
Abstract
Phosphatidylinositol 3-kinases (PI3Ks) have recently been implicated in apoptosis and ischemic cell death. We tested the efficacy of early intervention with a peptide PI3K activator in focal cerebral ischemia. After determining the most effective dose (24 μg/kg) and time window (2 h after MCAO) of treatment, a total of 48 rats were subjected to middle cerebral artery occlusion (MCAO). Diffusion weighted MRI (DWI) was performed 1 h after MCAO and rats with lesion sizes within a predetermined range were randomized to either PI3K activator or vehicle treatment arms. Fluid attenuated inversion recovery (FLAIR) MRI, neurological function, western blots, and immunohistochemistry were blindly assessed. Initial DWI lesion volumes were nearly identical between two groups prior to treatment. However, FLAIR showed significantly smaller infarct volumes in the PI3K activator group compared with vehicle (146 ± 81 mm3 and 211 ± 96 mm3, p = 0.045) at 48 h. The PI3K activator group also had better neurological function for up to 2 weeks. In addition, PI3K activator decreased the number of TUNEL-positive cells in the peri-infarct region compared with the control group. Western blot and immunohistochemistry showed increased expression of phosphorylated Akt (Ser473) and GSK-3β (Ser9) and decreased expression of cleaved caspase-9 and caspase-3. Our results suggest a neuroprotective role of early activation of PI3K in ischemic stroke. The use of DWI in the randomization of experimental groups may reduce bias.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Phosphatidylinositol 3-kinases (PI3Ks) are a family of enzymes characterized by protein and lipid kinase activities. Activation of PI3Ks along with Akt phosphorylation has been shown to contribute to cell growth and cell survival [1, 2]. This signaling pathway is also known to block mitochondrial cytochrome c release and caspase activity, which may inhibit apoptosis [3]. PI3K/Akt activation is an important mechanism in ischemic stroke implicated in the neuroprotective effects of growth factors, hypothermia, and ischemic preconditioning, as well as several neuroprotective agents such as statin and cilostazol [4–12]. However, to the best of our knowledge, the effect of direct activation of PI3K has not been previously explored.
PI3K isoforms can be divided into three classes (I–III) based on structural features and lipid substrate preference [1]. Among these, class I PI3Ks are the best understood and can be divided into a class IA group (p110α, p110β, and p110δ) and a class IB group (p110γ) [1]. Although PI3Ks are generally linked to Akt/PKB activation and neuroprotection during cerebral ischemia [3], subtypes such as PI3Kγ have been shown to be harmful in ischemic stroke models [13, 14]. Here, using a peptide PI3K activator (KKHTDDGYMPMSPGVA), so-called insulin receptor substrate 1 (IRS-1), we tested the efficacy of early PI3K activation in improving tissue and neurological outcome following experimental focal cerebral ischemia. This peptide binds to the insulin and insulin-like growth factor-1 receptors, which is then phosphorylated by tyrosine kinase receptors. This tyrosine-phosphorylated IRS-1 activates the PI3 kinase SH2 domain unique to class IA PI3Ks, and activates PI3Kα, PI3Kβ, and PI3Kδ isoforms [15, 16]. It is well-known that this peptide is more specific to the activation of the PI3K pathway [17–19]. Thus, this peptide is now used as a PI3K activator, while there are some reports suggesting that IRS-1 could activate the mitogen-activated protein (MAP) kinase pathway, as well [20].
In this study, we used a transient middle cerebral artery occlusion (MCAO) model in rats to test the efficacy of early activation of PI3K by IRS1P in improving tissue and neurological outcome after focal cerebral ischemia. To eliminate selection bias, we randomized rats into treatment arms based on diffusion-weighted MRI (DWI) lesion volumes during the hyperacute stage.
Materials and Methods
Animal Preparation and Experimental Protocols
All animal procedures were performed in accordance with the Hanyang University guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee (IACUC) of Hanyang University. Two-month-old male Sprague-Dawley (SD) rats weighing 270–309 g were used (Biogenomics Incorporated, Seoul, Korea). In accordance with the ARRIVE guidelines, all experiments were carried out in a strictly blinded fashion, inclusion and exclusion criteria predetermined, and attrition due to mortality and other causes reported. However, we could not calculate sample sizes a priori because anticipated differences in infarct size and estimated efficacy with PI3K activators were not available.
