
Abstract
Multiple sclerosis (MS) is an inflammatory demyelinating central nervous system (CNS) disease with an uncertain etiology. MS is heterogeneous, involving multiple clinical pathologies, including neurodegeneration, depression, fatigue and sleep disorders, migraine, osteoporosis and cerebral hemodynamic impairments. The underlying causes of these pathologies remain mostly unknown. Based on the accumulating evidence derived from our studies and those of other investigators, we propose that the dysregulation in the neurovisceral integration of cardiovascular modulation can lead to many MS-related clinical symptoms. We show that MS inflammatory and neurodegenerative processes are intertwined with the aforementioned clinical morbidities and are collectively the manifestations of cardiovascular autonomic nervous system (ANS) dysfunction. The strategies for improving sympathovagal balance would likely prevent and minimize many MS-related clinical symptoms, improving patients’ quality of life. Similar strategies could be applied to other autoimmune and neurodegenerative diseases where autonomic imbalance plays a role.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple sclerosis (MS) is a progressive, autoimmune, demyelinating disease of the central nervous system (CNS) in which chronic inflammation is thought to cause axonal injury and loss, resulting in deficits of motor and cognitive functions [1]. MS affects young adults between ages 20 and 40, with a population prevalence of approximately 400,000 in the USA and one million worldwide [2]. The exact etiology of MS remains unknown; however, it is thought that genetic [3] and epigenetic [4] factors interact with environmental factors, determining the initiation and progression of MS.
MS patients show dysregulation in the neurovisceral integration of cardiovascular modulation, indicated by the dysfunction of both sympathetic and parasympathetic autonomic nervous system (ANS) responses; this dysfunction correlates with MS disease activity and progression to disability [5]. In fact, cardiovascular ANS dysfunction more closely correlates with the level of clinical disability than the extended disability status score (EDSS) and MRI [6]. ANS dysfunction is observed in 45 % of MS patients, although the percentages greatly vary depending on one or a combination of tests. The sympathetic dysfunction is known to be more prevalent than parasympathetic dysfunction (30 vs.18 %), and the latter is often observed in patients with long disease duration and advanced clinical disability [7].
The sympathetic and parasympathetic neurons are the output of the central autonomic network, one of the CNS compartments, comprised of a number of structures, which are reciprocally interconnected. The central autonomic network regulates visceral, endocrine, and behavioral responses, with the information flowing bi-directionally [8]. The post-ganglionic sympathetic output is then mediated by the neurotransmitter norepinephrine, which acts through α- and β-adrenergic receptors, whereas the post-ganglionic parasympathetic nerves use acetylcholine (Ach) as transmitter, which functions through nicotinic and muscarinic receptors.
The output of the sympathetic and parasympathetic neurons at the sinoatrial node of the heart and the interactions between the two branches are manifested by a complex beat-to-beat variability in both frequency and amplitude. This heart rate variability (HRV) connects the central autonomic network output to cardiac autonomic tone [9]. HRV measures the frequency power spectral density analysis of heartbeat interval series, providing information on the distribution of the power as a function of frequency and as a mean to quantify autonomic balance at any given time [10].
The power spectral analysis shows the following two main oscillations; the high-frequency (HF) oscillation components (0.15~0.4 Hz), which correspond to the variation in respiration, also known as respiratory sinus arrhythmia, an index of parasympathetic tone, and the low-frequency (LF) oscillation components (0.04~0.15 Hz), which are the rhythms corresponding to the vasomotor tone, involving mainly the arterial pressure variabilities, an index of sympathetic tone [9, 11]. The ratio of LF to HF is the key indicator for HRV, and assumed to describe the sympathovagal balance, which can be affected by changes in LF, HF, or a combination of both [10, 12].
Brezinova et al. [13] observed reduction in the absolute LF and HF powers in MS patients, as compared to control subjects. The reduction in LF power was more significant (66 %) than reduction in HF power (49 %). Nevertheless, the existence of both sympathetic and parasympathetic dysfunctions in the same MS patient [14] suggests that the parasympathetic dysfunction may not be an independent phenomenon, but may result from an impaired interaction between a dysfunctional MS sympathetic branch, with the parasympathetic branch [9].
Among factors that could lead to MS sympathetic dysfunction is the damage to locus coeruleus [15]. Located in the pons structure of the brain stem, it is the principal site for brain norepinephrine synthesis and is involved in physiological responses to stress. Consistently, MS patients show reduced brain norepinephrine [15] and reduced intracellular catecholamine concentrations in the peripheral blood lymphocytes [16], suggesting the dysfunction of both central and peripheral sympathetic functions.
The results of our recent studies, which aimed to determine the prevalence of cardiovascular risk factors in MS patients, showed significantly lower systolic BP readings and plasma glucose levels, two variables which are determined by the sympathetic ANS function, in MS patients compared to non-MS patients [17, 18]. Among MS patients, 41 and 79 % presented with the systolic BP readings and plasma glucose levels below 120 mmHg and 100 mg/dL, respectively, as compared to 29 and 46 % for age- and gender-matched non-MS patients [17]. The gap between MS and non-MS patients further widened when disease-modifying drug (DMD)-naïve MS patients were compared to non-MS patients [18]. MS patients also exhibit hypothalamus pituitary adrenal (HPA) axis hypoactivity, both at rest and during physiological stress [19]. Sympathetic activation is known to be a prerequisite for the rise in the corticotropin-releasing hormone (CRH) and for the rise in ACTH secretion [20, 21].
Locus coeruleus has projections to many brain structures, including hypothalamus, brain stem and spinal cord, cerebral cortex, amygdala, and hippocampus, and it receives input from hypothalamus, prefrontal cortex, and cerebellum, among others. The far-reaching inputs to and outputs from locus coeruleus suggest that the damage to this structure may have widespread adverse effects on physiological processes of many organs. Locus coeruleus is known having roles in cognition, attention and memory, arousal, sleep-wake cycle, posture and balance, blood flow control, mood, and addiction [22–25]. The dysregulation in this system can lead to arrays of clinical symptoms often observed in MS patients, including cognitive dysfunction, sleep disorders, depression, migraine and reduced cerebral blood flow (see paragraphs below for detailed “Discussion”).
In addition, through sensory and immune-related stimuli to the brain, ANS also regulates the function of the immune system. This neurovisceral regulation extends beyond organs such as bone marrow, spleen, and thymus, into the regulation of cells involved in the innate and adaptive immunity [26], and achieved via the expression of adrenergic and cholinergic receptors on immune cells [27, 28]. Signaling through these receptors is known to influence immune cell proliferation and cytokine production, cellular inflammation, adhesion, and cell migration [29]. MS alterations in both adrenergic [30] and cholinergic [31] receptors and their correlation with clinical and MRI indicators of disease activity [32] suggest that ANS dysfunction is likely to play a role in MS immune dysregulation and promotion of inflammatory and neurodegenerative processes.
Thayer et al. [8] proposed dysregulation in the neurovisceral integration of cardiovascular modulation as contributing factor not only to cardiovascular disease but also to the related modifiable and unmodifiable risk factors. In their model, the dysregulation in the neurovisceral integration was related to sympathetic overactivity and parasympathetic underactivity. Predominance of sympathetic over parasympathetic has the potential leading to a reduction in ANS system flexibility and adaptability, which is a prerequisite for quickly responding to environmental demands. This impaired parasympathetic function has been attributed to a reduced prefrontal inhibitory control over sympathoexcitatory circuits in the brain stem [8, 33]. A similar model has been proposed for neurovisceral integration of emotions, suggesting that emotions, such as anxiety, are the by-products of faulty negative feedback, which then gives rise to unregulated and uninhibited positive feedback loops [34].
Both MS patients [13] and patients with cardiovascular disorders [35] present with reduced HRV values when compared to controls. The reduced HRV in patients with cardiovascular disease is due to high levels of sympathetic input, which is known to diminish HRV, whereas in MS patients, the reduced sympathetic outflow is the main contributor to the reduction in the HRV [36], suggesting that both sympathetic hyperactivity and hypoactivity may be associated with pathologies. However, unlike the neurovisceral dysregulation model which is based on the inhibition of vagal activity and dominance of sympathetic activity [8, 33, 34], we propose that lower-than-normal sympathetic output is the main cause for sympathovagal imbalance, leading to MS clinical morbidities.
The role of ANS dysfunction in the pathology of MS and its many clinical presentations is overlooked. Cardiovascular ANS dysfunction is not a benign phenomenon, but it may contribute to the many MS clinical presentations, with a significant negative impact on MS patients’ quality of life. We have recently discussed the role of ANS dysfunction in inflammation, neurodegeneration, and reduced response to immunomodulatory therapies [37], and its role in the high prevalence of osteoporosis often observed in MS patients (under peer review). Our assumption is based on the notion that sympathetic activation, and subsequent norepinephrine release and its binding to β2-adrenergic receptors on peripheral immune cells, has the potential upregulating cAMP, resulting in the inhibition of Th1-derived proinflammatory cytokines while promoting the production of Th2-dervided antiinflammatory cytokines [29, 38]. Similarly, the β2-adrenergic receptors on brain cells have a protective and supportive role [39], and their activation reduces memory impairment [40]. Therefore, the dysregulation in sympathetic activity could contribute to altered Th1/Th2 cytokines’ ratio and to heightened inflammatory state observed in MS patients [41], as well as to the MS cognitive impairments.
