706837
Koh SH, Park Y, Song CW, Kim JG, Kim K, Kim J, Kim MH, Lee SR et al (2004) The effect of PARP inhibitor on ischaemic cell death, its related inflammation and survival signals. Eur J Neurosci 20(6):1461–1472. https://doi.org/10.1111/j.1460-9568.2004.03632.x
Moroni F (2008) Poly(ADP-ribose)polymerase 1 (PARP-1) and postischemic brain damage. Curr Opin Pharmacol 8(1):96–103. https://doi.org/10.1016/j.coph.2007.10.005
Rom S, Zuluaga-Ramirez V, Dykstra H, Reichenbach NL, Ramirez SH, Persidsky Y (2015) Poly(ADP-ribose) polymerase-1 inhibition in brain endothelium protects the blood-brain barrier under physiologic and neuroinflammatory conditions. J Cereb Blood Flow Metab 35(1):28–36. https://doi.org/10.1038/jcbfm.2014.167
Takahashi K, Pieper AA, Croul SE, Zhang J, Snyder SH, Greenberg JH (1999) Post-treatment with an inhibitor of poly(ADP-ribose) polymerase attenuates cerebral damage in focal ischemia. Brain Res 829(1-2):46–54. https://doi.org/10.1016/S0006-8993(99)01335-9
Teng F, Beray-Berthat V, Coqueran B, Lesbats C, Kuntz M, Palmier B, Garraud M, Bedfert C et al (2013) Prevention of rt-PA induced blood-brain barrier component degradation by the poly(ADP-ribose)polymerase inhibitor PJ34 after ischemic stroke in mice. Exp Neurol 248:416–428. https://doi.org/10.1016/j.expneurol.2013.07.007
Teng F, Zhu L, Su J, Zhang X, Li N, Nie Z, Jin L (2016) Neuroprotective effects of poly(ADP-ribose)polymerase inhibitor olaparib in transient cerebral ischemia. Neurochem Res 41(7):1516–1526. https://doi.org/10.1007/s11064-016-1864-6
Koh SH, Yoo AR, Chang DI, Hwang SJ, Kim SH (2008) Inhibition of GSK-3 reduces infarct volume and improves neurobehavioral functions. Biochem Biophys Res Commun 371(4):894–899. https://doi.org/10.1016/j.bbrc.2008.05.006
Kim YS, Yoo A, Son JW, Kim HY, Lee YJ, Hwang S, Lee KY, Lee YJ et al (2017) Early activation of phosphatidylinositol 3-kinase after ischemic stroke reduces infarct volume and improves long-term behavior. Mol Neurobiol 54(7):5375–5384. https://doi.org/10.1007/s12035-016-0063-4
Chen TY, Goyagi T, Toung TJ, Kirsch JR, Hurn PD, Koehler RC, Bhardwaj A (2004) Prolonged opportunity for ischemic neuroprotection with selective kappa-opioid receptor agonist in rats. Stroke 35(5):1180–1185. https://doi.org/10.1161/01.STR.0000125011.93188.c6
Zhao X, Liu SJ, Zhang J, Strong R, Aronowski J, Grotta JC (2005) Combining insulin-like growth factor derivatives plus caffeinol produces robust neuroprotection after stroke in rats. Stroke 36(1):129–134. https://doi.org/10.1161/01.STR.0000149624.87661.18
Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M, Chopp M (2001) Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke 32(4):1005–1011. https://doi.org/10.1161/01.STR.32.4.1005
Sughrue ME, Mocco J, Komotar RJ, Mehra A, D’Ambrosio AL, Grobelny BT, Penn DL, Connolly ES Jr (2006) An improved test of neurological dysfunction following transient focal cerebral ischemia in rats. J Neurosci Methods 151(2):83–89. https://doi.org/10.1016/j.jneumeth.2005.04.023
Zhang RL, Chopp M, Zhang ZG, Jiang Q, Ewing JR (1997) A rat model of focal embolic cerebral ischemia. Brain Res 766(1-2):83–92. https://doi.org/10.1016/S0006-8993(97)00580-5
Haddad M, Beray-Berthat V, Coqueran B, Plotkine M, Marchand-Leroux C, Margaill I (2013) Combined therapy with PJ34, a poly(ADP-ribose)polymerase inhibitor, reduces tissue plasminogen activator-induced hemorrhagic transformations in cerebral ischemia in mice. Fundam Clin Pharmacol 27(4):393–401. https://doi.org/10.1111/j.1472-8206.2012.01036.x