Filament Middle Cerebral Artery Occlusion
After a 7-day period of adaptation and pre-training for neurological examination, the left middle cerebral arteries (MCA) were occluded for 2 h using the intraluminal filament technique described previously [21–23]. The rats were anesthetized with isoflurane (3 % for induction and 2 % for surgical procedure) in a mixture of oxygen/nitrous oxide (30 %/70 %). Body temperature was maintained at 36.6 ± 0.5 °C with a thermistor-controlled heating pad (Supplementary Table 1). Arterial pH, pCO2, pO2, and hematocrit were measured in 0.1 mL of arterial blood obtained from a right femoral catheter using a blood-analysis system (International Technidyne, NJ, USA) (Supplementary Table 1). Arterial pressure was monitored from the femoral catheter with a strain-gauge transducer (LIFE KIT DX-360; Nihon Kohden, Tokyo, Japan) and amplifier (MacLab Bridge Amplifier, AD Instruments Pty Ltd., Castle Hill, Australia). Phasic pressure, mean arterial pressure (MAP), and heart rate (HR) were recorded at a sampling rate of 200/s, using a data acquisition system and laboratory computer (MacLab 8 analog-to-digital converter and Macintosh computer) (Supplementary Table 1). After 2 h of occlusion, reperfusion was performed as described previously [21–23]. A sham surgery was performed by introducing and then immediately withdrawing a thread into the left common carotid artery. We measured regional cerebral blood flow (rCBF), using a laser Doppler flowmeter (ALF21; Advance, Tokyo, Japan) and a wire-type probe (0.3 mm diameter; Unique Medical, Tokyo, Japan). We inserted them through a small burr hole 2 mm lateral just to the bregma at the surface of the cortex to evaluate the ischemic core in the caudate and putamen (Supplementary Table 1).
Dose-Response and Therapeutic Time Window of Efficacy
The PI3K activator (1732.8 kDa) was purchased from Santa Cruz Biotech (Delaware, CA, USA). Additionally, LY294002 (Sigma, Saint Louis, MO, USA), a specific PI3K inhibitor, was used to directly block PI3K. Prior to experimental use, drugs were dissolved in distilled water and further diluted with normal saline to yield the desired final concentrations. In order to determine the most effective dose, rats were randomly divided into vehicle and 12, 24, and 48 μg/kg PI3K activator groups, administered intravenously at the time of reperfusion. To determine the therapeutic time window, rats were treated with the most effective dose (24 μg/kg) administered at 2, 6, or 24 h after MCAO compared to vehicle (n = 10 each). To confirm PI3K activation, LY294002 (24 μg/kg) was administered simultaneously with PI3K activator (24 μg/kg) at the time of reperfusion. A separate group was treated with LY294002 to control for its direct effects. All rats were sacrificed at 48 h after MCAO and infarct volume was calculated after staining with 2,3,5-triphenyltetrazolium chloride (TTC, Sigma, USA) as described previously [22]. Brain water content (%) was calculated by the wet and dry weight method using 100 × (wet weight−dry weight)/wet weight.
Diffusion-Weighted MRI-Based Randomized Cohort
After determining the optimal dose and therapeutic window, we studied a larger cohort randomized based on diffusion-weighted MRI (DWI, TR/TE = 4455/74 ms, slice thickness/gap = 1/1 mm, matrix = 64 × 63, FOV = 50 × 50 mm; 3-Tesla, Achieva, Philips, Best, The Netherlands) with a four-channel phased array coil (mouse coil, Shanghai Chenguang Medical Technologies Co., Ltd., Shanghai, China). All rats underwent DWI 1 h after MCAO and DWI lesion volumes were measured with MIPAV software (Medical Image Processing, Analysis, and Visualization; National Institutes of Health, Bethesda, Maryland, USA). The rats were randomized blindly to receive either PI3K activator (24 μg/kg) or vehicle (saline, 1 mL). Because rats with DWI lesions larger than 200 mm3 showed high mortality within 48 h, 15 rats were excluded in the final analysis. All rats included in the treatment groups were subjected to fluid attenuated inversion recovery (FLAIR) MRI (TR/TE/TI = 11,000/77/2800 ms, slice thickness/gap = 1/1 mm; matrix = 128 × 128, FOV = 50 × 50 mm) 48 h after MCAO. The final infarct volume was independently assessed with MIPAV by a neurologist and a radiologist (YSK and YJL) blinded to the experimental groups and was calculated from the averages of the two measurements.