MS patients show alterations in adrenergic influences of the immune cells, indicated by lower cellular norepinephrine levels [42, 43] and higher number and density of dysfunctional [30] β2-adrenergic receptors on immune cells [32, 43] These receptors are absent from the brain of chronic progressive MS patients [44], and their absence is associated with MS clinical and MRI indicators of disease activity [32, 45]. The low norepinephrine levels and the dysfunction of the β2-adrenergic receptors have the potential of activating the α2-adrenergic receptors on macrophages, upregulating inflammatory processes [46]. The activation of the same receptors in the brain is associated with neurodegenerative processes [47].
The association between low sympathetic output and inflammation is consistent with the reduction in the rate of clinical relapse in female MS patients during pregnancy, when a progressive rise in the production of norepinephrine plasma levels is observed [48], whereas the post-partum clinical relapse is characterized by an approximately 40 % reduction in the norepinephrine plasma levels and reduction in the number and the affinity of the β2-adrenergic receptors on peripheral blood mononuclear cells [49]. Furthermore, the reduction in the α2-adrenergic receptors during pregnancy and their upregulation during post-partum [50] suggest the involvement of these receptors in inflammation and their contribution to MS clinical relapse.
The neuroprotective effects of increasing CNS norepinephrine have been shown in clinical trials of MS patients treated with antidepressants [51], and in its animal model, the experimental autoimmune encephalomyelitis (EAE) [52]. These results are consistent with studies showing a correlation between LF domain of the HRV and verbal learning and memory [53].
Similarly, the activation of the cholinergic axis has the potential exerting antiinflammatory and neuroprotective effects [54, 55]. PET studies show reduced nAChRs in the frontal, cingulate, temporal, parietal, and occipital cortices, and cerebellum of MS patients, and the correlation between reduction in these receptors and impaired memory and attention [56]. MS patients also show alterations in muscarinic cholinergic receptors on peripheral immune cells [31], suggesting both central and peripheral parasympathetic dysfunctions.
ANS dysfunction could also reduce patients’ clinical response to immunomodulatory therapies. It is known that chronic progressive MS patients, who present with a more severe ANS dysfunction [5], are often the ones who have poor response to immunomodulatory therapies [57]. Monocytes derived from these patients show low intracellular norepinephrine levels and do not adequately respond to the IL-2-induced rise in β2-adrenergic receptors density [43].
The reduced and or loss of response to long-term immunomodulatory therapy may stem from the potential of immunomodulators to downregulate the sympathetic ANS function, either directly or by shifting the profile of Th1 to Th2 cells [58]. Th2 cells do not possess β2-adrenergic receptors and hence are unable to stimulate the sympathetic ANS function [59]. Analogously, chronic progressive MS patients, who have predominantly Th2 cytokines [59], do not respond favorably to treatment with agents that increase Th2 cytokines [60]. The adverse effects of immunomodulators on sympathetic ANS function may especially be apparent in older MS patients and those patients with longer disease duration [61, 62]. These patients are likely to have a more severe ANS dysfunction, resulting from both age-induced impairment in ANS function and from the MS-related disease burden [63, 64].
The reduced response to immunomodulatory therapies depends on the duration, and often occurs within 2 years of use, correlating closely with patients’ clinical disability [65]. For example, an acute interferon beta (IFN-β) administration enhances lymphocytes’ production of norepinephrine in IFN-β-naïve MS patients [66]. But, lymphocytes derived from long-term IFN-β users show reduced cytokine response to IFN-β treatment [67], as well as reduced baseline and post-IFN-β-induced rise in intracellular norepinephrine [16]. The association between ANS dysfunction and reduced response to immunomodulatory therapy is supported by the result of a clinical study showing improved clinical efficacy of glatiramer acetate when combined with the β-adrenergic agonist, albuterol [68].
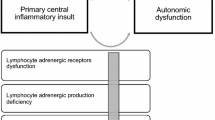
In this evidence-based review, we propose that the dysregulation of the neurovisceral integration of cardiovascular modulation (Fig. 1) cannot only promote inflammatory and neurodegenerative processes, but it can also lead to many other MS-related morbidities, including depression, fatigue and sleep disorders, migraine, and cerebral hemodynamic impairments; the latter can lead to cerebral venous remodeling and subsequent stenosis. We demonstrate that these clinical presentations are intertwined with MS inflammatory and neurodegenerative processes.

The Clinical effects of impaired neurovisceral integration of cardiovascular modulation. The diagram shows that an impaired neurovisceral integration of cardiovascular modulation has widespread adverse clinical effects. It can promote inflammation/neurodegeneration, reduce the response to immunomodulatory therapies, and contribute to the clinical presentations of fatigue/sleep disorders, depression, migraine, osteoporosis, and HPA axis hypo-responsiveness; It can lead to cerebral hemodynamic impairments, indicated by reduced cerebral blood flow, diminished cerebral autoregulation, and altered cerebrospinal fluid dynamics; It can lead to hypoxia-induced-TCR response diminution, with further adverse effects on cerebral blood flow; It can influence cerebral venous microvascular and macrovascular structures, indicated by venous remodeling and compression/stenosis respectively. HPA hypothalamus pituitary adrenal; TCR trigemino-cardiac reflex
Discussion
ANS Dysfunction in MS Clinical Morbidities
Depression
Depression is a common clinical presentation in MS, affecting up to 50 % MS patients during their lifetime [69]. MS patients are twice as likely to be on antidepressants as compared to non-MS patients (35 vs.18 %) [17]. The pathophysiology of depression is complex, but it is known to partly involve reduced brain norepinephrine and dopamine (the latter is metabolized to norepinephrine by the enzyme dopamine β-hydroxylase) [70]. Lymphocytes of individuals with depression show lower β-adrenergic receptor binding [71] and sensitivity [72] compared to those in healthy controls. Furthermore, a significant reduction in the parasympathetic-related HRV parameters have been observed in patients with clinical symptoms of depression [73], suggesting that both adrenergic and cholinergic dysfunctions may play roles in the pathology of MS depression.
Consistent with this conclusion, individual treatment with the antihypertensive drug reserpine, which depletes monoamine stores, induces depression [74], whereas monoamine oxidase inhibitors (MAOIs) and tricyclic antidepressants (TCAs), which are known to increase synaptic levels of CNS norepinephrine, reduce depressive symptoms. In addition, vagal afferent nerve fiber stimulation is known to benefit patients with depression [75]. However, this FDA-approved strategy is thought to exert beneficial effects, in part, by transmitting impulses to the locus coeruleus [75] and increasing CNS norepinephrine concentrations [76]. Similar beneficial effects of vagal stimulation have been shown in MS patients [77], who are known to have locus coeruleus damage [15].
In addition, lower brain-derived neurotrophic factor (BDNF) serum levels are known to be associated with depressive disorders [78]. This observation agrees with the reported low BDNF serum levels in MS patients. The levels correlate with neuroimaging indicators of disease activity and are increased after antidepressant therapy [79, 80]. BDNF is produced by both immune cells and neurons with a potential for exerting immunomodulatory and neuroprotective effects [81]. The increase in neuronal norepinephrine [82] and vagus nerve stimulation [76] are known to increase BDNF levels, suggesting a relationship between ANS function, BDNF levels, depression, and MS inflammatory and neurodegenerative processes.
Gender differences in cardiovascular ANS regulation have been reported, showing lower sympathetic function in females than males [64, 83]. Our own study showed a significantly higher percentage of DMD-naïve female MS patients with systolic BP below 120 mmHg, as compared to male patients (55 vs. 17 %). This trend was reversed for systolic BP above 130 mmHg (27 % of females vs. 50 % of males) [17].
These differences may contribute to a higher prevalence of depression observed in MS females, as compared to males [84]. In addition, MS patients with longer disease duration, and chronic progressive MS patients, show more severe clinical depression compared to patients with a shorter disease duration and patients with other form of MS [84, 85]. These observations are consistent with the greater severity of ANS dysfunction in chronic progressive MS patients and those with longer disease duration [5, 63].
The association between depression and ANS dysfunction in MS patients is mostly unknown. However, MS depression closely correlates with inflammation and neurodegeneration. For example, MS patients with depression show higher hyperintense and hypointense lesion volumes in the left medial inferior frontal region than patients without depression [86]. A diffusion tensor imaging of MS patients with depression shows reduced normal appearing white matter and elevated mean diffusivity in normal appearing grey matter in the temporal lobes [87]. Furthermore, an association between MS depressive symptoms and a reduced hippocampal volume has been observed [88]. These studies are in line with the reported negative influences of depression on MS cognitive function [89] and the role of ANS dysfunction in promoting neurodegeneration.
Fatigue-Sleep Disorders
Fatigue is one of the most common clinical symptoms affecting 80 % of MS patients, 50 % of whom are severely affected [90, 91], with a significant impact on the quality of life [92]. Fatigue is closely associated with depression [92, 93], with a higher prevalence in females than males [94]. MS patients can experience peripheral and or central fatigue. The peripheral fatigue is known as muscular fatigue, correlating with EDSS, whereas the pathophysiology of central fatigue involves impaired cognitive processing, memory, concentration, and deficits in attention and arousal [95, 96].