Zhang L, Zhang ZG, Zhang RL, Lu M, Adams J, Elliott PJ, Chopp M (2001) Postischemic (6-hour) treatment with recombinant human tissue plasminogen activator and proteasome inhibitor PS-519 reduces infarction in a rat model of embolic focal cerebral ischemia. Stroke 32(12):2926–2931. https://doi.org/10.1161/hs1201.100207
Schreiber V, Dantzer F, Ame JC, de Murcia G (2006) Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 7(7):517–528. https://doi.org/10.1038/nrm1963
Moroni F, Cozzi A, Chiarugi A, Formentini L, Camaioni E, Pellegrini-Giampietro DE, Chen Y, Liang S et al (2012) Long-lasting neuroprotection and neurological improvement in stroke models with new, potent and brain permeable inhibitors of poly(ADP-ribose) polymerase. Br J Pharmacol 165(5):1487–1500. https://doi.org/10.1111/j.1476-5381.2011.01666.x
Patel MR, Bhatt A, Steffen JD, Chergui A, Murai J, Pommier Y, Pascal JM, Trombetta LD et al (2014) Discovery and structure-activity relationship of novel 2,3-dihydrobenzofuran-7-carboxamide and 2,3-dihydrobenzofuran-3(2H)-one-7-carboxamide derivatives as poly(ADP-ribose)polymerase-1 inhibitors. J Med Chem 57(13):5579–5601. https://doi.org/10.1021/jm5002502
Yao H, Ji M, Zhu Z, Zhou J, Cao R, Chen X, Xu B (2015) Discovery of 1-substituted benzyl-quinazoline-2,4(1H,3H)-dione derivatives as novel poly(ADP-ribose)polymerase-1 inhibitors. Bioorg Med Chem 23(4):681–693. https://doi.org/10.1016/j.bmc.2014.12.071
Rosado MM, Bennici E, Novelli F, Pioli C (2013) Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology 139(4):428–437. https://doi.org/10.1111/imm.12099
Chen S, Wu H, Klebe D, Hong Y, Zhang J, Tang J (2013) Regulatory T cell in stroke: a new paradigm for immune regulation. Clin Dev Immunol 2013(689827):1–9. https://doi.org/10.1155/2013/689827
O’Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW (2006) 1,026 experimental treatments in acute stroke. Ann Neurol 59(3):467–477. https://doi.org/10.1002/ana.20741
Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, Lo EH, Group, S (2009) Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke 40(6):2244–2250. https://doi.org/10.1161/STROKEAHA.108.541128
Liu F, Lang J, Li J, Benashski SE, Siegel M, Xu Y, McCullough LD (2011) Sex differences in the response to poly(ADP-ribose) polymerase-1 deletion and caspase inhibition after stroke. Stroke 42(4):1090–1096. https://doi.org/10.1161/STROKEAHA.110.594861
Yuan M, Siegel C, Zeng Z, Li J, Liu F, McCullough LD (2009) Sex differences in the response to activation of the poly (ADP-ribose) polymerase pathway after experimental stroke. Exp Neurol 217(1):210–218. https://doi.org/10.1016/j.expneurol.2009.02.012
Funding
This study was funded by grant of the Korea Drug Development Fund (KDDF-201410-08) and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (A100453) for the clinical development of JPI-289.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
All national and institutional guidelines for the care and use of animals were followed.
Rights and permissions
About this article

Cite this article
Kim, Y., Kim, Y.S., Kim, H.Y. et al. Early Treatment with Poly(ADP-Ribose) Polymerase-1 Inhibitor (JPI-289) Reduces Infarct Volume and Improves Long-Term Behavior in an Animal Model of Ischemic Stroke. Mol Neurobiol 55, 7153–7163 (2018). https://doi.org/10.1007/s12035-018-0910-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-0910-6