Acute Neurological Outcome
A modified neurological severity score (mNSS) was performed before MCAO, just before reperfusion (2 h after MCAO), and just before sacrifice (48 h after MCAO) by an investigator who was blinded to the experimental groups. The distribution of rats to either the PI3K or control groups was based on pre-reperfusion mNSS with MRI data analysis. mNSS is based on consciousness (0, normal; 1, restless; 2, lethargic; 3, stuporous), gait (0, normal; 1, paw adduction; 2, unbalanced walking; 3, circling; 4, unable to stand; 5, no movement), and limb tone (0, normal; 1, spastic; 2, flaccid). Neurological function was graded on a scale of 0 to 10 (normal score, 0; maximal deficit score, 10) [24].
Long-Term Neurological Outcome
We studied long-term neurological outcome in a separate cohort with DWI lesion volumes of 80–120 mm3 to optimize the balance between deficits and survival. Sensorimotor function was assessed using beam walking, rotarod, and modified sticky-tape tests performed before MCAO and 1, 3, 7 and 14 days after MCAO by an investigator blinded to the experimental groups. For beam walking test, rats were trained 7 days before MCAO to walk on a wooden beam (2.5 × 2.5 × 80 cm) 60 cm above the floor to get back to their home cage. The test was scored as: 0, traverses the beam with no foot slip; 1, traverses by grasping the lateral side of the beam; 2, shows disability walking on the beam but can traverse; 3, takes a considerable amount of time to traverse the beam because of difficulty walking; 4, unable to traverse the beam; 5, unable to move the body or any limb on the beam; 6, the rat is unable to stay on the beam for 10 s [25]. For the rotarod test, rats were placed on an accelerating rotarod cylinder and the riding time that the animal remained on the rotarod was measured. The speed was slowly increased from 4 to 40 rpm within 5 min. A trial ended if the animal fell off the rungs or gripped to the device and spun around for two consecutive revolutions without attempting to walk on the rungs. The animals were trained 7 days before MCAO. Motor test data are presented as mean duration (3 trials) on the rotarod [26]. For the modified sticky-tape test, a sleeve was created using a 3-cm piece of green paper tape, 1 cm in width and was wrapped around the forepaw so that the tape attached to itself and that the fingers protrude slightly from the sleeve formed. The typical response was for the rat to vigorously attempt to remove the sleeve by either pulling at the tape with its mouth and/or brushing the tape with its contralateral paw. After attaching the sleeve, the rat is placed in its cage and observed for 30s. The timer was only turned on while the animal attempted to remove the taped sleeve. The data collected represents the fraction of the 30-s observation period that the animal spent attending to the stimulus. The contralateral and ipsilateral limbs were tested separately. The test was repeated three times per test day and the best two scores were averaged [27]. Motor test data are presented as mean duration (seconds) and right/left ratio.
Histology and Immunohistochemistry
All rats used for the evaluation of infarction volume with MRI were sacrificed at 48 h for histological and immunohistochemical analysis. Rats were transcardially perfused with phosphate-buffered saline (PBS), brains were quickly removed, and cooled in ice-cold artificial cerebrospinal fluid for 5 min. Subsequently, 2-mm-thick slices of brain matrix were prepared by sectioning in the coronal plane at approximately −0.4 mm from the bregma point on the skull, and were fixed overnight in 4 % paraformaldehyde solution and then cryoprotected in a 30 % sucrose solution for 3 days. The remaining brain tissues were stored at −80 °C for Western blotting. Each brain was mounted on a cold metal block using optimal cutting temperature compound (Leica, Wetzlar, Germany), and coronal brain sections (20 μm in thickness) were cut on a motorized cryostat (Leica, Wetzlar, Germany) at −20 °C. Three sections were placed on one “Muto” glass slide by opposing the slide onto the section as it came off the cryostat blade. Slides were stored at −80 °C and processed for hematoxylin and eosin (H&E) and terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) staining. Based on H&E staining, we were able to distinguish the peri-infarct region, and we counted TUNEL-positive cells in both PI3K and control groups. Immunohistochemical staining was performed as previously described [22, 23], using antibodies for pAkt (Ser473) (1:500; Cell Signaling, Beverly, MA, USA), cleaved caspase-3 (1:500, Cell Signaling, Beverly, MA, USA), anti-neuronal nucleus (NeuN) (1:100, Santa Cruz Biotech, CA, USA), and appropriate secondary antibodies conjugated to tetramethylrhodamine isothiocyanate (TRITC) (Invitrogen, CA, USA) or fluorescein isothiocyanate (FITC) (Invitrogen, CA, USA). Immunohistochemically positive cells were identified by fluorescence microscopy (Olympus, PA, USA). The number of positive cells in the peri-infarct area of the brain from the vehicle control group and in the corresponding area from the PI3K activator-treated group was expressed as the average number of positive cells per high power field (×200). As a negative control, the above procedures were repeated without a primary antibody, which showed no stained cells.