A number of clinical scales are used to measure fatigue in MS patients. These scales include Fatigue Severity Scale (FSS), Fatigue Impact Scale (FIS), Modified Fatigue Impact Scale (MFIS), and Neurological Fatigue Index (NFI). However, these clinical scales are based on self-reported questionnaires and do not always correlate with each other [97]. Fatigued MS patients are unable to adequately increase BP during handgrip exercise [98] and during stressful physical tests [99], suggesting the involvement of sympathetic dysfunction in MS fatigue. In addition, MS fatigue, as measured by the FSS and the MFIS, correlates with HRV [100]. The reduced HRV is especially apparent during the short-term memory test [99], suggesting a relationship between ANS dysfunction, cognitive impairment, and MS fatigue.
The perception of fatigue can be increased by comorbid conditions such as depression and sleeping disturbances [101]. In fact, similar to fatigue and depression, sleeping disturbances are three times more likely in MS patients than non-MS patients, and reduced sleep quality is twice as likely in MS patients than non-MS patient [102]. In addition, MS patients with fatigue are more likely to experience disrupted sleep, indicated by frequent awakening, than non-fatigued MS patients and healthy controls [103]. Cardiovascular ANS dysfunction, especially a reduced parasympathetic function, is known to contribute to MS sleep disturbance [104].
Strober et al. [101] proposed a model showing that fatigue, depression, and sleeping disorders are interrelated and each independently correlate with MS disease severity. This model agrees with the reported relationship between MS fatigue and higher white and grey matter atrophies, and between MS fatigue and higher T1 and T2 lesion loads [105, 106]. The relationship between fatigue and MS neurodegenerative processes is consistent with ANS dysfunction contributing to MS physical and cognitive disability [107]. The coexistence of fatigue and ANS dysfunction has been also demonstrated in other autoimmune [108] and neurodegenerative [109] diseases.
Migraine
Headache is another common MS clinical symptom, with a 50 % higher prevalence in MS patients than in control subjects [110]. MS patients present with primary headache, including migraine (with and without aura) and tension type headache, although many MS patients have unclassified headache [111]. Migraine is more prevalent in females and in young relapsing-remitting MS patients, correlating with disease exacerbations [112], whereas tension-type headache is more often observed in secondary progressive male MS patients [113].
Studies show that the relative risk of developing MS is significantly higher in migraineurs than in non-migraineurs, and the odds of being diagnosed with migraine is higher in women with MS than those without MS [114], suggesting that MS and migraine may be related at some level. Factors such as demyelinating brain stem lesions [115] and or treatment with DMDs [116] have been thought to contribute to the high prevalence of migraine in MS patients, but the role of ANS dysfunction in MS migraine has not been investigated.
The cranial circulation has both sympathetic and parasympathetic components [117], suggesting a role for ANS dysfunction in migraine. A wide variety of diagnostic tests indicate the presence of a sympathetic ANS dysfunction in migraineurs. Pupillary sympathetic hypofunction, orthostatic hypotension, a poor response to cold pressor test, a decreased overshoot in the Valsalva maneuver, and low levels of plasma norepinephrine have all been documented in patients with migraine and tension headache [118–121]. The reduced plasma norepinephrine level is also apparent in migraineurs during the headache-free period [121].
Patients with migraine and tension-type headache also show reduction in the HRV-related parameters of parasympathetic ANS activity, as well as reduced HR responses during sustained handgrip, and RR-interval oscillatory response to neck suction [122–124]. These results collectively show the involvement of both sympathetic and parasympathetic dysfunctions in the pathology of migraine. Similar to MS patients, migraineurs suffer from greater fatigue as well as depressive tendencies [122, 125], observations which are in line with the reported role of sympathetic impairment as the cause of migraine [126]. Consistent with the role of ANS dysfunction in neurodegeneration, migraineurs also exhibit increased serum markers of glial activation and neuronal damage, apparent even in migraine-free periods [127].
Cerebral Hemodynamic Impairments
The newer theory of vascular etiology of MS, which has been set forward by Zamboni, describes impairments in the principal pathways of extracranial venous drainage, affecting mostly the internal jugular veins (IJVs), the vertebral, and the azygos veins [128, 129]. However, the normal cerebrospinal fluid (CSF) ferritin levels in MS patients [130] exclude the role of venous reflux as the factor contributing to MS inflammatory and neurodegenerative processes. We have recently proposed that dysregulation in neurovisceral integration of cardiovascular modulation can lead to cerebral hemodynamic impairments, associated with the chronic cerebrospinal venous insufficiency (CCSVI). In this review, we provide additional evidence supporting our original proposal, showing a close association between venous stenosis and other clinical manifestations of ANS dysfunction.
ANS-dysfunction-induced arterial BP reduction [131] is among factors contributing to CCSVI occurrence. Our recent study shows that CCSVI-positive MS patients show deviations from normal ranges in arterial BP. Approximately, 50 % of patients present with systolic BP below 120 mmHg [132]. Furthermore, 91 % of MS patients present with three or more symptoms of ANS dysfunction, including cognitive impairment, fatigue, sleep disorders, and headache, and a significant statistical relationship between various clinical symptoms has been observed [132]. The coexistence of CCSVI and cardiovascular ANS dysfunction in MS, as well as in other autoimmune and neurological diseases [133, 134], further supports the notion that the two phenomena may be related.
Reduced arterial BP could also to a lower than normal cerebral perfusion pressure, which is determined by the differences in the pressure gradient between mean arterial BP and the intracranial pressure [135]. Although intracranial venous pressure has been shown to be normal [136], PET or single-photon emission CT techniques show hypoperfusion of MS brain parenchyma, involving both cerebral white and grey matters [137, 138]. These hemodynamic impairments correlate with CCSVI severity [139], with MS disease progression, with neuropsychological dysfunction, and with fatigue scores [137, 138, 140].
The Starling resistor hypothesis dictates that an increase in intracranial pressure and cerebral venous pressure would accompany a reduction in cerebral blood flow and cerebral perfusion pressure [141]. However, despite reduced cerebral blood flow [142, 143], MS patients show normal intracranial pressure, as measured by venous pressure [136]. This discrepancy may be due to differences in the degree of compliance of various brain regions, leading to an intracranial pressure gradient. Therefore, the level of the intracranial pressure may depend on the area of measurement [144].
The hypoperfusion is assumed to be the result of deficiency in astrocytic β2-adrenergic receptors and a reduced formation of cAMP [138], rather than the presence [138] or absence [137, 138] of CCSVI. These results confirm that sympathetic dysfunction, rather than venous obstruction, may contribute to the observed cerebral hypoperfusion. The improvement in cerebral blood flow post-balloon angioplasty [145] could be attributed to sympathetic activation [146] due to venous distention [147], rather than correction of the venous obstruction. The reported transient increase in cerebral blood flow, before lesion development [148], indicates sympathetic ANS activation in a setting of acute inflammation.
Despite alteration in arterial BP, little or no changes in cerebral blood flow is expected to occur as long as the perfusion pressure is greater than cerebral autoregulation, which is known to maintain constant cerebral blood flow despite fluctuations in the cerebral perfusion pressure. Nevertheless, in pathological states, cerebral blood flow could become pressure dependent due to impairment in cerebral autoregulation. Indeed, subsequent to the publication of our recent review, where we postulated the likelihood of the impairment of MS cerebral autoregulation, Mezei et al. [149] reported such impairment, indicated by changes in MS cerebral blood flow velocity as a function of changes in mean arterial BP. The impairment in cerebral autoregulation is likely to be the result of ANS dysfunction, since cerebral autoregulation is under autonomic neural control [150, 151].
The combination of low arterial BP and impairment in cerebral autoregulation, and high levels of nitric oxide, produced due to the chronic MS brain inflammatory processes [152], will ultimately reduce transmural pressure, promoting the critical closure pressure (CrCP) [153, 154]. Alterations in the CrCP contribute to the compression or the collapse of the cerebral venous system in general, and IJVs in particular, owing to their cytoarchitectionial construction. The IJVs collapsibility also explains their common tendency for restenosis after venous angioplasty [155]. Furthermore, IJVs reopening after their collapse due to orthostatic challenge may be hindered due to a suboptimal central venous pressure, the latter resulting from a low sympathetic outflow [156].
Phase contrast CSF flow imaging studies show reduced net CSF flow, correlating with CCSVI severity [157, 158]. It is known that CSF dynamic is coupled to cerebral arterial and venous systems through windkessel effect [159]. The intracranial venous flow is normal in MS patients, showing no sign of reflux, but the arterial flow is reduced [158], suggesting that the impairment in CSF flow may be the by-product of reduced arterial pressure rather than venous obstruction.
A dynamic secretion and resorption of CSF is required for maintaining intracranial pressure equilibria. Circulatory movement due to hydrostatic pressure gradients regulates both CSF secretion at the choroid plexuses and CSF resorption into the venous system at the arachnoid granulations. However, a dynamic CSF flow depends also on the vasomotor waves or Traube-Herring-Mayer (THM) waves which are cyclic changes in arterial BP brought about by oscillations in arterial baroreceptors [160]. The frequency and the peak occurrence of THM waves depend on the functions of both sympathetic and parasympathetic ANS [160].
THM waves along with increased HRV are considered to be markers of autonomic balance. MS patients present with baroreflex dysfunction, indicated by reduced systolic BP oscillations and reduced RR-interval oscillations in response to baroreceptor stimulation [14]. These alterations have the potential for reducing the THM wave. Whether the observed MS improvement in CSF dynamics [161] is the result of an increase in THM wave due to balloon angioplasty-induced sympathetic activation [146] is unknown and warrants further investigations.
