
Abstract
Intracerebral hemorrhage (ICH) is a subtype of stroke with high mortality and morbidity. When a diseased artery within the brain bursts, expansion and absorption of the resulting hematoma trigger a series of reactions that cause primary and secondary brain injury. Microglia are extremely important for removing the hematoma and clearing debris, but they are also a source of ongoing inflammation. This article discusses the role of microglial activation/polarization and related inflammatory mediators, such as Toll-like receptor 4, matrix metalloproteinases, high-mobility group protein box-1, nuclear factor erythroid 2-related factor 2, heme oxygenase, and iron, in secondary injury after ICH and highlights the potential targets for ICH treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Intracerebral hemorrhage (ICH) accounts for 15–20 % of all strokes and is associated with high morbidity and mortality [1]. During ICH, rapid accumulation of blood within the surrounding brain leads to primary injury [2]. This primary damage occurs within minutes to hours from the onset of the event, causes disruption of normal anatomy, and leads to high pressure in the local brain tissue [2]. Secondary damage is, for the most part, due to the presence and clearance of hematoma [1–3]. Inflammatory cells, including blood-derived leukocytes, microglia/macrophages, astrocytes, and mast cells, are vital for clearing the hematoma but can also augment the brain damage caused by ICH [3–5]. Microglia are considered to be the earliest inflammatory cells to react to ICH. Increasing evidence suggests that microglia are the major cell type responsible for secondary damage after ICH owing to their release of cytokines, chemokines, prostaglandins, proteases, ferrous iron, and other immunoactive molecules [6–9]. In this review, we will discuss the role of microglial activation/polarization and summarize recent progress in the study of inflammatory mediators, such as Toll-like receptor 4, matrix metalloproteinases, high-mobility group protein box-1, nuclear factor erythroid 2-related factor 2 (Nrf2), heme oxygenase (HO), and iron, which are released by microglia during the secondary injury phase after ICH.
Microglia
Microglial Activation After ICH
Microglia are the critical immune cells in the central nervous system (CNS) and are often referred to as the brain’s macrophage. They are involved in brain development, maintenance of the neural environment, and response to injury and repair [10–12]. Microglia can be activated by various types of brain damage and undergo phenotype and functional transformations to maintain tissue homeostasis. Specifically, activated microglia become enlarged, with rod-like, spherical, or amoeboid shapes, and phagocytic [13]. Because both microglia and macrophages originate from primitive myeloid progenitors, they express the same markers (CD11b, F4/80, Iba-1) [14]. For this reason, it is difficult to distinguish activated microglia from activated macrophages by immunohistochemistry. Therefore, most researchers customarily identify activated microglia as microglia/macrophage. However, studies with flow cytometry reported that microglia and macrophages can be separated based on the expression of CD45lowCD11b+ in microglia [15–17].
Microglia are the first immune cells to react to brain injury and can be activated by blood components, including red cells, heme, leukocytes, and plasma proteins. The activation of microglia is thought to have a dual role in ICH. The main role of activated microglia is to phagocytose the hematoma and cell debris, thereby maintaining tissue homeostasis, and to promote neurologic functional recovery. However, in this process, the activated microglia can also produce a variety of deleterious cytokines, such as proinflammatory factors, chemokines, reactive oxygen species (ROS), proteases, nitric oxide (NO) synthase, and prostaglandins, which increase brain damage after ICH [6, 18–23].
Knowledge of microglial activation during ICH comes mainly from preclinical animal models. Though many different animal models of ICH have been established [24], the most frequently used are the autologous blood-induced and collagenase-induced models in rodents [25]. Both models have limitations in their ability to reflect the exact clinical features of ICH, but they are currently the most suitable tools to study the mechanism of ICH [26]. The advantage of the autologous blood model is that only blood is injected into the striatum. This model is appropriate for studying mechanisms of neuronal damage induced by blood and blood catabolism. We modified this method by injecting the blood in two portions to prevent the blood from flowing back along the needle track [27]. However, this model does not mimic brain damage caused by hematoma growth, which occurs clinically in approximately 73 % of patients [28]. The second commonly used model, the collagenase injection model, is better able to mimic acute cerebrovascular injury and hematoma growth. However, the bleeding results from the breakdown of cerebral blood vessels rather than the rupture of a small, deep-penetrating artery, as is seen in patients [26, 29, 30]. Moreover, whether the collagenase can directly induce inflammation is still disputed [26, 29, 31]. Because the two models reflect different pathomechanisms of ICH, we use both to study the mechanisms and roles of genes and proteins and to evaluate the effectiveness of drugs [9, 21, 32].
Activated microglia can be detected within 1 h after collagenase-induced ICH, even earlier than the appearance of neutrophils, which are seen within 4–5 h after collagenase injection [33]. As in the collagenase model, activated microglia can be found in the perihematoma within 1–4 h in the autologous blood model [34]. In both models, the peak microglial activation is within 3–7 days [29, 35]. The number of microglia decreases after 7 days and returns to normal at 21 days [29], although some studies show that microglial activation will persist for 4 weeks [26, 36]. The time course of microglial activation may determine their different roles in the process of ICH. In the early stage of ICH, activated microglia have been shown to produce proinflammatory factors and the numbers of microglia in the ipsilateral hemisphere correlate with neurologic functional damage [37]. Additionally, inhibiting microglial activation can decrease brain injury and edema [38]. Compounds that regulate microglial activation, such as sinomenine [20], minocycline [39], microglia/macrophage inhibitory factors (tuftsin fragment 1–3) [29, 40], misoprostol [21], Chinese medicine (curcumin [41], sesamin [42]), and iron chelators deferoxamine [43, 44] and 2,2′-dipyridyl [45], reduce ICH-induced brain injury and improve neurologic function in rodents. In addition, cell therapies, including mesenchymal stem cells [46] and regulatory T cells [47], attenuate ICH-induced brain injury, which correlates with inhibition of microglial activation. However, the underlying signaling pathways involved in microglia-regulated inflammatory responses after ICH remain elusive.
Microglial Polarization
With appropriate stimulation, both microglia and macrophages can be activated to two polarization states: the M1, classically activated phenotype and the M2, alternatively activated phenotype. Stimulation with lipopolysaccharide or interferon gamma [48] causes microglia to be activated to the M1 phenotype and to produce proinflammatory mediators (interleukin (IL)-1β, IL-6, IL-12, IL-23, tumor necrosis factor alpha (TNF-α)), chemokines, redox molecules (NADPH oxidase, phagocyte oxidase, inducible NO synthase), costimulatory proteins (CD40), and major histocompatibility complex II (MHC-II) [49–53]. Microglia can also be activated by IL4/IL3 to the M2 polarization state, which produces anti-inflammatory mediators (IL-10, transforming growth factor beta (TGFβ), and glucocorticoids) [54, 55]. M2 microglia are also interpreted to be nerve repair cells, as they secrete anti-inflammatory factors and upregulate neuroprotective factors in CNS disease [56]. Because M1 and M2 microglia secrete different factors, several markers have been used to distinguish the two phenotypes. MHC-II, CD16, CD32, CD80 (B7-1), CD86 (B7-2), and CD40 (TNFR) are commonly used as markers for M1 microglia [16, 57–64]. The alternatively activated M2 cells also present several specific antigens, including Ym-1 (chitinase 3-like 3), CD206 (mannose receptor), CD68, and arginase-1 [16, 65].
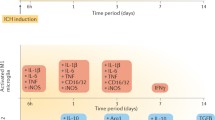
Although the complexity of the microglial phenotype shift after injury requires additional research to decipher, it appears that that one phenotype can convert to the other. After permanent middle cerebral artery occlusion in mice, M2 markers CD206 and Ym-1 were found exclusively in the ischemic core at 24 h, but M1 markers CD16, CD32, and CD86 increased over time beginning on day 3 and outnumbered the M2 cells during the second week [66]. These results reveal a dynamic change of M2 to M1 phenotype over time in the ischemic brain. The early recruitment of M2 microglia/macrophages may be an endogenous response to clear toxic waste products and protect the ischemic brain. But, this change is transient. In time, the M1 microglia/macrophages become dominant in the injury area and exacerbate brain damage by promoting an inflammatory response. This change from an M2 to an M1 phenotype has also been reported in models of traumatic brain injury [67] and spinal cord injury [68]. Moreover, induction of microglia/macrophage M2 polarization is considered to be beneficial for the damaged brain. For example, IL-4 administration can promote M2 polarization and reduce ischemic lesion volume [69] and lipopolysaccharide preconditioning improves spinal cord injury by facilitating M2 activation [70]. Although promoting microglia/macrophage M2 polarization could be beneficial for brain recovery, the phenotypic change of microglia during ICH has not been confirmed. Owing to their phenotypic duality, it is thought that microglia may also have both proinflammatory and neuroprotective roles after ICH (Fig. 1).