Western Blotting
Frozen brain tissues of two rats in the sham group, three in the vehicle group, and six in the PI3K group were rapidly micro-dissected on an ice-chilled plate. The peri-infarct region in the control group and the corresponding area in the PI3K activator group, which were confirmed based on the MRI finding, were used in Western blot analysis. The micro-dissected tissues were homogenized using T 10 basic homogenizer (IKA laboratory, Wilmington, NC, USA) and a type B pestle in 10:1 volume/weight buffer containing 10 mM Tris (pH 7.4), 10 mM EGTA, 250 mM sucrose, 2 μg/mL aprotinin, 5 μg/mL leuperptin, 2 μg/mL pepstatin, and 1 mM phenylmethylsulphonyl fluoride. The protein concentration of the tissue lysate was determined using a Bio-Rad protein assay kit. An equal amount (20 μg) of protein was resolved by 10 % sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Amersham Pharmacia Biotech, Piscataway, NJ, USA). The membranes were blocked with 5 % skim milk and sequentially incubated with the same antibodies for Akt (1:2000, Cell Signaling, Beverly, MA, USA), phosphorylated Akt (ser473) (1:500, Cell Signaling), glycogen synthase kinase (GSK)-3β (1:2000, Santa Cruz Biotech, Dallas, TX, USA), phosphorylated GSK-3β (Ser9) (1:1000, Cell Signaling), caspase-9 (1:2000, Cell Signaling), cleaved caspase-9 (1:500, Cell Signaling), caspase-3 (1:2000, Cell Signaling), and cleaved caspase-3 (1:2000, Cell Signaling). The membranes were washed with Tris-buffered saline containing 0.05 % Tween-20 (TBST) and processed using horseradish peroxidase-conjugated anti-rabbit antibody (Jackson ImmunoResearch Laboratories INC, West Grove, PA, USA), followed by ECL detection (GenDEPOT, Katy, TX, USA). The results of Western blots were quantified with image analyzer (Bio-Rad, Quantity One-4,2,0, Hercules, CA, USA).
Statistical Analysis
Data are presented as mean ± SEM from five or more independent experiments. Normality was tested by Kolmogorov-Smirnov test. Statistical comparisons between two groups were performed using the t test. Analyses of three or more data sets were performed using one-way ANOVA followed by Tukey’s post hoc comparisons. Two-tailed p values less than 0.05 were considered statistically significant. All statistical analyses were performed using the SPSS 17.0 software package for Windows (SPSS, Seoul, Korea).
Results
Reduction of Infarct Volume and Water Content by PI3K Activator
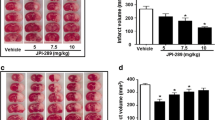
Administration of PI3K activator at reperfusion 2 h after MCAO attenuated infarct volumes at 48 h. The most effective dose of the PI3K activator was 24 μg/kg (Fig. 1a) and this dose was used in all subsequent experiments. When administered 6 or 24 h after MCAO, PI3K activator treatment was ineffective (Fig. 1b). The water content of the ischemic hemisphere was significantly lower in the PI3K activator treated group (62 ± 2 %) compared to that of vehicle control group (71 ± 8 %, p = 0.039) (Fig. 1c). Just before reperfusion, the mean mNSS was 4 in both the vehicle control group and the PI3K activator treated group. At 48 h post-MCAO, the mean mNSS was 3.6 in the vehicle control group and 3.3 in the PI3K activator-treated group (p = 0.188). Simultaneous treatment with LY294002 (24 μg/kg), PI3 kinase inhibitor, abolished the protective effect of PI3K activator (24 μg/kg) treated group on cerebral infarction confirming selective targeting of PI3K by the peptide and LY294002 treatment alone did not alter the infarct volume (Fig. 1d).

Dose and therapeutic window screening. TTC staining shows cerebral infarction in several groups (a). Administration of PI3K activator had beneficial effects on infarct volume reduction and the most effective dose was 24 μg/kg (n = 10/group) (b). Treatment with PI3K activator was only effective when administered at reperfusion (n = 10/group) (c). The water content of the ischemic brain hemisphere was significantly reduced with PI3K activator (n = 10/group) (d). Simultaneous treatment with PI3K inhibitor (LY294002) almost completely blocked the protective effect of PI3K activator and LY294002 itself did not showed toxic effect (n = 10/group) (e). The data are presented as mean ± SEM. *p < 0.05 and **p < 0.01 (vs. control group in b, c and e; vs. contralateral in d); # p < 0.05 (vs. ipsilateral hemisphere of the control group)
Efficacy of PI3K Activator after Randomization Based on DWI Lesion Volume
The number of rats randomized into either PI3K activator-treated group or vehicle control groups based on DWI lesion volumes 1 h after MCAO is shown in Fig. 2a. Based on initial DWI at 1 h after MCAO, 33 rats with less than 200 mm3 on DWI were included in the final analysis (PI3K activator-treated group, n = 17; vehicle control group n = 16) (Fig. 2a). FLAIR MRI 48 h after MCAO showed significantly smaller infarct volume in the PI3K activator-treated group compared to that of the vehicle control group (Fig. 2c; p = 0.044). Neurologically, PI3K activator-treated group showed consistently better long-term outcomes than the vehicle control group (Fig. 3).

