
Abstract
Carnosic acid (CA) and carnosol are the major diterpenes found in Rosmarinus officinalis (rosemary), a culinary spice. CA and carnosol account for over 90 % of its anti-oxidant activity in rosemary leaves. The diterpenes exert anti-oxidant, anti-inflammatory, and anti-carcinogenic activities, and present neuroprotective effects in both in vitro and in vivo experimental models. In some cases, CA exerted protective effects upon neuronal cells more intensely than resveratrol or sulforaphane. Therefore, CA and carnosol demonstrate a potential pharmacological role for rosemary diterpenes in ameliorating mammalian brain redox status, among other parameters, as for instance the modulation of neuroinflammation. The aim of this review is to discuss the biological effects of CA and carnosol on neuronal and glial cells with focus on the mechanism of action of such diterpenes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Carnosic acid (CA; C20H28O4, MW 332.43392 g/mol), an ortho-diphenolic diterpene containing an abietane carbon skeleton, is found in Rosmarinus officinalis (rosemary) and exerts several protective effects on mammalian cells, as demonstrated in both in vitro and in vivo experimental models [1]. CA is an anti-oxidant molecule and possesses anti-inflammatory and anti-carcinogenic properties [2–8].
CA is an ortho-dihydroquinone-type of compound that becomes electrophilic (an ortho-quinone form) when reacts with free radicals produced in biological systems. Therefore, CA is a pro-electrophilic molecule that becomes electrophilic after oxidative conversion to a quinone form, which is able to activate the nuclear factor erythroid 2-related factor 2 (Nrf2) transcription factor, the major regulator of the cellular redox homeostasis in mammalian animals [9]. Moreover, the direct CA anti-oxidant effects associated to its chemical structure (due to two O-phenolic hydroxyl moieties present at C11 and C12—the so-called catechol moiety) were seen in different in vitro experimental models [4, 10]. In this context, CA has been viewed as a potent neuroprotective agent due to its ability to modulate glutathione (GSH) synthesis and to downregulate the levels of neurotrophins (as for instance brain-derived neurotrophic factor (BDNF)) in neuronal cells in experimental model of chemically induced oxidative stress, for example [5, 11–13].
Carnosol (C20H26O4, MW 330.4 g/mol), an ortho-diphenolic diterpene with an abietane carbon skeleton, is found in R. officinalis as a derivative of CA degradation and also presents several protective effects in studies utilizing mammalian cells and experimental animals [1, 14]. Carnosol has been known as a promising anti-inflammatory and anti-carcinogenic agent [14–17]. Additionally, carnosol blocked the redox-dependent inactivation of the human brain muscarinic acetylcholine receptor elicited by an endogenous inhibitor able to induce oxidative damage to biomolecules [18], among other cytoprotective effects that will be discussed in the present work.
CA and carnosol are found in rosemary leaves (approximately 5 % CA more carnosol dry weight) and account for over 90 % of its anti-oxidant activity [2] (Figs. 1, 2, and 3). Furthermore, Tamaki et al. reported that CA and carnosol (5 μM for 24 h) were the most effective rosemary polyphenols (when compared with genkwanin, rosmarinic acid, luteolin, verbenone, or caffeic acid) in causing an increase in total GSH contents, in the activation of transcription, and protecting HT22 neuronal cells against glutamate [19]. CA is present mainly in rosemary aerial tissues related to photosynthesis in the following order: leaves > sepals > petals [1, 20, 21].