After intracerebral hemorrhage (ICH), microglia can be activated/polarized to two phenotypes, M1 and M2. The M1 phenotype can produce proinflammatory mediators (interleukin (IL)-1β, IL-6, tumor necrosis factor alpha (TNF-α)), chemokines, redox molecules (NADPH oxidase (NOX), phagocyte oxidase (PHOX), inducible nitric oxide synthase (iNOS)), and heme oxygenase-1 (HO-1). These factors lead to neuroinflammation, iron accumulation, and reactive oxygen species (ROS) production and finally cause brain damage. The M2 microglia can produce anti-inflammatory mediators (IL-10, transforming growth factor beta (TGFβ)) and promote hematoma clearance through phagocytosis and promotion of angiogenesis. The Nrf2 signaling pathway may be involved in this process. Because they can be activated to either an M1 or M2 phenotype, microglia may have both proinflammatory and anti-inflammatory roles after ICH
Inflammatory Response Mediated by Microglia After ICH
Recently, Sansing et al. [71] showed that activated microglia express high levels of Toll-like receptor (TLR) 4, which leads to neuroinflammation after ICH. TLRs belong to a pattern-recognition receptor family and play an important role in innate immunity and inflammatory responses [72, 73]. To date, ten functional TLRs have been found in humans and 13 in mice. TLRs can recognize distinct pathogen-associated molecular patterns from viruses, bacteria, mycobacteria, fungi, and parasites. TLR4 in particular contributes to inflammatory injury in CNS pathologies such as cerebral ischemia and ICH [74–76]. Treatment with the TLR4 inhibitor ethyl (6R)-6-[N-(2-chloro-4-fluorophenyl) sulfamoyl]-cyclohex-1- ene-1-carboxylate (TAK-242) or genetic deletion of TLR4 in mice decreases inflammation-mediated brain damage and improves neurologic function after ICH [71, 77–80]. Recent clinical studies also have shown that increased expression of TLR4 is associated with poorer functional outcome and greater residual volume in ICH patients [81]. Compared with wild-type (WT) mice, TLR4 knockout (−/−) mice exhibited less water content and fewer neurologic deficits [71]. What is more, infiltrating inflammatory cells (macrophages, leukocytes, and monocytes) and inflammatory cytokines (TNF-α, IL-1β, and IL-6) were decreased around the perihematomal region in TLR4−/− mice [71, 77, 79].
TLRs bind various ligands through the leucine-rich repeats in their extracellular ectodomains and then recruit intracellular adaptor proteins, including myeloid differentiation factor 88 (MyD88), Toll-interleukin 1 receptor (TIR) domain-containing adaptor-inducing interferons (TRIFs), TIR domain-containing adaptor protein (TIRAP), and TRIF-related adaptor molecule (TRAM), by their intracellular TIR domains [78]. These adaptors then upregulate transcription factors such as NF-κB to promote microglial secretion of IL-6, TNF-α, and IL-1β [72, 78]. Studies have shown that MyD88 and TRIF are the main signaling pathways of TLR4-induced inflammatory response [78]. Both MyD88 and TRIF signals participate in the regulation of inflammatory factors after ICH. MyD88 or TRIF gene deficiency causes a decrease in the secretion of inflammatory factors and infiltration of microglia/macrophages after ICH [77]. The expression of MyD88 and TRIF is also decreased after ICH in TLR4−/− mice [77]. In short, the activation of microglia may mediate an inflammatory response in the early stage of ICH through TLR4 and its downstream signaling. A deeper understanding of the TLR4 signaling pathways should enable development of potential therapeutic targets for prevention and treatment of ICH.
Matrix metalloproteinases (MMPs) are a large family of ubiquitous zinc-dependent endopeptidases. Currently, 24 MMP genes and 23 MMP proteins have been identified in humans, with two identical genes on chromosome 1 that encode MMP-23 [82]. Under normal conditions, the MMP expression is low but it can be activated in response to various brain diseases [82, 83]. Studies of MMPs in ICH have focused mainly on MMP-2 and MMP-9, which increase in mouse brain within 2–3 days after ICH [84, 85]. The elevations in expression or activation of MMP-2 and MMP-9 correlate highly with brain edema, inflammation, and blood–brain barrier disruption, and these effects can be reduced by MMP inhibition [21, 84, 85]. In acute stroke, activation of MMP-2 initially disrupts the extracellular matrix proteins in the basal lamina and then attacks the tight junction proteins. If the hypoxic stress does not continue, or MMP is not further activated, the basal lamina integrity can be restored [83]. If damage continues, MMP-9 can be activated by neuroinflammatory cytokines (TNF-α and IL-1β) and free radicals. MMP-9 then degrades the basal lamina and tight junctions of endothelia cells, causing BBB disruption and vasogenic edema [83, 84]. Although the exact relation between MMP-9 and microglia is still unclear, microglia are one of the major sources of MMP-9. The cytokines (TNF-α and IL-1β) and free radicals secreted by microglia can also activate MMP-9, which mediates microglia-induced brain damage after ICH.
High-mobility group protein box-1 (HMGB1) is a highly conserved nonhistone DNA-binding protein that can be actively released into the cytoplasm and is involved in many inflammatory diseases [86–88]. Recent studies have shown a correlation between HMGB1 and acute ICH. In one study, HMGB1 was released from the nucleus into the cytoplasm in the ipsilateral brain as early as 1 h after ICH induction [89]. Moreover, ethyl pyruvate, which inhibits HMGB1 expression, significantly ameliorated ICH-induced neuroinflammation [89]. Similar effects were observed in ICH animals treated with glycyrrhizin (another nonspecific HMGB1 inhibitor) [86]. These findings suggest that HMGB1 can promote neuroinflammation after ICH. In the brain, many cells can express HMGB1, including microglia. In vitro, TNF-α stimulated cultured microglia to release large amounts of HMGB1 [90]. Recombinant human HMGB1 (rhHMGB1) also activated microglia, as demonstrated by increased NF-κB activity, increased production of NO, and upregulated transcription of cyclooxygenase (COX)-2, TNF-α, and IL-1β [91]. However, these effects were lost in TLR4−/− microglia treated with rhHMGB1 [91]. These observations suggest that HMGB1 can trigger microglial activation through the TLR4 receptor. Thus, microglial activation may be an important switch for HMGB1-mediated neuroinflammation after ICH. A recent study also has reported that long-term inhibition of HMGB1 reduced the recovery of neurologic function, decreased vascular endothelial growth factor and nerve growth factor levels in the ipsilateral striatum, and decreased the number of 5-bromo-2-deoxyuridine (BrdU)- and doublecortin-positive cells around the hematoma [92]. These results indicate that HMGB1 may promote angiogenesis and neurogenesis in the late phase of ICH. Thus, HMGB1-mediated neuroinflammation may be involved in angiogenesis and neurogenesis, suggesting a new therapeutic target for treating ICH.
Neuroprotection by Microglia After ICH
Although cumulative data suggest that inhibiting microglial activation could be beneficial to patients with ICH, long-term inhibition may be harmful because potentially neuroprotective functions of microglia, such as phagocytosis, could be inhibited [40]. The activation of microglia can promote hematoma and cell debris absorption and induce neurogenesis and angiogenesis. Several receptors on the surface of microglia are related to phagocytosis [93]. CD36, a class II scavenger receptor of microglia/macrophages, has been reported to bind to phosphatidylserine, phosphatidylinositol, modified lipids, thrombospondin on sickle red blood cells, symmetric red cell ghosts, and apoptotic neutrophils to mediate phagocytosis. In vitro, transfection of CD36 into nonphagocytic cells confers the capacity for phagocytosis [94]. Inducing the expression of CD36 by peroxisome proliferator-activated receptor-γ (PPARγ) promotes hematoma absorption in the autologous blood injection model of ICH in mice [34]. Furthermore, a clinical study revealed that the absorption of hematoma is slower in CD36−/− ICH patients than in patients with normal CD36 expression [78]. Though the underlying mechanism by which CD36 promotes hematoma absorption needs additional investigation, promoting CD36 expression may represent a potential therapeutic option for ICH.