MRI-based randomization study flow and the effect of early activation of PI3K on infarct volume. A study flow chart is shown in (a). Rats were classified into either control or PI3K group based on DWI infarct volume which were smaller than 200mm3 (a). Infarct volume measurement with MIPAV is demonstrated (b). Although baseline infarct volume based on DWI was not different between two groups, final infarct volume at 24 h of MCAO on FLAIR MRI was significantly reduced in PI3K group (c). The data are presented as mean ± SEM. *p < 0.05 (vs. control group)

The effect of early activation of PI3K on behavioral function tests. Behavioral function tests (beam walking, rotarod, modified sticky-tape) before and after MCAO showed beneficial effects of early PI3K activation on motor function of rats (a–d). The data are presented as mean ± SEM. *p < 0.05 and **p < 0.01 (vs. control group)
Effect of PI3K Activator on Expression TUNEL-Positive Cells
Following FLAIR MRI, rats were sacrificed and brain tissues were sectioned and analyzed for TUNEL staining. The number of TUNEL-positive cells in the ischemic peri-infarct region of PI3K activator-treated group was less than that of the vehicle control group (51 ± 10 cells/HPF in the PI3K activator-treated group and 78 ± 12 cells/HPF in the vehicle control group) (p = 0.016; Fig. 4a). When TUNEL-positive cells were analyzed by cell type, PI3K activator treated-group was consistently associated with fewer TUNEL-positive cells (Fig. 4b).

TUNEL staining and TUNEL cell counting. Microscopic observation of TUNEL staining shows reduced TUNEL-positive cells in the PI3K group (a). Absolute counts of TUNEL-positive cells were significantly reduced in the PI3K group regardless of cell type (n = 10/group) (b). The data are presented as means ± SEM from five independent experiments. *p < 0.05 (vs. control group)
Effect of PI3K on Intracellular Signals
To investigate the mechanisms underlying PI3K-induced neuroprotection following ischemic stroke, the immunoreactivities of phosphorylated Akt (pAkt at Ser473), phosphorylated GSK-3β (pGSK-3β at Ser9), cleaved caspase-9 and cleaved caspase-3 were measured in the peri-infarct region. Immunoreactivity for survival-related proteins pAkt and pGSK-3β were significantly increased in the PI3K activator-treated group compared to that of vehicle control group. In addition, cleaved caspase-9 and cleaved caspase-3, two pro-apoptotic proteins, were relatively decreased after PI3K activation (Fig. 5). Immunohistochemistry confirmed increased pAkt at Ser473 and decreased cleaved caspase-3 after PI3K activation (Fig. 6).

The effect of PI3K on intracellular signaling proteins in brains injured by ischemic stroke. Administration of PI3K activator (24 μg/kg) increased pAkt (ser473) and pGSK-3β (Ser9) (a, b) and decreased cleaved caspase-9 and cleaved caspase-3 (c, d) compared with the control group. The data are means (% of control) ± SEM from five independent experiments. *p < 0.05 (vs. sham group) and # p < 0.05 (vs. control group)

Immunohistochemical staining. Immunohistochemical staining shows increased pAkt (Ser473) (b) and decreased cleaved caspase-3 (d) in PI3K group when compared with the control group (a and c, respectively)
Discussion
The present study demonstrates that early activation of PI3K during hyperacute stroke reduces infarct volume and ischemic edema and restores long-term neurological function. This protective effect is completely blocked by simultaneous treatment with the PI3K inhibitor LY294002. PI3K activator stimulated neuroprotective pathways, as shown by increased phosphorylation of Akt at Ser473 and GSK-3β at Ser9, and suppressed cleavage of pro-apoptotic caspase-9 and caspase-3.