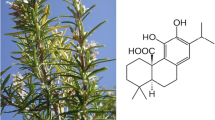
Rosmarinus officinalis L. and the chemical structure of its main anti-oxidant components carnosic acid (CA) and carnosol. CA and carnosol accounts for more than 90 % of the anti-oxidant activity of R. officinalis leaves, as discussed in the text

A summary of the mechanisms by which CA elicits protective effects upon mammalian brain cells. CA exerts anti-oxidant effects by activation of the PI3K/Akt/Nrf2 signaling pathway, leading to increased expression of anti-oxidant enzymes and neuronal differentiation (as assessed through increased neurotrophin production and morphologically through neurite outgrowth visualization). Furthermore, CA upregulates the PI3K/Akt/NF-κB axis culminating in increased expression of phase II enzyme GSTP, as discussed in the text. CA also exhibits anti-inflammatory activity by downregulating the secretion of the pro-inflammatory cytokines IL-1β and IL-6, for example. Moreover, CA reduces NO production in some experimental models dealing with activated microglia. An anti-apoptotic role is mediated through suppressing JNK1/2 and/or p38 phosphorylation and abrogating caspase-3 activation. Please see text for a more detailed information

A graphical abstract summarizing the anti-oxidant and anti-inflammatory effects exhibited by carnosol in different experimental models. Carnosol upregulates the PI3K/Akt/Nrf2 axis by a similar way when compared with CA. Nrf2/ARE activation causes increased expression of the anti-oxidant enzyme HO-1 and augmented levels of the non-enzymatic anti-oxidant GSH in different experimental models. The anti-inflammatory actions elicited by carnosol involve reduced secretion of pro-inflammatory cytokines and decreased NO generation, as well as suppression of PGE2 production. Please, see text for more a detailed information
CA has access to the mammalian brain by penetrating the blood-brain barrier, as previously demonstrated [22]. CA exerts its beneficial effects to both neuronal and glial cells through a myriad of mechanisms, as will be discussed in the present work. Carnosol may also pass through the blood-brain barrier and accumulates in mammalian brain, where it acts as a neuroprotective agent [23]. Doolaege et al. reported that CA bioavailability was 40.1 % after 360 min (CA at 64.3 ± 5.8 mg/kg orally administrated) [24]. The authors found CA in rat plasma, intestine, liver, and muscle. The major route for CA excretion was described as the fecal route. Romo Vaquero et al. found that female Zucker rats that received a rosemary extract enriched in CA (40 %, w/w) presented CA, CA metabolites, CA derivatives, and carnosol in samples obtained from rat plasma, small intestine, caecum, colon, liver, and brain [23]. Authors reported that CA metabolites were detected in the animal samples initially after 25 min administration. In this way, CA and carnosol may be widely distributed in the mammalian organism, eliciting its effects according to its dosage and other conditions, as we will see here.
Therefore, the aim of this work is to discuss about the protective effects exerted by CA and carnosol, two dietary compounds, on neuronal and glial cells regarding its redox environment and inflammation-related factors. Furthermore, the mechanism of action of these polyphenols will be addressed here.
The In Vitro Effects of CA and Carnosol on Neuronal and Glial Cells
The neuroprotective effects of CA and carnosol have been investigated in in vitro experimental models using different cell types in order to examine the mechanism by which such diterpenes elicit such benefits on mammalian brain cells. The complete mechanism still remains to be completely understood but involves the intrinsic anti-oxidant capacity of CA and carnosol and the activation of signaling pathways that modulate anti-oxidant defenses in brain cells. Furthermore, the mechanisms by which CA and carnosol exert its anti-inflammatory actions were partially elucidated. In spite of this, further research is necessary to better understand exactly how these diterpenes protects brain cells from different insults. It is important to note that more studies regarding bioavailability of CA and carnosol are needed in order to verify whether the concentrations of such diterpenes utilized in in vitro experimental models may be reached in vivo.