A recent study also has suggested that inflammatory factors can inhibit the expression of CD36. In TLR4−/− mice, CD36 expression is upregulated in perihematomal tissues after ICH [95]. Studies also have shown that inflammatory factors that are induced by TLR signaling, such as TNF-α, can regulate the expression of CD36 in macrophages [95, 96]. CD36 expression and hematoma absorption were significantly increased in TLR4−/− and MyD88−/− mice. What is more, the inflammatory factors TNF-α and IL-1β were induced by activation of microglia, and TLR4 signaling inhibited CD36 expression and hematoma absorption [78, 81]. These results suggest that TLR4 can regulate CD36-mediated hematoma absorption after ICH. Therefore, in addition to blocking the acute detrimental effects of microglial activation, stimulating microglial phagocytosis might offer therapeutic potential by enhancing recovery.
In addition to phagocytosis, microglia can promote neurogenesis and angiogenesis. Conditioned medium from microglia promotes neural precursor cell proliferation, migration, and differentiation [97]. When stimulated with IL-4, microglia promote insulin-like growth factor-1 expression, which causes neural precursor cell neurogenesis in a coculture system [55]. Some other factors secreted by microglia, such as protease serine 2, brain-derived neurotrophic factor, and glial cell line-derived neurotrophic factor, can also promote neurogenesis [98]. Furthermore, microglia upregulate endothelial ephrin-A3 and ephrin-A4 expression to facilitate in vitro angiogenesis of brain endothelial cells [99]. These studies provide evidence that microglia may promote neurogenesis and angiogenesis, which are beneficial for brain function recovery.
Microglia-Correlated Inflammatory Mediators
Nrf2 is a transcriptional factor that regulates transcription of genes involved in antioxidative pathways, NADPH regeneration, and heme and iron metabolism [100]. It is regarded as a brain protector that can decrease microglial activation and oxidative injury after ICH [32, 101]. HO, which is downstream of Nrf2 signaling, can be activated by the heme released when hemoglobin is degraded. The HO isoforms (HO-1 and HO-2) then degrade heme to produce biliverdin, carbon monoxide, and iron. Increasing evidence suggests that accumulation of iron in brain can increase microglial activation and the production of free radicals that cause injury [26].
Nrf2
Recent genome-wide analysis showed that Nrf2 regulates hundreds of genes that are involved in the cytoprotective response against oxidative stress [102]. In normal, unstressed cells, Nrf2 is degraded by kelch-like ECH-associated protein 1 (Keap1). Keap1 can bind to the evolutionarily conserved N-terminal Neh2 regulatory domain of Nrf2 to suppress its transcriptional activity [102]. However, when cells experience oxidative stress, the cysteine residues in Keap1 are oxidized, leading to a conformational change that releases the suppression of Nrf2 activation [103]. In the brain, Nrf2 is expressed mainly in astrocytes, neurons, and microglia. It is thought to protect brain tissue from damage [104]. Accumulating evidence indicates that Nrf2 protects neurons from oxidative stress injuries after ischemic stroke in vivo and in vitro [105–107]. Compared with WT mice, Nrf2−/− mice suffered more brain damage in a cerebral ischemia model [107, 108]. In recent preclinical studies, sulforaphane [109], ursolic acid [110], erythropoietin [111], and (−)-epigallocatechin gallate [112] were shown to protect against brain damage after ischemic stroke by activating the Nrf2 pathway. However, Nrf2 has not been as well studied in ICH as it has been in ischemic stroke. We were the first to show that Nrf2 has a neuroprotective effect after the collagenase-induced ICH model [113]. Our study showed that ICH-induced brain damage is exacerbated in Nrf2−/− mice because of increased leukocyte infiltration and microglial activation (Fig. 2). This protective role of Nrf2 was confirmed by Zhao et al. [114] in an autologous blood model of ICH. A time course study showed that Nrf2 was significantly increased at 2 h and reached a peak at 24 h after ICH [115]. Treatment with Nrf2-inducer sulforaphane decreased the neutrophil count, oxidative damage, and behavioral deficits in WT mice but not in Nrf2−/− mice [114]. Additionally, (−)-epicatechin was shown to reduce early brain damage via synergistic Nrf2 pathways in collagenase and autologous blood models of ICH [32]. Although accumulating data suggest that Nrf2 has a neuroprotective role after ICH, the temporal and spatial distribution of Nrf2 is still unclear. Shang et al. [115] reported that Nrf2 is expressed chiefly in neuronal cells but not in glial cells. However, their assessment of Nrf2 distribution was limited to 2 h after ICH, whereas the peak of Nrf2 expression is at 24 h. Nevertheless, the activation of Nrf2 in microglia can induce antioxidative defense components, reduce peroxide formation, and upregulate CD36 expression to enhance red blood cell phagocytosis in vitro [116]. The Nrf2 inducer sulforaphane can also induce CD36 expression to increase hematoma clearance in WT mice, but not Nrf2−/− mice, subjected to the autologous blood injection model of ICH [116]. Together, these data suggest that Nrf2 can regulate functional changes in microglia after ICH.

Deletion of Nrf2 increases leukocyte infiltration but does not affect microglial activation in mice subjected to intracerebral hemorrhage (ICH). a–d Infiltrating neutrophils (MPO-positive cells; scale bar 40 μm) and activated microglia (Iba1-positive cells; scale bar 20 μm) were apparent in or around the injury site in Nrf2−/− and wild-type (WT) mice 24 h post-ICH. e Quantification analysis indicated that Nrf2−/− mice had significantly more infiltrating neutrophils than did WT mice at 24 h post-ICH; the number of activated microglia around the injury site was similar in Nrf2−/− and WT mice (both n = 3/group, *p < 0.05) [113]
HO
After ICH, hemoglobin and its degradation products are highly toxic to brain tissue [77, 116]. Experimental evidence suggests that free heme liberated mainly from hemoglobin degradation contributes to the production of ROS and inflammation [117]. Because the extracellular free heme cannot be recycled in the brain, heme metabolism is critical to recovery after ICH. HO is the rate-limiting enzyme in the metabolism of heme to equimolar iron, carbon monoxide, and biliverdin, which is then converted to bilirubin by biliverdin reductase [118]. Two HO isoenzymes are present in mammalian cells: HO-1, the inducible isoform, and HO-2, the constitutive isoform. In the CNS, HO-1, also known as heat-shock protein 32 (HSP-32) [119], is expressed at a low level but can be rapidly induced by hemin [33], oxidative stress [120], heat stress [121], proinflammatory cytokines such as TNF-α and IL-1α [122], and anti-inflammatory cytokines such as IL-10 [123]. After ICH, HO-1 is induced mainly in microglia, whereas HO-2 is constitutively expressed in neuronal cells [26]. Though both HO-1 and HO-2 catalyze the same chemical reaction, their expression in different cell types suggests that they may have distinct roles during ICH.
The Effect of HO-2 After ICH
The unavailability of HO inhibitors has necessitated the use of genetically modified mice in mechanistic studies. HO-2−/− mice have been used to study the effect of HO-2 after ICH in autologous blood and collagenase models. However, the results in these two models differed. In the collagenase induced-ICH model, HO-2 deletion increased brain swelling, neuronal death, ROS, inflammation, injury volume, and neurologic deficits compared with those in WT mice [124, 125]. Moreover, HO-2 deletion did not appear to alter HO-1 expression from that in WT mice [124]. These data suggest that HO-2 may play a neuroprotective role in the collagenase-induced ICH model.
Whereas data from the collagenase-induced model suggested that HO-2 had a protective effect after ICH, data from the autologous blood induced-ICH model suggested a deleterious effect. Compared with outcomes in WT mice, HO-2 knockout attenuated perihematomal neuron loss at 4 and 8 days after blood injection and had a weak and variable effect on neurologic outcome [126]. Additionally, HO-2−/− mice exhibited a reduction in oxidative cell injury [127]. However, in this model, the expression of HO-1, which is thought to increase brain damage [26, 33], was significantly lower in the HO-2−/− mice than in the WT mice [127]. These distinct results may be caused by the different injury mechanisms in the collagenase and autologous blood injection models of ICH as well as the differences in HO-1 expression.