The hypoperfusion of MS brain parenchyma [137, 138] could lead to hypoxia, and an increase in the expression of hypoxia inducible factor-1α (HIF-1α), promoting myelin-associated glycoprotein degradation and CNS damage [162], as well as compromise the blood–brain barrier integrity [163]. Oligodendrocytes are known to be particularly sensitive to ischemic insults [164], and a mild chronic hypoperfusion in the range of 15 % is able to lead to the disruption of axon-myelin integrity [165].
Contrast-enhanced MRI and the use of echo planar technique, which measured parameters of brain perfusion, such as cerebral blood flow, cerebral blood volume, and vascular mean transit time, observed a 50 % reduction in cerebral blood flow and more than a twofold prolonged mean transit time and reduction in cerebral blood volume in normal appearing white matter (NAWM) in relapsing-remitting MS compared with controls [142, 143]. The chronic focal lesions in MS white matter showed a more decreased perfusion when compared with contralateral NAWM [142]. Similar results were observed also in primary progressive MS patients [143]. Such significant reduction in cerebral blood flow has the potential causing microglial activation, white matter damage, and axon sparing [166].
Cerebral hypoxic ischemia has the potential for fast and reversible activation of the oxygen-conserving reflexes, similar to their activation during hypoxic cerebral ischemia of stroke. Also termed trigeminocardiac reflexes (TCRs), these protective sympathetically mediated vasomotor responses originate from reticulospinal neurons of the rostroventrolateral medulla, projecting to the upper brain stem and/or thalamus, culminating in the activation of a population of neurons in the cortex. The activation of these cortical centers can in turn modulate cardiovascular centers, increasing cerebral vasodilation and blood flow, without changing cerebral oxygen or glucose metabolism, hence minimizing hypoxia-induced ischemic brain damage [167, 168]. However, the suboptimal MS sympathetic output may compromise TCRs activation and their downstream neuroprotective effects, leading to reduction in MS cerebral blood flow [142, 143], in the absence of optimal TCR corrective measures.
In addition, hemodynamic impairments due to ANS dysfunction can also lead to morphological and metabolic changes in the venous system [169], with the potential for promoting venous compression and or closure. A series of studies by Zamboni and colleagues [170–172] report morphological changes including IJV calcification patterns [170], absence of endothelial cells in the intraluminal obstacles and their replacement with fibrous lamina [171], and an increase in the proportions of type III to type I collagen within the adventitia [172]. These pathological processes in the venous system mirror those in the arterial system, where hemodynamic impairments could lead to arterial remodeling, arterial hardening, and stenosis, a process known as atherosclerosis.
One such example is the hemodynamic alterations in the surgically created arteriovenous (AV) fistula that often leads to vessel remodeling and AV graft stenosis [173]. In this model, immune cell infiltration into the AV fistula occurs early and transiently in the stenosis process, and the levels correlate with the vessel wall shear stress [174]. These results suggest that the absence of immune cells observed in the diseased IJVs [172] may be due to early and transient infiltration of immune cells into the diseased IJVs and or due to a very low physiological shear stress in these vessels compared to the shear stress in the arteries (1 dyne/cm2 in the venous system vs. 15 dyne/cm2 in arteries) [175]. The efficacy of 25-hydroxy vitamin D in MS could be related to its ability to beneficially influence ANS function [176], preventing the subsequent venous remodeling [177].
Compared to cerebral arteries, the cerebral venous system presents with a complex three-dimensional structure and extensive anatomic variabity; these could in turn influence the biophysics and hemodynamics of the cerebral venous system, differentiating them from those parameters, known to govern in the arterial system [178]. Although the cerebral venous occlusion may be partially compensated by collateral circulation, this occlusion often leads to venous infarction and neurological deterioration, with an underlying pathology that differs from cerebral arterial infarction [179]. Assuming that ANS dysfuntion plays a role in venous occlusion, the improvement in the function of this system has the potential for reducing the high rate of MS cerebral venous thromboembolic incidences [180, 181]. In contrast, MS low sympathetic output may contribute to reduced risk of ischemic stroke [182], ischemic heart disease, and myocardial infarction [183].
Concluding Remarks
We have shown through our studies and those of others that impaired neurovisceral integration of cardiovascular modulation plays a role in MS pathology, with the potential for promoting not only inflammation and neurodegeneration but also many other MS-related morbidities, including depression, fatigue and sleep disorders, migraine, and cerebral hemodynamic impairments; the latter could result in venous compression and or stenosis, in the absence of corrective measures. We have also recently describe the contribution of ANS dysfunction to the high prevalene of osteoporosis often observed in these patients (under peer review).
The close association between ANS dysfunction and MS pathological processes suggests that the routine ANS function testing should be conducted and used as a tool for improving the assessment of MS disease stage and the rate of its progression. Such data could be also used for evaluating the clinical efficacy of DMDs.
In a sequel to this review, we have described the pharmacological, non-pharmacological, and surgical strategies that could be adopted to improve neurovisceral integration of cardiovascular modulation, promoting a state of sympathovagal balance [184], which would likely have a positive impact on MS patients’ quality of life. Nevertheless, the corrective strategies should take into account the individual differences in the nature, whether sympathetic or parasympathetic, and the degree of the autonomic imbalance. In addition, genetic, epigenetic, and factors can also modulate ANS functional activity. These factors interact with environmental factors in a complex manner. This complexity is further enhanced in MS patients due to MS disease processes, disease duration, and the chronic use of many MS-related and unrelated drugs. Therefore, an optimal corrective strategy(s) in MS patients should take into consideration these additional factors and the interactions among them [185].
These strategies could be also applied to other autoimmune and neurodegenerative diseases where autonomic dysfunction plays a role.
References
Sturm D, Gurevitz SL, Turner A (2014) Multiple sclerosis: a review of the disease and treatment options. Consult Pharm: J Am Soc Consult Pharm 29(7):469–479
Kantarci O, Wingerchuk D (2006) Epidemiology and natural history of multiple sclerosis: new insights. Curr Opin Neurol 19(3):248–254
Willer CJ, Dyment DA, Risch NJ, Sadovnick AD, Ebers GC, Canadian Collaborative Study G (2003) Twin concordance and sibling recurrence rates in multiple sclerosis. Proc Natl Acad Sci U S A 100(22):12877–12882
Burrell AM, Handel AE, Ramagopalan SV, Ebers GC, Morahan JM (2011) Epigenetic mechanisms in multiple sclerosis and the major histocompatibility complex (MHC). Disc Med 11(58):187–196
Flachenecker P, Reiners K, Krauser M, Wolf A, Toyka KV (2001) Autonomic dysfunction in multiple sclerosis is related to disease activity and progression of disability. Mult Scler 7(5):327–334
Nasseri K, Uitdehaag BM, van Walderveen MA, Ader HJ, Polman CH (1999) Cardiovascular autonomic function in patients with relapsing remitting multiple sclerosis: a new surrogate marker of disease evolution? Eur J Neurol: Off J Eur Fed Neurol Soc 6(1):29–33
Kodounis A, Stamboulis E, Constantinidis TS, Liolios A (2005) Measurement of autonomic dysregulation in multiple sclerosis. Acta Neurol Scand 112(6):403–408
Thayer JF, Lane RD (2007) The role of vagal function in the risk for cardiovascular disease and mortality. Biol Psychol 74(2):224–242
Malliani A, Pagani M, Lombardi F, Cerutti S (1991) Cardiovascular neural regulation explored in the frequency domain. Circulation 84(2):482–492