CA and Neuronal Cells
Satoh et al. observed that CA and a CA-related compound (CAD1; diacetyl form) rescued HT22 neuronal cells from glutamate-induced toxicity in a dose-dependent fashion (0.01–10 μM for 20 h) [25]. In the same work, authors found that CA is much less toxic than carnosol and also presents increased neuroprotective action when compared with its derivatives. The CA derivatives were not effective in protecting primary cortical neurons at the same concentration of CA (3 μM), demonstrating the need for the existence of free carboxylic acid and catechol hydroxyl moieties as found in CA molecule to it exerts neuroprotection. In another study, Satoh et al. demonstrated that CA binds to Keap1 domains (BTB and IVR) and activates the translocation of Nrf2 to the nucleus of COS7 cells [22]. Moreover, CA also did bind to Kelch-like ECH-associated protein 1 (Keap1) in PC12h cells, a dopaminergic cell line, leading to anti-oxidant response element (ARE) activation. The authors also found that CA treatment (10 μM) leaded to increased γ-glutamyl cysteine ligase (γ-GCL) light chain, γ-GCL heavy chain, heme oxygenase-1 (HO-1), and NAD(P)H:quinone oxidoreductase 1 (NQO1) expression (after 6–24 h exposure). Then, CA activated the Nrf2/ARE pathway resulting in increased expression of genes related to phase II. Such enzymes afford protection against oxidative damage and also participate in detoxification reactions [26–28]. To test whether CA would exert a protective role in neuronal cells against oxidative damage, authors treated PC12h cells with CA (0.1–1 μM for 1 h) prior exposure of glutamate (for additional 20 h). CA prevented cell death in a dose-dependent manner by activation of the Nrf2/ARE, since the beneficial effects elicited by CA were suppressed in PC12hD5D cells (with low ARE activity). Interestingly, CA was found to exert a protective effect on PC12h cells to a much greater extent when compared with sulforaphane, a known electrophilic compound that activates Nrf2 potently [29, 30]. In the same work, CA (3 μM) pretreatment (60 min) protected primary immature cortical neurons (that do not express functional glutamate/NMDA receptors yet, but are targets of the glutamate-induced oxidative stress due to inhibition of cysteine influx, an event that lead to decreased synthesis of GSH) against glutamate (2 mM) and rotenone (300 nM) [23]. In a subsequent set of analyses, CA (3 μM) pretreatment (60 min) protected also primary mature cerebrocortical neurons (which express NMDA receptors) against NMDA-induced excitotoxicity by activation of Nrf2. CA accumulated in both neuronal and glial cells, demonstrating its potential to prevent both redox impairments and neuroinflammation. Therefore, CA was effective in protecting both cell lines and primary neurons against different neurotoxic agents that are known to cause deleterious effects, at least in part, by a mechanism involving redox impairment.
Tamaki et al. published similar data demonstrating that CA (10 μM for 24 h) increased the expression of phase II enzymes, including NQO1 and glutathione S-transferase (GST) through a mechanism dependent on the Nrf2/ARE pathway in HT22 neuronal cells (a cell line originated from the hippocampus of mouse) [19]. Authors also found that CA (5 μM) treatment (24 h) increased GSH content and prevented the toxic effects of 5 mM glutamate on such cells in a dose-dependent fashion (from 0.1 to 10 μM, 24 h). Accordingly, Chen et al. reported that CA (0.1–5 μM) pretreatment (12 h) prevented the decrease induced by 6-hydroxydopamine (6-OHDA) in SH-SY5Y neuroblastoma cells viability [31]. CA at 1 μM induced a stronger protective effect when compared with other CA concentrations (0.5 and 3 μM) regarding the ability to inhibit 6-OHDA-induced apoptosis. CA pretreatment decreased the immunocontent of cleaved caspase-3, consequently reducing the levels of cleaved poly-(ADP ribose) polymerase (PARP), a target of active caspase-3 during the apoptotic process. Authors demonstrated that CA prevented the increase in reactive oxygen species (ROS) production and alleviated the effect of 6-OHDA on the phosphorylation of both JNK1/2 and p38, which are protein kinases involved in the induction of apoptosis, among other events. CA increased the content of both γ-glutamylcysteine ligase catalytic subunit (GCLC) and γ-glutamylcysteine ligase modifier subunit (GCLM), leading to augmented GSH levels in a time-dependent manner (from 6 to 24 h). GCLC and GCLM are subunits of γ-glutamylcysteine ligase (γ-GCL), the rate-limiting enzyme in the synthesis of GSH [32, 33]. Importantly, CA induced an early activation of Nrf2 (reaching a peak at 1 h after CA administration), which strongly correlated with increased ARE binding activity. Treatment of SH-SY5Y cells with