The Effect of HO-1 After ICH
Unlike the constitutive expression of HO-2, which accounts for most HO activity under normal conditions, HO-1 is expressed at a low level in normal brain and can be rapidly induced after ICH. Immunofluorescence shows that HO-1 is expressed in vascular-like structures [33] in the normal mouse brain (Fig. 3). HO-1 protein increases significantly after ICH and reaches a peak on day 3 [128]. We reported that HO-1 deficiency in mice reduced ICH-induced leucocyte infiltration, microglial/macrophage activation, and free radical levels compared with those in WT mice, but that brain water content did not change significantly [33]. These findings suggested that HO-1 increases brain damage in the early stage of collagenase-induced ICH in mice. No reports have described the use of HO-1−/− mice in the autologous blood-induced ICH model. Porphyrin HO inhibitors also have been used to study the role of HO-1 after ICH. These studies have consistently shown that porphyrin HO inhibitors are neuroprotective in the blood-injection ICH model [129, 130]. These findings suggest that HO-1 mediates brain injury after ICH and raise the possibility that HO-1 may be a therapeutic target for early-stage treatment of ICH.

Cellular localization of heme oxygenase-1 (HO-1) in normal mouse brain. a In normal mouse brain, HO-1 immunoreactivity was observed in vascular-like structures. b–d HO-1 immunoreactivity (in green) was colocalized with CD31-positive cells (in red, an endothelial marker). Scale bar 50 μm
HO-1 expression correlates significantly with the antioxidant copper–zinc superoxide dismutase (Cu/Zn-SOD) on days 1 to 3 after ICH, but no definitive correlation has been identified on days 7 and 14. Conversely, HO-1 correlates significantly with the prooxidant malondialdehyde on days 7 and 14 after ICH but not on days 1 to 3 [128]. Recent studies also have shown that hemin-induced HO-1 expression in perivascular cells before ICH can attenuate blood–brain barrier disruption after ICH [131]. These data suggest that HO-1 may have both antioxidative and oxidative roles in the pathophysiology of ICH (Fig. 4).

In the early stage of intracerebral hemorrhage (ICH), activated microglia express high levels of heme oxygenase-1 (HO-1). The elevated HO-1 may affect cell function, promote iron accumulation, and increase inflammation. These changes result in high reactive oxygen species (ROS) production and an increase in brain damage (blood–brain barrier (BBB) disruption, brain edema, white matter injury, cell death, and neurologic deficits). In the late stage of ICH, high HO-1 expression in microglia and vascular endothelial cells may contribute to hematoma absorption and angiogenesis, thereby promoting neurologic recovery
Iron
Iron, one of the heme (hemin) degradation products, can accumulate in the brain parenchyma for months after ICH and is very damaging [44, 132–135]. In the brain, microglia regulate iron homeostasis by sequestering and storing it within ferritin [136]. However, overaccumulation of iron can influence the physiologic properties of microglia [136, 137]. When exposed to lipopolysaccharide, iron-loaded primary microglial cells promote secretion of proinflammatory cytokines, such as TNF-α and IL-1β [136]. Furthermore, HO-1, the key enzyme for heme degradation, is mainly expressed in microglia after ICH. The high expression of HO-1 can also exacerbate brain damage by promoting microglial activation and iron deposition [32, 33].
Another potential mechanism by which iron causes brain damage is the generation of free radicals. In the Fenton/Haber-Weiss reaction, iron reacts with lipid hydroperoxides to produce free radicals that attack DNA and cause oxidative brain injury [135, 138, 139]. The excess iron in brain tissue after ICH can cause brain edema, neuronal death, brain atrophy, and poor neurologic outcomes [45, 135, 140]. Chelators can bind “free” iron to form a stable complex and prevent the iron from entering into the Fenton/Haber–Weiss reaction. Therefore, removing excess iron with iron chelators is a common practice. Many studies, including our own, have shown that deferoxamine, a ferric iron chelator, decreases microglial activation, oxidative stress, brain edema, and neuronal death and improves functional outcome after ICH [43, 44, 141–143]. However, others claim that although deferoxamine can reduce iron content in the brain, it cannot attenuate injury or improve neurologic function [144–146]. In one recent study, ferrous iron chelator (clioquinol), but not ferric iron chelator (deferiprone), improved brain outcomes after ICH [147]. Ferric iron chelator is more likely to facilitate the Fenton reaction process and increase free radical production. Though the use of iron chelators to reduce brain injury needs additional investigation before it can be applied clinically, decreasing iron accumulation/toxicity in the brain remains an important goal for treatment of patients with ICH. Deferoxamine is now under phase II clinical trial for treating ICH patients [148].
Conclusion
The variable effects of microglial activation/polarization in models relevant to ICH suggest that they are challenging therapeutic targets. In the early stage of ICH, microglia can be activated by blood components and tend to acquire the M1 phenotype. M1 microglia express high levels of TLR4 and HO-1 to clear the hematoma, but they also increase inflammation, iron content, and free radical production, which exacerbate brain injury in the early stage after ICH. With hematoma resolution, the inflammatory reaction decreases and microglia change to an M2 phenotype, which promotes hematoma absorption and neurologic functional recovery. Inhibition of M1 microglial activation at the early stage of ICH and promotion of a shift from M1 to M2 phenotype in the late stage could be a potential therapeutic strategy for ICH treatment.
References
Qureshi AI, Mendelow AD, Hanley DF (2009) Intracerebral haemorrhage. Lancet 373(9675):1632–1644. doi:10.1016/s0140-6736(09)60371-8
Aronowski J, Zhao X (2011) Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke; a journal of cerebral circulation 42(6):1781–1786. doi:10.1161/STROKEAHA.110.596718
Wu H, Zhang Z, Li Y, Zhao R, Li H, Song Y, Qi J, Wang J (2010) Time course of upregulation of inflammatory mediators in the hemorrhagic brain in rats: correlation with brain edema. Neurochemistry international 57(3):248–253. doi:10.1016/j.neuint.2010.06.002
Donnan GA, Hankey GJ, Davis SM (2010) Intracerebral haemorrhage: a need for more data and new research directions. Lancet neurology 9(2):133–134. doi:10.1016/S1474-4422(10)70001-6
Wu H, Zhang Z, Hu X, Zhao R, Song Y, Ban X, Qi J, Wang J (2010) Dynamic changes of inflammatory markers in brain after hemorrhagic stroke in humans: a postmortem study. Brain research 1342:111–117. doi:10.1016/j.brainres.2010.04.033
Wang J, Dore S (2007) Inflammation after intracerebral hemorrhage. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 27(5):894–908. doi:10.1038/sj.jcbfm.9600403
Mracsko E, Veltkamp R (2014) Neuroinflammation after intracerebral hemorrhage. Frontiers in cellular neuroscience 8:388. doi:10.3389/fncel.2014.00388
Mohan S, Ahmad AS, Glushakov AV, Chambers C, Dore S (2012) Putative role of prostaglandin receptor in intracerebral hemorrhage. Frontiers in neurology 3:145. doi:10.3389/fneur.2012.00145
Zhao X, Wu T, Chang CF, Wu H, Han X, Li Q, Gao Y, Li Q et al (2015) Toxic role of prostaglandin E2 receptor EP1 after intracerebral hemorrhage in mice. Brain, behavior, and immunity 46:293–310. doi:10.1016/j.bbi.2015.02.011