Pagani M, Lombardi F, Guzzetti S, Rimoldi O, Furlan R, Pizzinelli P et al (1986) Power spectral analysis of heart rate and arterial pressure variabilities as a marker of sympatho-vagal interaction in man and conscious dog. Circ Res 59(2):178–193
Eckberg DL (1997) Sympathovagal balance: a critical appraisal. Circulation 96(9):3224–3232
Pagani M, Lombardi F, Guzzetti S, Sandrone G, Rimoldi O, Malfatto G et al (1984) Power spectral density of heart rate variability as an index of sympatho-vagal interaction in normal and hypertensive subjects. J Hypertens Suppl: Off J Int Soc Hypertens 2(3):S383–S385
Brezinova M, Goldenberg Z, Kucera P (2004) Autonomic nervous system dysfunction in multiple sclerosis patients. Bratisl Lek Listy 105(12):404–407
Sanya EO, Tutaj M, Brown CM, Goel N, Neundorfer B, Hilz MJ (2005) Abnormal heart rate and blood pressure responses to baroreflex stimulation in multiple sclerosis patients. Clin Auton Res: Off J Clin Auton Res Soc 15(3):213–218
Polak PE, Kalinin S, Feinstein DL (2011) Locus coeruleus damage and noradrenaline reductions in multiple sclerosis and experimental autoimmune encephalomyelitis. Brain: J Neurol 134(Pt 3):665–677
Rajda C, Bencsik K, Fuvesi J, Seres E, Vecsei L, Bergquist J (2006) The norepinephrine level is decreased in the lymphocytes of long-term interferon-beta-treated multiple sclerosis patients. Mult Scler 12(3):265–270
Sternberg Z, Leung C, Sternberg D, Li F, Karmon Y, Chadha K et al (2013) The prevalence of the classical and non-classical cardiovascular risk factors in multiple sclerosis patients. CNS Neurol Disord Drug Targets 12(1):104–111
Sternberg Z, Leung C, Sternberg D, Yu J, Hojnacki D (2014) Disease modifying therapies modulate cardiovascular risk factors in patients with multiple sclerosis. Cardiovasc Ther 32(2):33–39
Arata M, Sternberg Z (2015) Neuroendocrine responses to transvascular autonomic modulation: a modified balloon angioplasty in multiple sclerosis patients. Horm Metab Res = Hormon- und Stoffwechselforschung = Horm Metab
Plotsky PM, Cunningham ET Jr, Widmaier EP (1989) Catecholaminergic modulation of corticotropin-releasing factor and adrenocorticotropin secretion. Endocr Rev 10(4):437–458
Plotsky PM (1987) Facilitation of immunoreactive corticotropin-releasing factor secretion into the hypophysial-portal circulation after activation of catecholaminergic pathways or central norepinephrine injection. Endocrinology 121(3):924–930
Sara SJ, Bouret S (2012) Orienting and reorienting: the locus coeruleus mediates cognition through arousal. Neuron 76(1):130–141
Sara SJ (2009) The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci 10(3):211–223
Takahashi K, Kayama Y, Lin JS, Sakai K (2010) Locus coeruleus neuronal activity during the sleep-waking cycle in mice. Neuroscience 169(3):1115–1126
McGregor R, Siegel JM (2010) Illuminating the locus coeruleus: control of posture and arousal. Nat Neurosci 13(12):1448–1449
Nance DM, Sanders VM (2007) Autonomic innervation and regulation of the immune system (1987–2007). Brain Behav Immun 21(6):736–745
Kohm AP, Sanders VM (2001) Norepinephrine and beta 2-adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacol Rev 53(4):487–525
Kawashima K, Fujii T, Moriwaki Y, Misawa H (2012) Critical roles of acetylcholine and the muscarinic and nicotinic acetylcholine receptors in the regulation of immune function. Life Sci 91(21–22):1027–1032
Sanders VM, Straub RH (2002) Norepinephrine, the beta-adrenergic receptor, and immunity. Brain Behav Immun 16(4):290–332
Giorelli M, Livrea P, Trojano M (2004) Post-receptorial mechanisms underlie functional disregulation of beta2-adrenergic receptors in lymphocytes from multiple sclerosis patients. J Neuroimmunol 155(1–2):143–149
Anlar B, Karaszewski JW, Reder AT, Arnason BG (1992) Increased muscarinic cholinergic receptor density on CD4+ lymphocytes in progressive multiple sclerosis. J Neuroimmunol 36(2–3):171–177
Zoukos Y, Thomaides TN, Kidd D, Cuzner ML, Thompson A (2003) Expression of beta2 adrenoreceptors on peripheral blood mononuclear cells in patients with primary and secondary progressive multiple sclerosis: a longitudinal six month study. J Neurol Neurosurg Psychiatry 74(2):197–202
Thayer JF, Hansen AL, Saus-Rose E, Johnsen BH (2009) Heart rate variability, prefrontal neural function, and cognitive performance: the neurovisceral integration perspective on self-regulation, adaptation, and health. Ann Behav Med: Publ Soc Behav Med 37(2):141–153
Thayer JF, Lane RD (2000) A model of neurovisceral integration in emotion regulation and dysregulation. J Affect Disord 61(3):201–216
Kamath MV, Fallen EL (1993) Power spectral analysis of heart rate variability: a noninvasive signature of cardiac autonomic function. Crit Rev Biomed Eng 21(3):245–311
Malik M, Camm AJ (1993) Components of heart rate variability—what they really mean and what we really measure. Am J Cardiol 72(11):821–822
Sternberg Z (2012) Sympathetic nervous system dysfunction in multiple sclerosis, linking neurodegeneration to a reduced response to therapy. Curr Pharm Des 18(12):1635–1644
Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES (2000) The sympathetic nerve—an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev 52(4):595–638
Laureys G, Clinckers R, Gerlo S, Spooren A, Wilczak N, Kooijman R et al (2010) Astrocytic beta(2)-adrenergic receptors: from physiology to pathology. Prog Neurobiol 91(3):189–199
Zarrindast MR, Khodjastehfar E, Oryan S, Torkaman-Boutorabi A (2001) Baclofen-impairment of memory retention in rats: possible interaction with adrenoceptor mechanism(s). Eur J Pharmacol 411(3):283–288
Amedei A, Prisco D, D’Elios MM (2012) Multiple Sclerosis: the role of cytokines in pathogenesis and in therapies. Int J Mol Sci 13(10):13438-13460. doi:10.3390/ijms131013438
Rajda C, Bencsik K, Vecsei LL, Bergquist J (2002) Catecholamine levels in peripheral blood lymphocytes from multiple sclerosis patients. J Neuroimmunol 124(1–2):93–100
Karaszewski JW, Reder AT, Anlar B, Kim WC, Arnason BG (1991) Increased lymphocyte beta-adrenergic receptor density in progressive multiple sclerosis is specific for the CD8+, CD28- suppressor cell. Ann Neurol 30(1):42–47
Zeinstra E, Wilczak N, De Keyser J (2000) [3H]dihydroalprenolol binding to beta adrenergic receptors in multiple sclerosis brain. Neurosci Lett 289(1):75–77
Zoukos Y, Kidd D, Woodroofe MN, Kendall BE, Thompson AJ, Cuzner ML (1994) Increased expression of high affinity IL-2 receptors and beta-adrenoceptors on peripheral blood mononuclear cells is associated with clinical and MRI activity in multiple sclerosis. Brain: J Neurol 117(Pt 2):307–315
Szelenyi J, Kiss JP, Vizi ES (2000) Differential involvement of sympathetic nervous system and immune system in the modulation of TNF-alpha production by alpha2- and beta-adrenoceptors in mice. J Neuroimmunol 103(1):34–40
Marien MR, Colpaert FC, Rosenquist AC (2004) Noradrenergic mechanisms in neurodegenerative diseases: a theory. Brain Res Brain Res Rev 45(1):38–78
Shetty DN, Pathak SS (2002) Correlation between plasma neurotransmitters and memory loss in pregnancy. J Reprod Med 47(6):494–496
Aune B, Vartun A, Oian P, Sager G (2000) Evidence of dysfunctional beta2-adrenoceptor signal system in pre-eclampsia. BJOG: Int J Obstet Gynaecol 107(1):116–121
Tschada R, Hettenbach A, Melchert F, Alken P (1991) Pregnancy-induced changes in adrenergic receptors. Z Geburtshilfe Perinatol 195(4):182–186
Puri BK, Bydder GM, Chaudhuri KR, Al Saffar BY, Curati WL, White SJ et al (2001) MRI changes in multiple sclerosis following treatment with lofepramine and L-phenylalanine. Neuroreport 12(9):1821–1824
Simonini MV, Polak PE, Sharp A, McGuire S, Galea E, Feinstein DL (2010) Increasing CNS noradrenaline reduces EAE severity. J Neuroimmune Pharm: Off J Soc NeuroImmune Pharmacol 5(2):252–259
Shah AJ, Su S, Veledar E, Bremner JD, Goldstein FC, Lampert R et al (2011) Is heart rate variability related to memory performance in middle-aged men? Psychosom Med 73(6):475–482
Egea J, Buendia I, Parada E, Navarro E, Leon R, Lopez MG (2015) Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol
Kawamata J, Suzuki S, Shimohama S (2011) Enhancement of nicotinic receptors alleviates cytotoxicity in neurological disease models. Ther Adv Chron Dis 2(3):197–208
Meyer PKK, Thomae E, Becker G, Schildan A, Patt M, Lobsien D, Hesse S, Then Bergh F et al (2010) Reduced cortical {alpha}4β2* nicotinic acetylcholine receptor binding and its relationship to impaired memory and attention in early stage multiple sclerosis: a 2-[18F]F-A-85380 (2FA) PET study. Abstr-J Nucl Med 51(Suppl 2):1792