l-buthionine sulfoximine (BSO) abolished the beneficial effects of CA counteracting 6-OHDA-triggered oxidative damage. BSO is an inhibitor of GSH synthesis and such results demonstrate a major role for GSH metabolism in mediating the protective effects of CA on SH-SY5Y cells. Then, CA probably upregulated GSH metabolism by activating the Nrf2/ARE pathway, causing protection of SH-SY5Y dopaminergic cells against 6-OHDA-induced toxicity. In the work by Lin et al., CA (1 μM) pretreatment (18 h) prevented the toxic effects elicited by 6-OHDA on SH-SY5Y neuroblastoma cells by activating phosphatidylinositol 3-kinase (PI3K), protein kinase B (PKB, also known as Akt), and the transcription factor called nuclear factor κ B (NF-κB) resulting in increased expression and activity of the Pi form of GST (GSTP) [34]. The effects of CA were suppressed by the administration of LY294002, an inhibitor of the PI3K/Akt pathway. CA did not activate c-Jun N-terminal kinase (JNK1/2), extracellular signal-regulated kinases (ERK1/2), or p38 kinases, which are regulatory proteins that play a role in activating NF-κB, as published elsewhere [35]. Therefore, CA protected SH-SY5Y by activating the PI3K/Akt/NF-κB signaling pathway leading to increased GSTP expression and activity and ameliorating cell survival due to detoxifying role of GST against 6-OHDA. Interestingly, authors also reported that CA treatment (20 mg/kg body weight orally three times a week during 3 weeks) restored GSTP levels also in vivo in an experimental model of hemi-parkinsonism induced by 6-OHDA injection in the striatum of Wistar rats [34]. GSTP (which is a GSH consuming enzyme) plays a role counteracting the toxic effects elicited by other neurotoxins, as for instance 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), as previously reported [36, 37]. Then, CA regulates GSH metabolism in neuronal cells either directly or indirectly by modulating different signaling pathways resulting in neuronal protection against chemical stressors.
Hou et al. found that a treatment with CA (0.1–1 μM) prevented the deleterious effects induced by hypoxia (for 1 h) in PC12 cells by inhibiting the phosphorylation of MAPK, as for example, JNK1/2, ERK1/2, and p38 [38]. CA exerted a concentration-dependent effect downregulating the expression of cyclooxygenase-2 (COX-2), a pro-inflammatory enzyme, in PC12 cell line. Consequently, CA led to decreased prostaglandin E2 (PGE2) production in hypoxic cells. CA (1 μM) also exerted anti-oxidant effects in hypoxic PC12 cells by decreasing ROS production and the levels of lipid peroxidation. Additionally, CA upregulated SOD activity, which was decreased by hypoxia induction. CA modulated the intracellular levels of calcium ions (Ca2+) in PC12 cells, decreasing it and protecting cells from Ca2+-induced injury. CA prevented apoptosis in PC12 cells by modulating the pro-apoptotic signaling and by suppressing deleterious alterations in the redox environment elicited by hypoxia. COX-2 inhibition is an important step towards redox maintenance, since COX-2 is able to produce reactive molecules that may impair redox homeostasis in neuronal and glial cells [39]. Furthermore, PGE2 upregulates the pro-inflammatory signal and favor redox disturbances perpetuation due to microglial activation [40–42]. Indeed, authors found that CA at 5 μM (but not 1 or 2 μM) prevented nitric oxide (NO) production and interleukin (IL)-1β and IL-6 secretion in stimulated BV-2 microglial cells [38]. Then, CA protected neuronal and glial cells by regulation of both redox and immune aspects.
Wu et al. found that CA pretreatment (1 μM for 8 h) counteracted the effects of 6-OHDA on SH-SY5Y neuroblastoma cells by upregulating the expression of the anti-oxidant enzymes GCLC and GCLM subunits, glutathione reductase (GR), and superoxide dismutase (SOD) [43]. As mentioned above, γ-GCL is the rate-limiting enzyme in GSH synthesis, and GR is important to reduce GSSH converting it to two molecules of GSH by utilizing NADPH [32, 33]. Therefore, CA improved the cellular ability to produce and to recycle GSH, the major non-enzymatic anti-oxidant in mammalian cells. Additionally, SOD is the enzyme that converts superoxide anion radical (O2 −·) to H2O2, decreasing the chance of that free radical to react with biomolecules [44]. Furthermore, CA exerted an anti-apoptotic effect on SH-SY5Y cells by decreasing caspase-3 activation and, consequently, reducing PARP cleavage. The exact link between the redox protection elicited by CA and its ability to inhibit neuroblastoma cells apoptosis was not investigated in that work. However, it is possible that CA inhibited apoptosis by modulating the redox environment through Nrf2 activation, which has been associated to anti-apoptotic effects [22, 31, 45].