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML et al (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nature neuroscience 8(6):752–758. doi:10.1038/nn1472
Kim SU, de Vellis J (2005) Microglia in health and disease. Journal of neuroscience research 81(3):302–313. doi:10.1002/jnr.20562
Ransohoff RM, Cardona AE (2010) The myeloid cells of the central nervous system parenchyma. Nature 468(7321):253–262. doi:10.1038/nature09615
Wang J, Tsirka SE (2005) Contribution of extracellular proteolysis and microglia to intracerebral hemorrhage. Neurocritical care 3(1):77–85. doi:10.1385/NCC:3:1:077
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ et al (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330(6005):841–845. doi:10.1126/science.1194637
Campanella M, Sciorati C, Tarozzo G, Beltramo M (2002) Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke; a journal of cerebral circulation 33(2):586–592
Patel AR, Ritzel R, McCullough LD, Liu F (2013) Microglia and ischemic stroke: a double-edged sword. International journal of physiology, pathophysiology and pharmacology 5(2):73–90
Mracsko E, Javidi E, Na SY, Kahn A, Liesz A, Veltkamp R (2014) Leukocyte invasion of the brain after experimental intracerebral hemorrhage in mice. Stroke; a journal of cerebral circulation 45(7):2107–2114. doi:10.1161/STROKEAHA.114.005801
Shiratori M, Tozaki-Saitoh H, Yoshitake M, Tsuda M, Inoue K (2010) P2X7 receptor activation induces CXCL2 production in microglia through NFAT and PKC/MAPK pathways. Journal of neurochemistry 114(3):810–819. doi:10.1111/j.1471-4159.2010.06809.x
Matsushita H, Hijioka M, Ishibashi H, Anan J, Kurauchi Y, Hisatsune A, Seki T, Shudo K et al (2014) Suppression of CXCL2 upregulation underlies the therapeutic effect of the retinoid Am80 on intracerebral hemorrhage in mice. Journal of neuroscience research 92(8):1024–1034. doi:10.1002/jnr.23379
Yang Z, Liu Y, Yuan F, Li Z, Huang S, Shen H, Yuan B (2014) Sinomenine inhibits microglia activation and attenuates brain injury in intracerebral hemorrhage. Molecular immunology 60(2):109–114. doi:10.1016/j.molimm.2014.03.005
Wu H, Wu T, Hua W, Dong X, Gao Y, Zhao X, Chen W, Cao W et al (2015) PGE2 receptor agonist misoprostol protects brain against intracerebral hemorrhage in mice. Neurobiology of aging 36(3):1439–1450. doi:10.1016/j.neurobiolaging.2014.12.029
Wu T, Wu H, Wang J, Wang J (2011) Expression and cellular localization of cyclooxygenases and prostaglandin E synthases in the hemorrhagic brain. Journal of neuroinflammation 8:22. doi:10.1186/1742-2094-8-22
Han X, Lan X, Li Q, Gao Y, Zhu W, Cheng T, Maruyama T, Wang J (2015) Inhibition of prostaglandin E2 receptor EP3 mitigates thrombin-induced brain injury. Journal of cerebral blood flow and metabolism. (in press). doi: 10.1177/0271678X15606462
James ML, Warner DS, Laskowitz DT (2008) Preclinical models of intracerebral hemorrhage: a translational perspective. Neurocritical care 9(1):139–152. doi:10.1007/s12028-007-9030-2
MacLellan CL, Silasi G, Auriat AM, Colbourne F (2010) Rodent models of intracerebral hemorrhage. Stroke; a journal of cerebral circulation 41(10 Suppl):S95–98. doi:10.1161/strokeaha.110.594457
Wang J (2010) Preclinical and clinical research on inflammation after intracerebral hemorrhage. Progress in neurobiology 92(4):463–477. doi:10.1016/j.pneurobio.2010.08.001
Wang J, Fields J, Dore S (2008) The development of an improved preclinical mouse model of intracerebral hemorrhage using double infusion of autologous whole blood. Brain research 1222:214–221. doi:10.1016/j.brainres.2008.05.058
Davis SM, Broderick J, Hennerici M, Brun NC, Diringer MN, Mayer SA, Begtrup K, Steiner T (2006) Hematoma growth is a determinant of mortality and poor outcome after intracerebral hemorrhage. Neurology 66(8):1175–1181. doi:10.1212/01.wnl.0000208408.98482.99
Wang J, Rogove AD, Tsirka AE, Tsirka SE (2003) Protective role of tuftsin fragment 1–3 in an animal model of intracerebral hemorrhage. Annals of neurology 54(5):655–664. doi:10.1002/ana.10750
Tang J, Liu J, Zhou C, Alexander JS, Nanda A, Granger DN, Zhang JH (2004) Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutant mice. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 24(10):1133–1145. doi:10.1097/01.WCB.0000135593.05952.DE
Chu K, Jeong SW, Jung KH, Han SY, Lee ST, Kim M, Roh JK (2004) Celecoxib induces functional recovery after intracerebral hemorrhage with reduction of brain edema and perihematomal cell death. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 24(8):926–933. doi:10.1097/01.WCB.0000130866.25040.7D
Chang CF, Cho S, Wang J (2014) (−)-Epicatechin protects hemorrhagic brain via synergistic Nrf2 pathways. Annals of clinical and translational neurology 1(4):258–271. doi:10.1002/acn3.54
Wang J, Dore S (2007) Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain : a journal of neurology 130(Pt 6):1643–1652. doi:10.1093/brain/awm095
Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, Grotta JC, Aronowski J (2007) Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Annals of neurology 61(4):352–362. doi:10.1002/ana.21097
Gong C, Hoff JT, Keep RF (2000) Acute inflammatory reaction following experimental intracerebral hemorrhage in rat. Brain research 871(1):57–65
Yabluchanskiy A, Sawle P, Homer-Vanniasinkam S, Green CJ, Motterlini R (2010) Relationship between leukocyte kinetics and behavioral tests changes in the inflammatory process of hemorrhagic stroke recovery. The International journal of neuroscience 120(12):765–773. doi:10.3109/00207454.2010.523129
Wu J, Yang S, Xi G, Song S, Fu G, Keep RF, Hua Y (2008) Microglial activation and brain injury after intracerebral hemorrhage. Acta neurochirurgica Supplement 105:59–65
Gao Z, Wang J, Thiex R, Rogove AD, Heppner FL, Tsirka SE (2008) Microglial activation and intracerebral hemorrhage. Acta neurochirurgica Supplement 105:51–53
Zhao F, Hua Y, He Y, Keep RF, Xi G (2011) Minocycline-induced attenuation of iron overload and brain injury after experimental intracerebral hemorrhage. Stroke; a journal of cerebral circulation 42(12):3587–3593. doi:10.1161/strokeaha.111.623926
Wang J, Tsirka SE (2005) Tuftsin fragment 1–3 is beneficial when delivered after the induction of intracerebral hemorrhage. Stroke; a journal of cerebral circulation 36(3):613–618. doi:10.1161/01.STR.0000155729.12931.8f
Yang Z, Zhao T, Zou Y, Zhang JH, Feng H (2014) Curcumin inhibits microglia inflammation and confers neuroprotection in intracerebral hemorrhage. Immunology letters 160(1):89–95. doi:10.1016/j.imlet.2014.03.005
Ohnishi M, Monda A, Takemoto R, Matsuoka Y, Kitamura C, Ohashi K, Shibuya H, Inoue A (2013) Sesamin suppresses activation of microglia and p44/42 MAPK pathway, which confers neuroprotection in rat intracerebral hemorrhage. Neuroscience 232:45–52. doi:10.1016/j.neuroscience.2012.11.057
Miao X, Liu X, Yue Q, Qiu N, Huang W, Wang J, Xu Y, Zhang Y et al (2012) Deferoxamine suppresses microglia activation and protects against secondary neural injury after intracerebral hemorrhage in rats. Nan fang yi ke da xue xue bao = Journal of Southern Medical University 32(7):970–975
Wu H, Wu T, Xu X, Wang J, Wang J (2011) Iron toxicity in mice with collagenase-induced intracerebral hemorrhage. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 31(5):1243–1250. doi:10.1038/jcbfm.2010.209
Wu H, Wu T, Li M, Wang J (2012) Efficacy of the lipid-soluble iron chelator 2,2′-dipyridyl against hemorrhagic brain injury. Neurobiology of disease 45(1):388–394. doi:10.1016/j.nbd.2011.08.028