Panitch H, Miller A, Paty D, Weinshenker B, North American Study Group on Interferon beta-1b in Secondary Progressive MS (2004) Interferon beta-1b in secondary progressive MS: results from a 3-year controlled study. Neurology 63(10):1788–1795
Krakauer M, Sorensen PS, Khademi M, Olsson T, Sellebjerg F (2006) Dynamic T-lymphocyte chemokine receptor expression induced by interferon-beta therapy in multiple sclerosis. Scand J Immunol 64(2):155–163
Sanders VM, Baker RA, Ramer-Quinn DS, Kasprowicz DJ, Fuchs BA, Street NE (1997) Differential expression of the beta2-adrenergic receptor by Th1 and Th2 clones: implications for cytokine production and B cell help. J Immunol 158(9):4200–4210
Esposito F, Radaelli M, Martinelli V, Sormani MP, Martinelli Boneschi F, Moiola L et al (2010) Comparative study of mitoxantrone efficacy profile in patients with relapsing-remitting and secondary progressive multiple sclerosis. Mult Scler 16(12):1490–1499
Villoslada P, Oksenberg JR, Rio J, Montalban X (2004) Clinical characteristics of responders to interferon therapy for relapsing MS. Neurology 62(9):1653, author reply
Waubant E, Vukusic S, Gignoux L, Dubief FD, Achiti I, Blanc S et al (2003) Clinical characteristics of responders to interferon therapy for relapsing MS. Neurology 61(2):184–189
Gunal DI, Afsar N, Tanridag T, Aktan S (2002) Autonomic dysfunction in multiple sclerosis: correlation with disease-related parameters. Eur Neurol 48(1):1–5
Liao D, Barnes RW, Chambless LE, Simpson RJ Jr, Sorlie P, Heiss G (1995) Age, race, and sex differences in autonomic cardiac function measured by spectral analysis of heart rate variability—the ARIC study. Atherosclerosis risk in communities. Am J Cardiol 76(12):906–912
Rio J, Nos C, Tintore M, Tellez N, Galan I, Pelayo R et al (2006) Defining the response to interferon-beta in relapsing-remitting multiple sclerosis patients. Ann Neurol 59(2):344–352
Zaffaroni M, Marino F, Bombelli R, Rasini E, Monti M, Ferrari M et al (2008) Therapy with interferon-beta modulates endogenous catecholamines in lymphocytes of patients with multiple sclerosis. Exp Neurol 214(2):315–321
Hamamcioglu K, Reder AT (2007) Interferon-beta regulates cytokines and BDNF: greater effect in relapsing than in progressive multiple sclerosis. Mult Scler 13(4):459–470
Khoury SJ, Healy BC, Kivisakk P, Viglietta V, Egorova S, Guttmann CR et al (2010) A randomized controlled double-masked trial of albuterol add-on therapy in patients with multiple sclerosis. Arch Neurol 67(9):1055–1061
Minden SL, Schiffer RB (1990) Affective disorders in multiple sclerosis. Review and recommendations for clinical research. Arch Neurol 47(1):98–104
Lambert G, Johansson M, Agren H, Friberg P (2000) Reduced brain norepinephrine and dopamine release in treatment-refractory depressive illness: evidence in support of the catecholamine hypothesis of mood disorders. Arch Gen Psychiatry 57(8):787–793
Srivastava N, Barthwal MK, Dalal PK, Agarwal AK, Nag D, Seth PK et al (2002) A study on nitric oxide, beta-adrenergic receptors and antioxidant status in the polymorphonuclear leukocytes from the patients of depression. J Affect Disord 72(1):45–52
Mann JJ, Brown RP, Halper JP, Sweeney JA, Kocsis JH, Stokes PE et al (1985) Reduced sensitivity of lymphocyte beta-adrenergic receptors in patients with endogenous depression and psychomotor agitation. N Engl J Med 313(12):715–720
Yang AC, Tsai SJ, Yang CH, Kuo CH, Chen TJ, Hong CJ (2011) Reduced physiologic complexity is associated with poor sleep in patients with major depression and primary insomnia. J Affect Disord 131(1–3):179–185
RS D (1999) The neurochemistry of mood disorders: preclinical studies. New York: Oxford University Press. In Charney DS, Nestler EJ, Bunney BS, editors. (Neurobiology of mental illness): 333–47
O’Reardon JP, Cristancho P, Peshek AD (2006) Vagus nerve stimulation (VNS) and treatment of depression: to the brainstem and beyond. Psychiatry 3(5):54–63
Follesa P, Biggio F, Gorini G, Caria S, Talani G, Dazzi L et al (2007) Vagus nerve stimulation increases norepinephrine concentration and the gene expression of BDNF and bFGF in the rat brain. Brain Res 1179:28–34
Marrosu F, Maleci A, Cocco E, Puligheddu M, Barberini L, Marrosu MG (2007) Vagal nerve stimulation improves cerebellar tremor and dysphagia in multiple sclerosis. Mult Scler 13(9):1200–1202
Lang UE, Hellweg R, Gallinat J (2004) BDNF serum concentrations in healthy volunteers are associated with depression-related personality traits. Neuropsychopharmacol: Off Publ Am Coll Neuropsychopharmacol 29(4):795–798
Comini-Frota ER, Rodrigues DH, Miranda EC, Brum DG, Kaimen-Maciel DR, Donadi EA et al (2012) Serum levels of brain-derived neurotrophic factor correlate with the number of T2 MRI lesions in multiple sclerosis. Braz J Med Biol Res = Rev Bras Pesquisas Med Biol / Soc Bras Biofisica [et al] 45(1):68–71
Shimizu E, Hashimoto K, Okamura N, Koike K, Komatsu N, Kumakiri C et al (2003) Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol Psychiatry 54(1):70–75
Stadelmann C, Kerschensteiner M, Misgeld T, Bruck W, Hohlfeld R, Lassmann H (2002) BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells? Brain: J Neurol 125(Pt 1):75–85
Chen MJ, Nguyen TV, Pike CJ, Russo-Neustadt AA (2007) Norepinephrine induces BDNF and activates the PI-3K and MAPK cascades in embryonic hippocampal neurons. Cell Signal 19(1):114–128
Evans JM, Ziegler MG, Patwardhan AR, Ott JB, Kim CS, Leonelli FM et al (2001) Gender differences in autonomic cardiovascular regulation: spectral, hormonal, and hemodynamic indexes. J Appl Physiol 91(6):2611–2618
Jones KH, Ford DV, Jones PA, John A, Middleton RM, Lockhart-Jones H et al (2012) A large-scale study of anxiety and depression in people with multiple sclerosis: a survey via the web portal of the UK MS Register. PLoS One 7(7), e41910
Beal CC, Stuifbergen AK, Brown A (2007) Depression in multiple sclerosis: a longitudinal analysis. Arch Psychiatr Nurs 21(4):181–191
Feinstein A, Roy P, Lobaugh N, Feinstein K, O’Connor P, Black S (2004) Structural brain abnormalities in multiple sclerosis patients with major depression. Neurology 62(4):586–590
Feinstein A, O’Connor P, Akbar N, Moradzadeh L, Scott CJ, Lobaugh NJ (2010) Diffusion tensor imaging abnormalities in depressed multiple sclerosis patients. Mult Scler 16(2):189–196
Kiy G, Lehmann P, Hahn HK, Eling P, Kastrup A, Hildebrandt H (2011) Decreased hippocampal volume, indirectly measured, is associated with depressive symptoms and consolidation deficits in multiple sclerosis. Mult Scler 17(9):1088–1097
Arnett PA, Higginson CI, Voss WD, Wright B, Bender WI, Wurst JM et al (1999) Depressed mood in multiple sclerosis: relationship to capacity-demanding memory and attentional functioning. Neuropsychology 13(3):434–446
Giovannoni G (2006) Multiple sclerosis related fatigue. J Neurol Neurosurg Psychiatry 77(1):2–3
Putzki N, Katsarava Z, Vago S, Diener HC, Limmroth V (2008) Prevalence and severity of multiple-sclerosis-associated fatigue in treated and untreated patients. Eur Neurol 59(3–4):136–142
Pittion-Vouyovitch S, Debouverie M, Guillemin F, Vandenberghe N, Anxionnat R, Vespignani H (2006) Fatigue in multiple sclerosis is related to disability, depression and quality of life. J Neurol Sci 243(1–2):39–45
Koch M, Mostert J, Heerings M, Uyttenboogaart M, De Keyser J (2009) Fatigue, depression and disability accumulation in multiple sclerosis: a cross-sectional study. Eur J Neurol: Off J Eur Fed Neurol Soc 16(3):348–352
Fejelstad C, Brittian D, Fejelstad AS, Pardo G (2010) Fatigue and thermo sensitivity affect physical activity in multiple sclerosis. J Appl Res 10(3):108–115
Rotstein D, O’Connor P, Lee L, Murray BJ (2012) Multiple sclerosis fatigue is associated with reduced psychomotor vigilance. Can J Neurol Sci Le J Can Sci Neurol 39(2):180–184
Fernandez-Munoz JJ, Moron-Verdasco A, Cigaran-Mendez M, Munoz-Hellin E, Perez-de-Heredia-Torres M, Fernandez-de-Las-Penas C (2015) Disability, quality of life, personality, cognitive and psychological variables associated with fatigue in patients with multiple sclerosis. Acta Neurol Scand
Flachenecker P, Kumpfel T, Kallmann B, Gottschalk M, Grauer O, Rieckmann P et al (2002) Fatigue in multiple sclerosis: a comparison of different rating scales and correlation to clinical parameters. Mult Scler 8(6):523–526
Lebre AT, Mendes MF, Tilbery CP, Almeida AL, Scatolini NA (2007) Relation between fatigue and autonomic disturbances in multiple sclerosis. Arq Neuropsiquiatr 65(3A):663–668