A different mechanism by which CA was demonstrated to elicit protective effects on neuronal cells was investigated in the work by El Omri et al. [46]. The authors showed that CA (20 μM for 6–12 h) downregulated the expression of proteins associated to stress response, namely heat shock protein 90α (Hsp90), transitional endoplasmic reticulum ATPase (VCP/p97), nucleoside diphosphate kinase (NDK), and hypoxia upregulated protein 1 (HYOU1) in PC12 cells. Such proteins participate in several process associated to protein metabolism in different mammalian cells. In neurons, the expression of Hsp90, for example, is associated to neuronal protection against the accumulation of toxic protein aggregates [47]. Additionally, HYOU1 mediates neuronal protection in cases of ischemia or excitotoxicity [48]. Interestingly, the modulation of NDK may affect cell fate, since that protein plays a role as a molecular switch between cellular proliferation and differentiation due to extracellular stimuli [49]. Taken together, such data may indicate a role for CA in inducing thermotolerance in neuronal cells, causing beneficial effects against neurodegenerative processes.
In addition to playing a role in the neuronal redox control, CA also modulates neuronal activity and plasticity, as investigated elsewhere. El Omri et al. found that CA (6.8 μg/mL for 48 h) induced differentiation of PC12 cells, leading to a morphology similar to sympathetic neurons [50]. Furthermore, CA induced a concentration-dependent (2.4–6.8 μg/mL) increase in acetylcholinesterase (AChE) activity. CA (6.8 μg/mL for 24 h) augmented the amounts of both total choline and acetylcholine in PC12 cells. The effects of CA on PC12 cells activity were blocked by U0126 (an ERK1/2 inhibitor). Nevertheless, U0126 only partially inhibited the effects of CA at the higher concentration tested in that work (6.8 μg/mL) regarding AChE activity, showing that, depending on the dosage, CA would exert its effects by different signaling pathways. Then, CA modulated neuronal morphology and activity, at least in part, by activation of ERK1/2 pathway, causing neuronal differentiation and increased metabolism of acetylcholine (both synthesis and degradation were regulated by CA).
In another work, CA promoted neurite outgrowth through activation of Nrf2 in rat pheochromocytoma PC12h cell line, as reported by Kosaka et al. [51]. CA induced a concentration-dependent (5–20 μM for 24 h) increase in the number of neurite-bearing cells and in the activity of ARE. The effect of CA on ARE was stronger than that observed to nerve growth factor (NGF; 1, 3, and 10 ng/mL) on PC12h cells. Moreover, CA upregulated Nrf2 and the markers of neural differentiation neurofilament M (NF-M) and neurofilament H (NF-H). Nrf2 knockdown (by utilization of small interfering RNA (siRNA) against Nrf2) did lead to decreased NF-H, NF-M, and MAP2 immunocontents in PC12h cells. Additionally, Nrf2 knockdown resulted in decreased neurite numbers in CA-treated cells. Authors also investigated by which signaling pathway CA exerted its effects on neuronal morphology. CA increased Akt and ERK1/2 phosphorylation. ERK1/2 activation was independent from tropomyosin-related kinase (TrkA) and Nrf2/ARE axis, since K252a (a TrkA inhibitor) and siRNA against Nrf2 did not affect ERK1/2 phosphorylation levels, respectively. On the other hand, PI3K activation was dependent on TrkA and Nrf2, since K252a treatment and Nrf2 knockdown caused decreased PI3K phosphorylation, respectively. The effects of CA on Nrf2 in PC12h were modulated by different kinases, mainly PI3K and ERK1/2 (extracellular signal-regulated kinase1/2), since specific inhibitors (namely LY294002 and U0126, inhibitors of PI3K and MEK1/2, respectively) decreased the effects of CA on Nrf2. Authors then analyzed by which program CA-induced neuronal differentiation in PC12h cells. CA increased the expression of nf-M, early growth response protein-1 (EGR-1), p62/zip (PKC-ζ-interacting protein), and musculoaponeurotic fibrosarcoma oncogene homolog K (mafK) genes. However, CA (15 μM) elicited distinct effects when compared with NGF (10 ng/mL) treatment, since the effect of CA on the expression of p62/zip and mafK genes was superior when compared