Following ischemic brain damage, it is believed that pAkt increases within a few hours and begins to decrease after 24 h [28, 29]. However, using an in vitro oxygen-glucose deprivation model, we previously showed that pAkt at Ser473 significantly decreases for around 2 h after ischemic insult before increasing over time [30]. This transient decrease in pAkt was associated with ischemic neuronal cell death. However, it remains unclear if the increase in pAkt represents an effort by the dying cells to rescue themselves after stroke, or if it is simply a marker of cell damage [3]. It has been reported that activation of PI3K with various compounds reduces ischemic damage and decreases pAkt activation [4–12]. Elevated pAkt inactivates downstream effectors such as forkhead in rhabodomyosarcoma (FKHR), GSK-3β, and Bad, resulting in cell survival [3]. It also decreases the activation of caspases, which are mediators of apoptotic cell death [31, 32]. It is well established that activation of PI3K is associated with the prevention of neuronal cell death; however, there have not yet been studies investigating the effects of direct and early activation of the PI3K pathway in ischemic stroke models. It is possible that previously described compounds that activate PI3K may also have other neuroprotective mechanisms and that the direct activation of PI3K may allow its unique role in neuroprotection in ischemic stroke to be elucidated.
In previous experimental neuroprotection studies in acute ischemic stroke, study quality and publication bias had substantial effects on the estimates of drug efficacy [33]. Poor study quality score based on STAIR I criteria was associated with overestimated efficacy [34], attributable in part to lack of randomization and blinding [34]. Moreover, a meta-analysis of 13 putative neuroprotectant experiments revealed that the presence or absence of randomization to a treatment group, blinding of drug assignment during stroke induction, and blinding of outcome assessments were among the most powerful determinants of outcome [34]. We therefore believe that randomization objectively stratified based on DWI lesion volume 1 h after MCAO minimized selection bias in our study, an important obstacle in successful clinical translation [33, 35, 36]. Indeed, the efficacy of PI3K activator appeared higher in our initial non-stratified dose-finding cohort than the DWI-stratified cohort. Exclusion of animals with a very large acute infarct (>200 mm3) was inevitable because they likely had smaller volumes of penumbra (i.e., viable tissue at risk) and carried higher mortality diminishing the predictive value of the stroke model.
There are some limitations in the present study. First of all, there is no data about pharmacokinetics and pharmacodynamics of IRS-1 after the intravenous injection, so we cannot estimate how much portion of the injected IRS-1 can be reached to the brain. Second, we cannot explain whether IRS-1 can enter the brain across the blood-brain barrier (BBB). Since there are many reports showing that endogenous IRS-1 exists in the brain and it plays important roles in cell survival [37], we believe that intravenously injected IRS-1 could enter the brain especially when the BBB breaks down due to ischemic stroke. Third, it is apparent that no single animal model can directly translate to ischemic stroke in humans. Thus, future studies of stroke models using different animals are warranted. Finally, molecular experiments were not performed early after the reperfusion in the present study. Because our experiments were mainly aimed to confirm the effect of the early activation of PI3K on cerebral infarction based on FLAIR MRI undertaken at 24 h of transient MCAO, our molecular experiments were performed at 48 h of transient MCAO. However, considering the previous finding that pAkt is increased after 4 h of reperfusion [28] and our in vitro result that pAkt was increased after 4 h of hypoxia [30], it seems that the exact mechanisms of PI3K activation in the early phase of cerebral infarction need to be more clearly demonstrated in the further experiments.
In summary, we demonstrated improved tissue and neurological outcome with early selective PI3K activation in an acute ischemic stroke model. Stratified and blinded DWI-based randomization improved our confidence in this finding. We believe acute stage MRI-based randomization is an important quality control that should be implemented whenever possible, its higher cost and effort notwithstanding, in order to improve the predictive value of preclinical translational studies.