Bao XJ, Liu FY, Lu S, Han Q, Feng M, Wei JJ, Li GL, Zhao RC et al (2013) Transplantation of Flk-1+ human bone marrow-derived mesenchymal stem cells promotes behavioral recovery and anti-inflammatory and angiogenesis effects in an intracerebral hemorrhage rat model. International journal of molecular medicine 31(5):1087–1096. doi:10.3892/ijmm.2013.1290
Yang Z, Yu A, Liu Y, Shen H, Lin C, Lin L, Wang S, Yuan B (2014) Regulatory T cells inhibit microglia activation and protect against inflammatory injury in intracerebral hemorrhage. International immunopharmacology 22(2):522–525. doi:10.1016/j.intimp.2014.06.037
Martinez FO, Gordon S (2014) The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime reports 6:13. doi:10.12703/P6-13
Ponomarev ED, Veremeyko T, Weiner HL (2013) MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia 61(1):91–103. doi:10.1002/glia.22363
Hanisch UK, Kettenmann H (2007) Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nature neuroscience 10(11):1387–1394. doi:10.1038/nn1997
Henkel JS, Beers DR, Zhao W, Appel SH (2009) Microglia in ALS: the good, the bad, and the resting. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology 4(4):389–398. doi:10.1007/s11481-009-9171-5
Varnum MM, Ikezu T (2012) The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Archivum immunologiae et therapiae experimentalis 60(4):251–266. doi:10.1007/s00005-012-0181-2
Boche D, Perry VH, Nicoll JA (2013) Review: activation patterns of microglia and their identification in the human brain. Neuropathology and applied neurobiology 39(1):3–18. doi:10.1111/nan.12011
Orihuela R, McPherson CA, Harry GJ (2015) Microglial M1/M2 polarization and metabolic states. British journal of pharmacology. doi:10.1111/bph.13139
Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, Martino G, Schwartz M (2006) Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Molecular and cellular neurosciences 31(1):149–160. doi:10.1016/j.mcn.2005.10.006
Pan J, Jin JL, Ge HM, Yin KL, Chen X, Han LJ, Chen Y, Qian L et al (2015) Malibatol A regulates microglia M1/M2 polarization in experimental stroke in a PPARgamma-dependent manner. Journal of neuroinflammation 12:51. doi:10.1186/s12974-015-0270-3
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308(5726):1314–1318. doi:10.1126/science.1110647
Ponomarev ED, Shriver LP, Maresz K, Dittel BN (2005) Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. Journal of neuroscience research 81(3):374–389. doi:10.1002/jnr.20488
Dawson DA, Martin D, Hallenbeck JM (1996) Inhibition of tumor necrosis factor-alpha reduces focal cerebral ischemic injury in the spontaneously hypertensive rat. Neuroscience letters 218(1):41–44
Lambertsen KL, Biber K, Finsen B (2012) Inflammatory cytokines in experimental and human stroke. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 32(9):1677–1698. doi:10.1038/jcbfm.2012.88
Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U (2002) Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. The Journal of neuroscience : the official journal of the Society for Neuroscience 22(7):RC216, doi:20026253
Iadecola C, Zhang F, Xu S, Casey R, Ross ME (1995) Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 15(3):378–384. doi:10.1038/jcbfm.1995.47
Loihl AK, Asensio V, Campbell IL, Murphy S (1999) Expression of nitric oxide synthase (NOS)-2 following permanent focal ischemia and the role of nitric oxide in infarct generation in male, female and NOS-2-deficient mice. Brain research 830(1):155–164
Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME (1997) Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. The Journal of neuroscience : the official journal of the Society for Neuroscience 17(23):9157–9164
Perego C, Fumagalli S, De Simoni MG (2011) Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. Journal of neuroinflammation 8:174. doi:10.1186/1742-2094-8-174
Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J (2012) Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke; a journal of cerebral circulation 43(11):3063–3070. doi:10.1161/STROKEAHA.112.659656
Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X, Liou AK, Leak RK et al (2013) Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 33(12):1864–1874. doi:10.1038/jcbfm.2013.146
Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG (2009) Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. The Journal of neuroscience : the official journal of the Society for Neuroscience 29(43):13435–13444. doi:10.1523/JNEUROSCI.3257-09.2009
Zhao X, Wang H, Sun G, Zhang J, Edwards NJ, Aronowski J (2015) Neuronal Interleukin-4 as a Modulator of Microglial Pathways and Ischemic Brain Damage. 35 (32):11281–11291. doi:10.1523/jneurosci.1685-15.2015
Hayakawa K, Okazaki R, Morioka K, Nakamura K, Tanaka S, Ogata T (2014) Lipopolysaccharide preconditioning facilitates M2 activation of resident microglia after spinal cord injury. Journal of neuroscience research 92(12):1647–1658. doi:10.1002/jnr.23448
Sansing LH, Harris TH, Welsh FA, Kasner SE, Hunter CA, Kariko K (2011) Toll-like receptor 4 contributes to poor outcome after intracerebral hemorrhage. Annals of neurology 70(4):646–656. doi:10.1002/ana.22528
Kong Y, Le Y (2011) Toll-like receptors in inflammation of the central nervous system. International immunopharmacology 11(10):1407–1414. doi:10.1016/j.intimp.2011.04.025
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124(4):783–801. doi:10.1016/j.cell.2006.02.015
Beutler BA (2009) TLRs and innate immunity. Blood 113(7):1399–1407. doi:10.1182/blood-2008-07-019307
Zhu J, Mohan C (2010) Toll-like receptor signaling pathways—therapeutic opportunities. Mediators of inflammation 2010:781235. doi:10.1155/2010/781235
Zhou Y, Wang Y, Wang J, Anne Stetler R, Yang QW (2014) Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Progress in neurobiology 115:25–44. doi:10.1016/j.pneurobio.2013.11.003
Lin S, Yin Q, Zhong Q, Lv FL, Zhou Y, Li JQ, Wang JZ, Su BY et al (2012) Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. Journal of neuroinflammation 9:46. doi:10.1186/1742-2094-9-46
Fang H, Wang PF, Zhou Y, Wang YC, Yang QW (2013) Toll-like receptor 4 signaling in intracerebral hemorrhage-induced inflammation and injury. Journal of neuroinflammation 10:27. doi:10.1186/1742-2094-10-27
Teng W, Wang L, Xue W, Guan C (2009) Activation of TLR4-mediated NFkappaB signaling in hemorrhagic brain in rats. Mediators of inflammation 2009:473276. doi:10.1155/2009/473276
Wang YC, Wang PF, Fang H, Chen J, Xiong XY, Yang QW (2013) Toll-like receptor 4 antagonist attenuates intracerebral hemorrhage-induced brain injury. Stroke; a journal of cerebral circulation 44(9):2545–2552. doi:10.1161/strokeaha.113.001038
Rodriguez-Yanez M, Brea D, Arias S, Blanco M, Pumar JM, Castillo J, Sobrino T (2012) Increased expression of Toll-like receptors 2 and 4 is associated with poor outcome in intracerebral hemorrhage. Journal of neuroimmunology 247(1–2):75–80. doi:10.1016/j.jneuroim.2012.03.019
Kim YS, Joh TH (2012) Matrix metalloproteinases, new insights into the understanding of neurodegenerative disorders. Biomolecules & therapeutics 20(2):133–143. doi:10.4062/biomolther.2012.20.2.133
Yang Y, Rosenberg GA (2015) Matrix metalloproteinases as therapeutic targets for stroke. Brain research 1623:30–38. doi:10.1016/j.brainres.2015.04.024
Chang JJ, Emanuel BA, Mack WJ, Tsivgoulis G, Alexandrov AV (2014) Matrix metalloproteinase-9: dual role and temporal profile in intracerebral hemorrhage. Journal of stroke and cerebrovascular diseases : the official journal of National Stroke Association 23(10):2498–2505. doi:10.1016/j.jstrokecerebrovasdis.2014.07.005