Yu F, Bilberg A, Dalgas U, Stenager E (2013) Fatigued patients with multiple sclerosis can be discriminated from healthy controls by the recordings of a newly developed measurement system (FAMOS): a pilot study. Disabil Rehabil Assist Technol 8(1):77–83
Flachenecker P, Rufer A, Bihler I, Hippel C, Reiners K, Toyka KV et al (2003) Fatigue in MS is related to sympathetic vasomotor dysfunction. Neurology 61(6):851–853
Strober LB, Arnett PA (2005) An examination of four models predicting fatigue in multiple sclerosis. Arch Clin Neuropsychol: Off J Ntnl Acad Neuropsychol 20(5):631–646
Lobentanz IS, Asenbaum S, Vass K, Sauter C, Klosch G, Kollegger H et al (2004) Factors influencing quality of life in multiple sclerosis patients: disability, depressive mood, fatigue and sleep quality. Acta Neurol Scand 110(1):6–13
Attarian HP, Brown KM, Duntley SP, Carter JD, Cross AH (2004) The relationship of sleep disturbances and fatigue in multiple sclerosis. Arch Neurol 61(4):525–528
Ferini-Strambi L, Rovaris M, Oldani A, Martinelli V, Filippi M, Smirne S et al (1995) Cardiac autonomic function during sleep and wakefulness in multiple sclerosis. J Neurol 242(10):639–643
Tedeschi G, Dinacci D, Lavorgna L, Prinster A, Savettieri G, Quattrone A et al (2007) Correlation between fatigue and brain atrophy and lesion load in multiple sclerosis patients independent of disability. J Neurol Sci 263(1–2):15–19
Calabrese M, Rinaldi F, Grossi P, Mattisi I, Bernardi V, Favaretto A et al (2010) Basal ganglia and frontal/parietal cortical atrophy is associated with fatigue in relapsing-remitting multiple sclerosis. Mult Scler 16(10):1220–1228
Acevedo AR, Nava C, Arriada N, Violante A, Corona T (2000) Cardiovascular dysfunction in multiple sclerosis. Acta Neurol Scand 101(2):85–88
Jones DE, Hollingsworth K, Fattakhova G, MacGowan G, Taylor R, Blamire A et al (2010) Impaired cardiovascular function in primary biliary cirrhosis. Am J Physiol Gastrointest Liver Physiol 298(5):G764–G773
Nakamura T, Hirayama M, Hara T, Hama T, Watanabe H, Sobue G (2011) Does cardiovascular autonomic dysfunction contribute to fatigue in Parkinson’s disease? Mov Disord: Off J Mov Disord Soc 26(10):1869–1874
D’Amico D, La Mantia L, Rigamonti A, Usai S, Mascoli N, Milanese C et al (2004) Prevalence of primary headaches in people with multiple sclerosis. Cephalalgia: Int J Headache 24(11):980–984
Mohrke J, Kropp P, Zettl UK (2013) Headaches in multiple sclerosis patients might imply an inflammatorial process. PLoS One 8(8), e69570
Tabby D, Majeed MH, Youngman B, Wilcox J (2013) Headache in multiple sclerosis: features and implications for disease management. Int J MS Care 15(2):73–80
Villani V, Prosperini L, Ciuffoli A, Pizzolato R, Salvetti M, Pozzilli C et al (2008) Primary headache and multiple sclerosis: preliminary results of a prospective study. Neurol Sci: Off J Ital Neurol Soc Ital Soc Clin Neurophysiol 29(Suppl 1):S146–S148
Kister I, Munger KL, Herbert J, Ascherio A (2012) Increased risk of multiple sclerosis among women with migraine in the Nurses’ Health Study II. Mult Scler 18(1):90–97
Gee JR, Chang J, Dublin AB, Vijayan N (2005) The association of brainstem lesions with migraine-like headache: an imaging study of multiple sclerosis. Headache 45(6):670–677
Khromov A, Segal M, Nissinoff J, Fast A (2005) Migraines linked to interferon-beta treatment of multiple sclerosis. Am J Phys Med Rehabil Assoc Acad Physiatrists 84(8):644–647
Goadsby PJ (2013) Autonomic nervous system control of the cerebral circulation. Handb Clin Neurol 117:193–201
Takeshima T, Takao Y, Takahashi K (1987) Pupillary sympathetic hypofunction and asymmetry in muscle contraction headache and migraine. Cephalalgia: Int J Headache 7(4):257–262
Mikamo K, Takeshima T, Takahashi K (1989) Cardiovascular sympathetic hypofunction in muscle contraction headache and migraine. Headache 29(2):86–89
Takeshima T, Takao Y, Urakami K, Nishikawa S, Takahashi K (1989) Muscle contraction headache and migraine. Platelet activation and plasma norepinephrine during the cold pressor test. Cephalalgia: Int J Headache 9(1):7–13
Gotoh F, Komatsumoto S, Araki N, Gomi S (1984) Noradrenergic nervous activity in migraine. Arch Neurol 41(9):951–955
Gass JJ, Glaros AG (2013) Autonomic dysregulation in headache patients. Appl Psychophysiol Biofeedback 38(4):257–263
Tabata M, Takeshima T, Burioka N, Nomura T, Ishizaki K, Mori N et al (2000) Cosinor analysis of heart rate variability in ambulatory migraineurs. Headache 40(6):457–463
Pogacnik T, Sega S, Pecnik B, Kiauta T (1993) Autonomic function testing in patients with migraine. Headache 33(10):545–550
Rubin LS, Graham D, Pasker R, Calhoun W (1985) Autonomic nervous system dysfunction in common migraine. Headache 25(1):40–48
Peroutka SJ (2004) Migraine: a chronic sympathetic nervous system disorder. Headache 44(1):53–64
Yilmaz N, Karaali K, Ozdem S, Turkay M, Unal A, Dora B (2011) Elevated S100B and neuron specific enolase levels in patients with migraine-without aura: evidence for neurodegeneration? Cell Mol Neurobiol 31(4):579–585
Zamboni P, Galeotti R, Menegatti E, Malagoni AM, Tacconi G, Dall’Ara S et al (2009) Chronic cerebrospinal venous insufficiency in patients with multiple sclerosis. J Neurol Neurosurg Psychiatry 80(4):392–399
Zamboni P (2006) The big idea: iron-dependent inflammation in venous disease and proposed parallels in multiple sclerosis. J R Soc Med 99(11):589–593
Worthington V, Killestein J, Eikelenboom MJ, Teunissen CE, Barkhof F, Polman CH et al (2010) Normal CSF ferritin levels in MS suggest against etiologic role of chronic venous insufficiency. Neurology 75(18):1617–1622
Joyner MJ, Charkoudian N, Wallin BG (2008) A sympathetic view of the sympathetic nervous system and human blood pressure regulation. Exp Physiol 93(6):715–724
Sternberg Z, Grewal P, Cen S, Debarge-Igoe F, Yu J, Arata M (2013) Blood pressure normalization post-jugular venous balloon angioplasty. Phlebol Venous Forum R Soc Med
Zivadinov R, Marr K, Cutter G, Ramanathan M, Benedict RH, Kennedy C et al (2011) Prevalence, sensitivity, and specificity of chronic cerebrospinal venous insufficiency in MS. Neurology 77(2):138–144
Dolic K, Weinstock-Guttman B, Marr K, Valnarov V, Carl E, Hagemeier J et al (2011) Risk factors for chronic cerebrospinal venous insufficiency (CCSVI) in a large cohort of volunteers. PLoS One 6(11), e28062
Reis DJ, Golanov EV (1997) Autonomic and vasomotor regulation. Int Rev Neurobiol 41:121–149
Meyer-Schwickerath R, Haug C, Hacker A, Fink F, Seidel D, Hartung HP et al (2011) Intracranial venous pressure is normal in patients with multiple sclerosis. Mult Scler 17(5):637–638
De Keyser J, Steen C, Mostert JP, Koch MW (2008) Hypoperfusion of the cerebral white matter in multiple sclerosis: possible mechanisms and pathophysiological significance. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab 28(10):1645–1651
Inglese M, Park SJ, Johnson G, Babb JS, Miles L, Jaggi H et al (2007) Deep gray matter perfusion in multiple sclerosis: dynamic susceptibility contrast perfusion magnetic resonance imaging at 3 T. Arch Neurol 64(2):196–202
Zamboni P, Menegatti E, Weinstock-Guttman B, Dwyer MG, Schirda CV, Malagoni AM et al (2011) Hypoperfusion of brain parenchyma is associated with the severity of chronic cerebrospinal venous insufficiency in patients with multiple sclerosis: a cross-sectional preliminary report. BMC Med 9:22
Inglese M, Adhya S, Johnson G, Babb JS, Miles L, Jaggi H et al (2008) Perfusion magnetic resonance imaging correlates of neuropsychological impairment in multiple sclerosis. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab 28(1):164–171
Mollanji R, Bozanovic-Sosic R, Zakharov A, Makarian L, Johnston MG (2002) Blocking cerebrospinal fluid absorption through the cribriform plate increases resting intracranial pressure. Am J Physiol Regul Integr Comp Physiol 282(6):R1593–R1599
Law M, Saindane AM, Ge Y, Babb JS, Johnson G, Mannon LJ et al (2004) Microvascular abnormality in relapsing-remitting multiple sclerosis: perfusion MR imaging findings in normal-appearing white matter. Radiology 231(3):645–652
Adhya S, Johnson G, Herbert J, Jaggi H, Babb JS, Grossman RI et al (2006) Pattern of hemodynamic impairment in multiple sclerosis: dynamic susceptibility contrast perfusion MR imaging at 3.0 T. NeuroImage 33(4):1029–1035
Schaller B, Graf R (2005) Different compartments of intracranial pressure and its relationship to cerebral blood flow. J Traumatol 59(6):1521–1531