References
Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B (2010) The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol 11(5):329–341
Koh SH, Lo EH (2015) The role of the PI3K pathway in the regeneration of the damaged brain by neural stem cells after cerebral infarction. J Clin Neurol 11(4):297–304
Zhao H, Sapolsky RM, Steinberg GK (2006) Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol Neurobiol 34(3):249–270
Jin G, Omori N, Li F, Nagano I, Manabe Y, Shoji M, Abe K (2003) Protection against ischemic brain damage by GDNF affecting cell survival and death signals. Neurol Res 25(3):249–253
Zhu C, Wang X, Xu F, Qiu L, Cheng X, Simbruner G, Blomgren K (2006) Intraischemic mild hypothermia prevents neuronal cell death and tissue loss after neonatal cerebral hypoxia-ischemia. Eur J Neurosci 23(2):387–393
Garcia L, Burda J, Hrehorovska M, Burda R, Martin ME, Salinas M (2004) Ischaemic preconditioning in the rat brain: effect on the activity of several initiation factors, Akt and extracellular signal-regulated protein kinase phosphorylation, and GRP78 and GADD34 expression. J Neurochem 88(1):136–147
Shehadah A, Chen J, Zacharek A, Cui Y, Ion M, Roberts C, Kapke A, Chopp M (2010) Niaspan treatment induces neuroprotection after stroke. Neurobiol Dis 40(1):277–283
Zhang L, Zhang ZG, Liu XS, Hozeska-Solgot A, Chopp M (2007) The PI3K/Akt pathway mediates the neuroprotective effect of atorvastatin in extending thrombolytic therapy after embolic stroke in the rat. Arterioscler Thromb Vasc Biol 27(11):2470–2475
Lee JH, Kim KY, Lee YK, Park SY, Kim CD, Lee WS, Rhim BY, Hong KW (2004) Cilostazol prevents focal cerebral ischemic injury by enhancing casein kinase 2 phosphorylation and suppression of phosphatase and tensin homolog deleted from chromosome 10 phosphorylation in rats. J Pharmacol Exp Ther 308(3):896–903
Yoshimoto T, Kanakaraj P, Ying Ma J, Cheng M, Kerr I, Malaiyandi L, Watson JA, Siesjo BK, et al. (2002) NXY-059 maintains Akt activation and inhibits release of cytochrome C after focal cerebral ischemia. Brain Res 947(2):191–198
Abe E, Fujiki M, Nagai Y, Shiqi K, Kubo T, Ishii K, Abe T, Kobayashi H (2010) The phosphatidylinositol-3 kinase/Akt pathway mediates geranylgeranylacetone-induced neuroprotection against cerebral infarction in rats. Brain Res 1330:151–157
Rau TF, Kothiwal A, Zhang L, Ulatowski S, Jacobson S, Brooks DM, Cardozo-Pelaez F, Chopp M, et al. (2011) Low dose methamphetamine mediates neuroprotection through a PI3K-AKT pathway. Neuropharmacology 61(4):677–686
Jin R, Song Z, Yu S, Piazza A, Nanda A, Penninger JM, Granger DN, Li G (2011) Phosphatidylinositol-3-kinase gamma plays a central role in blood-brain barrier dysfunction in acute experimental stroke. Stroke 42(7):2033–2044
Jin R, Yu S, Song Z, Quillin JW, Deasis DP, Penninger JM, Nanda A, Granger DN, et al. (2010) Phosphoinositide 3-kinase-gamma expression is upregulated in brain microglia and contributes to ischemia-induced microglial activation in acute experimental stroke. Biochem Biophys Res Commun 399(3):458–464
Garcia P, Shoelson SE, George ST, Hinds DA, Goldberg AR, Miller WT (1993) Phosphorylation of synthetic peptides containing Tyr-Met-X-Met motifs by nonreceptor tyrosine kinases in vitro. J Biol Chem 268(33):25146–25151
Rordorf-Nikolic T, Van Horn DJ, Chen D, White MF, Backer JM (1995) Regulation of phosphatidylinositol 3′-kinase by tyrosyl phosphoproteins. Full activation requires occupancy of both SH2 domains in the 85-kDa regulatory subunit. J Biol Chem 270(8):3662–3666
Bruning JC, Winnay J, Cheatham B, Kahn CR (1997) Differential signaling by insulin receptor substrate 1 (IRS-1) and IRS-2 in IRS-1-deficient cells. Mol Cell Biol 17(3):1513–1521
Amoui M, Craddock BP, Miller WT (2001) Differential phosphorylation of IRS-1 by insulin and insulin-like growth factor I receptors in Chinese hamster ovary cells. J Endocrinol 171(1):153–162
Myers MG Jr, Sun XJ, Cheatham B, Jachna BR, Glasheen EM, Backer JM, White MF (1993) IRS-1 is a common element in insulin and insulin-like growth factor-I signaling to the phosphatidylinositol 3′-kinase. Endocrinology 132(4):1421–1430
Liang L, Jiang J, Frank SJ (2000) Insulin receptor substrate-1-mediated enhancement of growth hormone-induced mitogen-activated protein kinase activation. Endocrinology 141(9):3328–3336