Power C, Henry S, Del Bigio MR, Larsen PH, Corbett D, Imai Y, Yong VW, Peeling J (2003) Intracerebral hemorrhage induces macrophage activation and matrix metalloproteinases. Annals of neurology 53(6):731–742. doi:10.1002/ana.10553
Ohnishi M, Katsuki H, Fukutomi C, Takahashi M, Motomura M, Fukunaga M, Matsuoka Y, Isohama Y et al (2011) HMGB1 inhibitor glycyrrhizin attenuates intracerebral hemorrhage-induced injury in rats. Neuropharmacology 61(5–6):975–980. doi:10.1016/j.neuropharm.2011.06.026
Hayakawa K, Qiu J, Lo EH (2010) Biphasic actions of HMGB1 signaling in inflammation and recovery after stroke. Annals of the New York Academy of Sciences 1207:50–57. doi:10.1111/j.1749-6632.2010.05728.x
Gao TL, Yuan XT, Yang D, Dai HL, Wang WJ, Peng X, Shao HJ, Jin ZF et al (2012) Expression of HMGB1 and RAGE in rat and human brains after traumatic brain injury. The journal of trauma and acute care surgery 72(3):643–649. doi:10.1097/TA.0b013e31823c54a6
Lei C, Lin S, Zhang C, Tao W, Dong W, Hao Z, Liu M, Wu B (2013) High-mobility group box1 protein promotes neuroinflammation after intracerebral hemorrhage in rats. Neuroscience 228:190–199. doi:10.1016/j.neuroscience.2012.10.023
Wang R, Zhang Q, Yang S, Guo Q (2015) [TNF-alpha induces the release of high mobility group protein B1 through p38 mitogen-activated protein kinase pathway in microglia]. Zhong nan da xue xue bao Yi xue ban = Journal of Central South University Medical sciences 40(9):967–972. doi:10.11817/j.issn.1672-7347.2015.09.004
Yang QW, Lu FL, Zhou Y, Wang L, Zhong Q, Lin S, Xiang J, Li JC et al (2011) HMBG1 mediates ischemia-reperfusion injury by TRIF-adaptor independent Toll-like receptor 4 signaling. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 31(2):593–605. doi:10.1038/jcbfm.2010.129
Lei C, Lin S, Zhang C, Tao W, Dong W, Hao Z, Liu M, Wu B (2013) Effects of high-mobility group box1 on cerebral angiogenesis and neurogenesis after intracerebral hemorrhage. Neuroscience 229:12–19. doi:10.1016/j.neuroscience.2012.10.054
Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC (2002) Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia 40(2):195–205. doi:10.1002/glia.10148
Ren Y, Silverstein RL, Allen J, Savill J (1995) CD36 gene transfer confers capacity for phagocytosis of cells undergoing apoptosis. The Journal of experimental medicine 181(5):1857–1862
Fang H, Chen J, Lin S, Wang P, Wang Y, Xiong X, Yang Q (2014) CD36-mediated hematoma absorption following intracerebral hemorrhage: negative regulation by TLR4 signaling. Journal of immunology 192(12):5984–5992. doi:10.4049/jimmunol.1400054
Zamora C, Canto E, Nieto JC, Angels Ortiz M, Juarez C, Vidal S (2012) Functional consequences of CD36 downregulation by TLR signals. Cytokine 60(1):257–265. doi:10.1016/j.cyto.2012.06.020
Ekdahl CT, Kokaia Z, Lindvall O (2009) Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience 158(3):1021–1029. doi:10.1016/j.neuroscience.2008.06.052
Loane DJ, Kumar A (2016) Microglia in the TBI brain: the good, the bad, and the dysregulated. Experimental neurology 275(Pt 3):316–327. doi:10.1016/j.expneurol.2015.08.018
Li Y, Liu DX, Li MY, Qin XX, Fang WG, Zhao WD, Chen YH (2014) Ephrin-A3 and ephrin-A4 contribute to microglia-induced angiogenesis in brain endothelial cells. Anatomical record 297(10):1908–1918. doi:10.1002/ar.22998
Dinkova-Kostova AT, Abramov AY (2015) The emerging role of Nrf2 in mitochondrial function. Free radical biology & medicine 88(Pt B):179–188. doi:10.1016/j.freeradbiomed.2015.04.036
Lee JM, Li J, Johnson DA, Stein TD, Kraft AD, Calkins MJ, Jakel RJ, Johnson JA (2005) Nrf2, a multi-organ protector? FASEB journal : official publication of the Federation of American Societies for Experimental Biology 19(9):1061–1066. doi:10.1096/fj.04-2591hyp
Itoh K, Mimura J, Yamamoto M (2010) Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxidants & redox signaling 13(11):1665–1678. doi:10.1089/ars.2010.3222
Itoh K, Ye P, Matsumiya T, Tanji K, Ozaki T (2015) Emerging functional cross-talk between the Keap1-Nrf2 system and mitochondria. Journal of clinical biochemistry and nutrition 56(2):91–97. doi:10.3164/jcbn.14-134
Sandberg M, Patil J, D’Angelo B, Weber SG, Mallard C (2014) NRF2-regulation in brain health and disease: implication of cerebral inflammation. Neuropharmacology 79:298–306. doi:10.1016/j.neuropharm.2013.11.004
Li M, Zhang X, Cui L, Yang R, Wang L, Liu L, Du W (2011) The neuroprotection of oxymatrine in cerebral ischemia/reperfusion is related to nuclear factor erythroid 2-related factor 2 (nrf2)-mediated antioxidant response: role of nrf2 and hemeoxygenase-1 expression. Biological & pharmaceutical bulletin 34(5):595–601
Liu Y, Zhang L, Liang J (2015) Activation of the Nrf2 defense pathway contributes to neuroprotective effects of phloretin on oxidative stress injury after cerebral ischemia/reperfusion in rats. Journal of the neurological sciences 351(1–2):88–92. doi:10.1016/j.jns.2015.02.045
Shih AY, Erb H, Murphy TH (2007) Dopamine activates Nrf2-regulated neuroprotective pathways in astrocytes and meningeal cells. Journal of neurochemistry 101(1):109–119. doi:10.1111/j.1471-4159.2006.04345.x
Shah ZA, Li RC, Ahmad AS, Kensler TW, Yamamoto M, Biswal S, Dore S (2010) The flavanol (−)-epicatechin prevents stroke damage through the Nrf2/HO1 pathway. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 30(12):1951–1961. doi:10.1038/jcbfm.2010.53
Alfieri A, Srivastava S, Siow RC, Cash D, Modo M, Duchen MR, Fraser PA, Williams SC et al (2013) Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood–brain barrier disruption and neurological deficits in stroke. Free radical biology & medicine 65:1012–1022. doi:10.1016/j.freeradbiomed.2013.08.190
Li L, Zhang X, Cui L, Wang L, Liu H, Ji H, Du Y (2013) Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain research 1497:32–39. doi:10.1016/j.brainres.2012.12.032
Meng H, Guo J, Wang H, Yan P, Niu X, Zhang J (2014) Erythropoietin activates Keap1-Nrf2/ARE pathway in rat brain after ischemia. The International journal of neuroscience 124(5):362–368. doi:10.3109/00207454.2013.848439
Han J, Wang M, Jing X, Shi H, Ren M, Lou H (2014) (−)-Epigallocatechin gallate protects against cerebral ischemia-induced oxidative stress via Nrf2/ARE signaling. Neurochemical research 39(7):1292–1299. doi:10.1007/s11064-014-1311-5
Wang J, Fields J, Zhao C, Langer J, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S et al (2007) Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free radical biology & medicine 43(3):408–414. doi:10.1016/j.freeradbiomed.2007.04.020
Zhao X, Sun G, Zhang J, Strong R, Dash PK, Kan YW, Grotta JC, Aronowski J (2007) Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke; a journal of cerebral circulation 38(12):3280–3286. doi:10.1161/STROKEAHA.107.486506
Shang H, Yang D, Zhang W, Li T, Ren X, Wang X, Zhao W (2013) Time course of Keap1-Nrf2 pathway expression after experimental intracerebral haemorrhage: correlation with brain oedema and neurological deficit. Free radical research 47(5):368–375. doi:10.3109/10715762.2013.778403
Kwon KJ, Kim JN, Kim MK, Kim SY, Cho KS, Jeon SJ, Kim HY, Ryu JH et al (2013) Neuroprotective effects of valproic acid against hemin toxicity: possible involvement of the down-regulation of heme oxygenase-1 by regulating ubiquitin-proteasomal pathway. Neurochemistry international 62(3):240–250. doi:10.1016/j.neuint.2012.12.019
Dutra FF, Bozza MT (2014) Heme on innate immunity and inflammation. Frontiers in pharmacology 5:115. doi:10.3389/fphar.2014.00115