Thibault P, Lewis W, Niblett S (2015) Objective duplex ultrasound evaluation of the extracranial circulation in multiple sclerosis patients undergoing venoplasty of internal jugular vein stenoses: a pilot study. Phlebol Venous Forum R Soc Med 30(2):98–104
Winklewski PJ, Frydrychowski AF (2013) Cerebral blood flow, sympathetic nerve activity and stroke risk in obstructive sleep apnoea. Is there a direct link? Blood Press 22(1):27–33
Cui J, McQuillan PM, Blaha C, Kunselman AR, Sinoway LI (2012) Limb venous distension evokes sympathetic activation via stimulation of the limb afferents in humans. Am J Physiol Heart Circ Physiol 303(4):H457–H463
Wuerfel J, Bellmann-Strobl J, Brunecker P, Aktas O, McFarland H, Villringer A et al (2004) Changes in cerebral perfusion precede plaque formation in multiple sclerosis: a longitudinal perfusion MRI study. Brain: J Neurol 127(Pt 1):111–119
Mezei Z, Olah L, Kardos L, Kovacs RK, Csiba L, Csepany T (2013) Cerebrovascular hemodynamic changes in multiple sclerosis patients during head-up tilt table test: effect of high-dose intravenous steroid treatment. J Neurol 260(9):2335–2342
Zhang R, Zuckerman JH, Iwasaki K, Wilson TE, Crandall CG, Levine BD (2002) Autonomic neural control of dynamic cerebral autoregulation in humans. Circulation 106(14):1814–1820
Azevedo E, Castro P, Santos R, Freitas J, Coelho T, Rosengarten B et al (2011) Autonomic dysfunction affects cerebral neurovascular coupling. Clin Aut Res: Off J Clin Aut Res Soc 21(6):395–403
Yuceyar N, Taskiran D, Sagduyu A (2001) Serum and cerebrospinal fluid nitrite and nitrate levels in relapsing-remitting and secondary progressive multiple sclerosis patients. Clin Neurol Neurosurg 103(4):206–211
Czosnyka M, Smielewski P, Piechnik S, Al-Rawi PG, Kirkpatrick PJ, Matta BF et al (1999) Critical closing pressure in cerebrovascular circulation. J Neurol Neurosurg Psychiatry 66(5):606–611
Ursino M, Lodi CA (1998) Interaction among autoregulation, CO2 reactivity, and intracranial pressure: a mathematical model. Am J Physiol 274(5 Pt 2):H1715–H1728
Zamboni P, Galeotti R, Menegatti E, Malagoni AM, Gianesini S, Bartolomei I et al (2009) A prospective open-label study of endovascular treatment of chronic cerebrospinal venous insufficiency. J Vasc Surg 50(6):1348–58.e1-3
Gisolf J, van Lieshout JJ, van Heusden K, Pott F, Stok WJ, Karemaker JM (2004) Human cerebral venous outflow pathway depends on posture and central venous pressure. J Physiol 560(Pt 1):317–327
Zamboni P, Menegatti E, Weinstock-Guttman B, Schirda C, Cox JL, Malagoni AM et al (2009) The severity of chronic cerebrospinal venous insufficiency in patients with multiple sclerosis is related to altered cerebrospinal fluid dynamics. Funct Neurol 24(3):133–138
ElSankari S, Baledent O, van Pesch V, Sindic C, de Broqueville Q, Duprez T (2013) Concomitant analysis of arterial, venous, and CSF flows using phase-contrast MRI: a quantitative comparison between MS patients and healthy controls. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab 33(9):1314–1321
Bateman GA, Levi CR, Schofield P, Wang Y, Lovett EC (2008) The venous manifestations of pulse wave encephalopathy: windkessel dysfunction in normal aging and senile dementia. Neuroradiology 50(6):491–497
Whedon JM, Glassey D (2009) Cerebrospinal fluid stasis and its clinical significance. Altern Ther Health Med 15(3):54–60
Zivadinov R, Magnano C, Galeotti R, Schirda C, Menegatti E, Weinstock-Guttman B et al (2013) Changes of cine cerebrospinal fluid dynamics in patients with multiple sclerosis treated with percutaneous transluminal angioplasty: a case–control study. J Vascular Int Radiol: JVIR 24(6):829–838
Aboul-Enein F, Rauschka H, Kornek B, Stadelmann C, Stefferl A, Bruck W et al (2003) Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol 62(1):25–33
Dux E, Temesvari P, Joo F, Adam G, Clementi F, Dux L et al (1984) The blood–brain barrier in hypoxia: ultrastructural aspects and adenylate cyclase activity of brain capillaries. Neuroscience 12(3):951–958
Jelinski SE, Yager JY, Juurlink BH (1999) Preferential injury of oligodendroblasts by a short hypoxic-ischemic insult. Brain Res 815(1):150–153
Reimer MM, McQueen J, Searcy L, Scullion G, Zonta B, Desmazieres A et al (2011) Rapid disruption of axon-glial integrity in response to mild cerebral hypoperfusion. J Neurosci: Off J Soc Neurosci 31(49):18185–18194
Farkas E, Donka G, de Vos RA, Mihaly A, Bari F, Luiten PG (2004) Experimental cerebral hypoperfusion induces white matter injury and microglial activation in the rat brain. Acta Neuropathol 108(1):57–64
Schaller B, Cornelius JF, Sandu N, Ottaviani G, Perez-Pinzon MA (2009) Oxygen-conserving reflexes of the brain: the current molecular knowledge. J Cell Mol Med 13(4):644–647
Sandu N, Cornelius J, Filis A, Nothen C, Rasper J, Kulinsky VI et al (2010) Cerebral hemodynamic changes during the trigeminocardiac reflex: description of a new animal model protocol. TheScientificWorldJOURNAL 10:1416–1423
Lee BB, Baumgartner I, Berlien P, Bianchini G, Burrows P, Gloviczki P et al (2014) Diagnosis and treatment of venous malformations consensus document of the International union of Phlebology (IUP): updated 2013. Int Angiol: J Int Union Angiol
Pascolo L, Gianoncelli A, Rizzardi C, Tisato V, Salome M, Calligaro C et al (2014) Calcium micro-depositions in jugular truncular venous malformations revealed by synchrotron-based XRF imaging. Sci Rep 4:6540
Zamboni P, Tisato V, Menegatti E, Mascoli F, Gianesini S, Salvi F et al (2014) Ultrastructure of internal jugular vein defective valves. Phlebol Venous Forum R Soc Med
Coen M, Menegatti E, Salvi F, Mascoli F, Zamboni P, Gabbiani G et al (2013) Altered collagen expression in jugular veins in multiple sclerosis. Cardiovasc Pathol: Off J Soc Cardiovasc Pathol 22(1):33–38
Fitts MK, Pike DB, Anderson K, Shiu YT (2014) Hemodynamic shear stress and endothelial dysfunction in hemodialysis access. Open Urol Nephrol J 7(Suppl 1 M5):33–44
Roy-Chaudhury P, Khan R, Campos B, Wang Y, Kurian M, Lee T et al (2014) Pathogenetic role for early focal macrophage infiltration in a pig model of arteriovenous fistula (AVF) stenosis. Journal Vasc Access 15(1):25–28
Schubert A, Cattaruzza M, Hecker M, Darmer D, Holtz J, Morawietz H (2000) Shear stress-dependent regulation of the human beta-tubulin folding cofactor D gene. Circ Res 87(12):1188–1194
Mann MC, Exner DV, Hemmelgarn BR, Sola DY, Turin TC, Ellis L et al (2013) Vitamin D levels are associated with cardiac autonomic activity in healthy humans. Nutrients 5(6):2114–2127
Varbiro S, Sara L, Antal P, Monori-Kiss A, Tokes AM, Monos E et al (2014) Lower-limb veins are thicker and vascular reactivity is decreased in a rat PCOS model: concomitant vitamin D3 treatment partially prevents these changes. Am J Physiol Heart Circ Physiol 307(6):H848–H857
Schaller B (2004) Physiology of cerebral venous blood flow: from experimental data in animals to normal function in humans. Brain Res Brain Res Rev 46(3):243–260
Schaller B, Graf R (2004) Cerebral venous infarction: the pathophysiological concept. Cerebrovasc Dis 18(3):179–188
Roshanisefat H, Bahmanyar S, Hillert J, Olsson T, Montgomery S (2014) Multiple sclerosis clinical course and cardiovascular disease risk—Swedish cohort study. Eur J Neurol: Off J Eur Fed Neurol Soc 21(11):1353–e88
Christiansen CF (2012) Risk of vascular disease in patients with multiple sclerosis: a review. Neurol Res 34(8):746–753
Sung EAL, Cen S, Krug A (2014) Sanossian N. Mult Scler Stroke Neurol 82(10):169
Allen NB, Lichtman JH, Cohen HW, Fang J, Brass LM, Alderman MH (2008) Vascular disease among hospitalized multiple sclerosis patients. Neuroepidemiology 30(4):234–238
Sternberg Z(2015) Promoting sympathovagal balance in multiple sclerosis; pharmacological, non-pharmacological, and surgical strategies. Autoimmunity Rev
Sternberg Z (2015) Genetic, epigenetic, and environmental factors influencing neurovisceral integration of cardiovascular modulation: focus on multiple sclerosis. Neuromol Med
Acknowledgments
The author thanks Prof. Bernhard Schaller for the intellectual input.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The author declares no competing interests.
Source of Funding
None
Rights and permissions
About this article

Cite this article
Sternberg, Z. Impaired Neurovisceral Integration of Cardiovascular Modulation Contributes to Multiple Sclerosis Morbidities. Mol Neurobiol 54, 362–374 (2017). https://doi.org/10.1007/s12035-015-9599-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9599-y