Koh SH, Chang DI, Kim HT, Kim J, Kim MH, Kim KS, Bae I, Kim H, et al. (2005) Effect of 3-aminobenzamide, PARP inhibitor, on matrix metalloproteinase-9 level in plasma and brain of ischemic stroke model. Toxicology 214(1–2):131–139
Koh SH, Park Y, Song CW, Kim JG, Kim K, Kim J, Kim MH, Lee SR, et al. (2004) The effect of PARP inhibitor on ischaemic cell death, its related inflammation and survival signals. Eur J Neurosci 20(6):1461–1472
Koh SH, Yoo AR, Chang DI, Hwang SJ, Kim SH (2008) Inhibition of GSK-3 reduces infarct volume and improves neurobehavioral functions. Biochem Biophys Res Commun 371(4):894–899
Koh SH, Kim KS, Choi MR, Jung KH, Park KS, Chai YG, Roh W, Hwang SJ, et al. (2008) Implantation of human umbilical cord-derived mesenchymal stem cells as a neuroprotective therapy for ischemic stroke in rats. Brain Res 1229:233–248
Urakawa S, Hida H, Masuda T, Misumi S, Kim TS, Nishino H (2007) Environmental enrichment brings a beneficial effect on beam walking and enhances the migration of doublecortin-positive cells following striatal lesions in rats. Neuroscience 144(3):920–933
Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M, Chopp M (2001) Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke 32(4):1005–1011
Sughrue ME, Mocco J, Komotar RJ, Mehra A, D’Ambrosio AL, Grobelny BT, Penn DL, Connolly ES Jr (2006) An improved test of neurological dysfunction following transient focal cerebral ischemia in rats. J Neurosci Methods 151(2):83–89
Noshita N, Lewen A, Sugawara T, Chan PH (2001) Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab 21(12):1442–1450
Kitagawa H, Warita H, Sasaki C, Zhang WR, Sakai K, Shiro Y, Mitsumoto Y, Mori T, et al. (1999) Immunoreactive Akt, PI3-K and ERK protein kinase expression in ischemic rat brain. Neurosci Lett 274(1):45–48
Noh MY, Kim YS, Lee KY, Lee YJ, Kim SH, HJ Y, Koh SH (2013) The early activation of PI3K strongly enhances the resistance of cortical neurons to hypoxic injury via the activation of downstream targets of the PI3K pathway and the normalization of the levels of PARP activity, ATP, and NAD(+). Mol Neurobiol 47(2):757–769
Manabat C, Han BH, Wendland M, Derugin N, Fox CK, Choi J, Holtzman DM, Ferriero DM, et al. (2003) Reperfusion differentially induces caspase-3 activation in ischemic core and penumbra after stroke in immature brain. Stroke 34(1):207–213
Le DA, Wu Y, Huang Z, Matsushita K, Plesnila N, Augustinack JC, Hyman BT, Yuan J, et al. (2002) Caspase activation and neuroprotection in caspase-3-deficient mice after in vivo cerebral ischemia and in vitro oxygen glucose deprivation. Proc Natl Acad Sci U S A 99(23):15188–15193
Sena E, van der Worp HB, Howells D, Macleod M (2007) How can we improve the pre-clinical development of drugs for stroke? Trends Neurosci 30(9):433–439
O’Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW (2006) 1,026 experimental treatments in acute stroke. Ann Neurol 59(3):467–477
Macleod MR, van der Worp HB, Sena ES, Howells DW, Dirnagl U, Donnan GA (2008) Evidence for the efficacy of NXY-059 in experimental focal cerebral ischaemia is confounded by study quality. Stroke 39(10):2824–2829
Savitz SI (2007) A critical appraisal of the NXY-059 neuroprotection studies for acute stroke: a need for more rigorous testing of neuroprotective agents in animal models of stroke. Exp Neurol 205(1):20–25
YF T, Jiang ST, Chow YH, Huang CC, Ho CJ, Chou YP (2016) Insulin receptor substrate-1 activation mediated p53 downregulation protects against hypoxic-ischemia in the neonatal brain. Mol Neurobiol 53(6):3658–3669
Acknowledgments
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2015R1A2A2A04004865) and by a grant from the NanoBio R&D Program of the Korea Science and Engineering Foundation, funded by the Ministry of Education, Science and Technology (2006-2004670).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Young Seo Kim, Arum Yoo, and Jeong Woo Son contributed equally to this work.
Electronic supplementary material
Supplementary Table 1
(DOC 36 kb)
Rights and permissions
About this article

Cite this article
Kim, Y.S., Yoo, A., Son, J.W. et al. Early Activation of Phosphatidylinositol 3-Kinase after Ischemic Stroke Reduces Infarct Volume and Improves Long-Term Behavior. Mol Neurobiol 54, 5375–5384 (2017). https://doi.org/10.1007/s12035-016-0063-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-0063-4