Abraham NG, Kappas A (2008) Pharmacological and clinical aspects of heme oxygenase. Pharmacological reviews 60(1):79–127. doi:10.1124/pr.107.07104
Raju VS, Maines MD (1994) Coordinated expression and mechanism of induction of HSP32 (heme oxygenase-1) mRNA by hyperthermia in rat organs. Biochimica et biophysica acta 1217(3):273–280
Dennery PA (2014) Signaling function of heme oxygenase proteins. Antioxidants & redox signaling 20(11):1743–1753. doi:10.1089/ars.2013.5674
Bloomer SA, Zhang HJ, Brown KE, Kregel KC (2009) Differential regulation of hepatic heme oxygenase-1 protein with aging and heat stress. The journals of gerontology Series A, Biological sciences and medical sciences 64(4):419–425. doi:10.1093/gerona/gln056
Terry CM, Clikeman JA, Hoidal JR, Callahan KS (1999) TNF-alpha and IL-1alpha induce heme oxygenase-1 via protein kinase C, Ca2+, and phospholipase A2 in endothelial cells. The American journal of physiology 276(5 Pt 2):H1493–1501
Mandal P, Park PH, McMullen MR, Pratt BT, Nagy LE (2010) The anti-inflammatory effects of adiponectin are mediated via a heme oxygenase-1-dependent pathway in rat Kupffer cells. Hepatology (Baltimore, Md) 51(4):1420–1429. doi:10.1002/hep.23427
Wang J, Dore S (2008) Heme oxygenase 2 deficiency increases brain swelling and inflammation after intracerebral hemorrhage. Neuroscience 155(4):1133–1141. doi:10.1016/j.neuroscience.2008.07.004
Wang J, Zhuang H, Dore S (2006) Heme oxygenase 2 is neuroprotective against intracerebral hemorrhage. Neurobiology of disease 22(3):473–476. doi:10.1016/j.nbd.2005.12.009
Chen-Roetling J, Cai Y, Regan RF (2014) Neuroprotective effect of heme oxygenase-2 knockout in the blood injection model of intracerebral hemorrhage. BMC research notes 7:561. doi:10.1186/1756-0500-7-561
Qu Y, Chen-Roetling J, Benvenisti-Zarom L, Regan RF (2007) Attenuation of oxidative injury after induction of experimental intracerebral hemorrhage in heme oxygenase-2 knockout mice. Journal of neurosurgery 106(3):428–435. doi:10.3171/jns.2007.106.3.428
Chen M, Regan RF (2007) Time course of increased heme oxygenase activity and expression after experimental intracerebral hemorrhage: correlation with oxidative injury. Journal of neurochemistry 103(5):2015–2021. doi:10.1111/j.1471-4159.2007.04885.x
Gong Y, Tian H, Xi G, Keep RF, Hoff JT, Hua Y (2006) Systemic zinc protoporphyrin administration reduces intracerebral hemorrhage-induced brain injury. Acta neurochirurgica Supplement 96:232–236
Wagner KR, Hua Y, de Courten-Myers GM, Broderick JP, Nishimura RN, Lu SY, Dwyer BE (2000) Tin-mesoporphyrin, a potent heme oxygenase inhibitor, for treatment of intracerebral hemorrhage: in vivo and in vitro studies. Cellular and molecular biology 46(3):597–608
Chen-Roetling J, Lu X, Regan RF (2015) Targeting heme oxygenase after intracerebral hemorrhage. Therapeutic targets for neurological diseases 2 (1). doi:10.14800/ttnd.474
Perez de la Ossa N, Sobrino T, Silva Y, Blanco M, Millan M, Gomis M, Agulla J, Araya P et al (2010) Iron-related brain damage in patients with intracerebral hemorrhage. Stroke; a journal of cerebral circulation 41(4):810–813. doi:10.1161/strokeaha.109.570168
Gu Y, Hua Y, He Y, Wang L, Hu H, Keep RF, Xi G (2011) Iron accumulation and DNA damage in a pig model of intracerebral hemorrhage. Acta neurochirurgica Supplement 111:123–128. doi:10.1007/978-3-7091-0693-8_20
Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF (2003) Heme and iron metabolism: role in cerebral hemorrhage. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 23(6):629–652. doi:10.1097/01.WCB.0000073905.87928.6D
Chen Z, Gao C, Hua Y, Keep RF, Muraszko K, Xi G (2011) Role of iron in brain injury after intraventricular hemorrhage. Stroke; a journal of cerebral circulation 42(2):465–470. doi:10.1161/STROKEAHA.110.602755
Zhang X, Surguladze N, Slagle-Webb B, Cozzi A, Connor JR (2006) Cellular iron status influences the functional relationship between microglia and oligodendrocytes. Glia 54(8):795–804. doi:10.1002/glia.20416
Rathnasamy G, Ling EA, Kaur C (2013) Consequences of iron accumulation in microglia and its implications in neuropathological conditions. CNS & neurological disorders drug targets 12(6):785–798
Labunskyy VM, Gladyshev VN (2013) Role of reactive oxygen species-mediated signaling in aging. Antioxidants & redox signaling 19(12):1362–1372. doi:10.1089/ars.2012.4891
Nakamura T, Keep RF, Hua Y, Hoff JT, Xi G (2005) Oxidative DNA injury after experimental intracerebral hemorrhage. Brain research 1039(1–2):30–36. doi:10.1016/j.brainres.2005.01.036
Gao C, Du H, Hua Y, Keep RF, Strahle J, Xi G (2014) Role of red blood cell lysis and iron in hydrocephalus after intraventricular hemorrhage. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. doi:10.1038/jcbfm.2014.56
Xie Q, Gu Y, Hua Y, Liu W, Keep RF, Xi G (2014) Deferoxamine attenuates white matter injury in a piglet intracerebral hemorrhage model. Stroke; a journal of cerebral circulation 45(1):290–292. doi:10.1161/strokeaha.113.003033
Hatakeyama T, Okauchi M, Hua Y, Keep RF, Xi G (2013) Deferoxamine reduces neuronal death and hematoma lysis after intracerebral hemorrhage in aged rats. Translational stroke research 4(5):546–553. doi:10.1007/s12975-013-0270-5
Hatakeyama T, Okauchi M, Hua Y, Keep RF, Xi G (2011) Deferoxamine reduces cavity size in the brain after intracerebral hemorrhage in aged rats. Acta neurochirurgica Supplement 111:185–190. doi:10.1007/978-3-7091-0693-8_31
Warkentin LM, Auriat AM, Wowk S, Colbourne F (2010) Failure of deferoxamine, an iron chelator, to improve outcome after collagenase-induced intracerebral hemorrhage in rats. Brain research 1309:95–103. doi:10.1016/j.brainres.2009.10.058
Chun HJ, Kim DW, Yi HJ, Kim YS, Kim EH, Hwang SJ, Jwa CS, Lee YK et al (2012) Effects of statin and deferoxamine administration on neurological outcomes in a rat model of intracerebral hemorrhage. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology 33(2):289–296. doi:10.1007/s10072-011-0733-y
Auriat AM, Silasi G, Wei Z, Paquette R, Paterson P, Nichol H, Colbourne F (2012) Ferric iron chelation lowers brain iron levels after intracerebral hemorrhage in rats but does not improve outcome. Experimental neurology 234(1):136–143. doi:10.1016/j.expneurol.2011.12.030
Wang G, Hu W, Tang Q, Wang L, Sun XG, Chen Y, Yin Y, Xue F et al (2015) Effect Comparison of Both Iron Chelators on Outcomes, Iron Deposit, and Iron Transporters After Intracerebral Hemorrhage in Rats. Molecular neurobiology. doi:10.1007/s12035-015-9302-3
Yeatts SD, Palesch YY, Moy CS, Selim M (2013) High dose deferoxamine in intracerebral hemorrhage (HI-DEF) trial: rationale, design, and methods. Neurocritical care 19(2):257–266. doi:10.1007/s12028-013-9861-y
Acknowledgments
This work was supported by the National Natural Science Foundation of China 81200885, Heilongjiang Postdoctoral Science-Research Foundation LBH-Q13120, the Natural Science Foundation of Heilongjiang Province of China LC2013C30, the Foundation of the First Clinical Hospital of Harbin Medical University 2012LX004, an American Heart Association grant 13GRNT15730001, and the National Institutes of Health (R01NS078026, R01AT007317). We thank Claire Levine for assistance with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Zhen Zhang and Ze Zhang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Zhang, Z., Zhang, Z., Lu, H. et al. Microglial Polarization and Inflammatory Mediators After Intracerebral Hemorrhage. Mol Neurobiol 54, 1874–1886 (2017). https://doi.org/10.1007/s12035-016-9785-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-9785-6