with NGF. On the other hand, NGF increased egr-1 expression more intensely than CA. Thus, CA and NGF triggered neuronal differentiation by slightly different mechanisms in PC12h cells. The effects of CA on p62/ZIP protein were dependent on Nrf2, since siRNA against Nrf2 did lead to p62/ZIP immunocontent in PC12h cells exposed to CA. Alternatively, knockdown of p62/ZIP impaired the neurite outgrowth induced by CA on PC12h cells. Thus, CA-triggered neurite outgrowth in PC12h cells by a mechanism that, at least in part, was associated to the Nrf2/ARE axis activation, as well as to the induction of protein kinases related to neuronal differentiation. Additionally, p62/ZIP exerted a fundamental role downstream Nrf2 in induction of neuronal differentiation. On the contrary, CA treatment elicited neuronal differentiation by a Nrf-2 independent mechanism when activated PI3K/Akt and ERK1/2 protein kinases. Therefore, CA-induced neuronal differentiation in PC12h cells occurred by an intricate mechanism associated to the master regulator of the redox environment and/or to protein kinases activation. The complete mechanism behind neuronal differentiation elicited by CA remains to be fully established. Vaka et al. found that intranasal administration of CA (2–4 mg/kg for 4 days) induced an increase in both NGF and BDNF levels in rat hippocampus [52]. Even though the mechanism underlying such effects were not investigated, it clearly shows that CA may be effective also in vivo regarding the upregulation of neurotrophins involved in control of neuronal and glial function. Additionally, CA would be an interesting strategy to treat neuronal disorders associated to alterations in the levels of neurotrophins.
CA also protected neuronal cells by decreasing the production of amyloid-β (Aβ) peptides, which may lead to redox impairment by interacting with biological membranes and other cellular compartments (including mitochondria) in biological systems [53–56]. Meng et al. found that CA (at 20 and 30 μM for 8 h and at 10, 20, and 30 μM for 24 h) decreased Aβ42 production in SH-SY5Y neuroblastoma cells [57]. CA (10–30 μM for 24 h) also downregulated Aβ40 production in such cells. CA increased (5–30 μM for 24 h) α-cleaved soluble fragment of amyloid precursor protein (sAPPα) but decreased (10–30 μM for 24 h) sAPPβ levels in SH-SY5Y cells. CA upregulated the α-secretase called tumor necrosis factor-converting enzyme/a disintegrin and metalloproteinase 17 (TACE/ADAM17), ADAM10, and presenilin-1 (PS1) messenger RNA (mRNA) levels in a concentration-dependent manner. The effect of CA on TACE, ADAM10, and PS1 was not mediated by mitogen-activated protein kinases (MAPK) proteins or by the PI3K/Akt/NF-κB signaling pathway, since specific inhibitors did not alter the effects of CA on TACE, ADAM10, and PS1. On the other hand, TACE was involved in the decrease in Aβ levels induced by CA, since siRNA against TACE resulted in increased Aβ42 levels in SH-SY5Y cells exposed to CA. Alternatively, CA did not increase sAPPα in SH-SY5Y cells treated with TACE siRNA. Additionally, ADAM10 silencing in CA-treated cells also did lead to decreased sAPPα levels but did not alter Aβ42 amounts in such experimental model. Knockdown of Nrf2 did not mediate the effect of CA on Aβ42, showing that Nrf2-dependent signaling was not necessary to CA exerts its protective effect on SH-SY5Y cells regarding Aβ peptide production. CA also augmented insulin-degrading enzyme (IDE) expression levels, but IDE did not participate in the regulation of Aβ production in CA-exposed cells, since Aβ42 content did not differ between IDE knockdown cells and control in the CA-treated group. Therefore, authors demonstrated that CA modulated the levels of secretases involved in Aβ peptides metabolism, leading to decreased production of Aβ42 through upregulation of α- and γ-secretases (TACE/ADAM17 and ADAM10, and PS1, respectively) and downregulation of β-secretase (BACE1, β-site APP-cleaving enzyme-1) expression, leading to increased sAPPα and decreased Aβ42 amounts. Then, CA did exert an indirect anti-oxidant effect by regulating Aβ peptide synthesis, which may disrupt the physiologic redox status by interacting with several biomolecules and organelles. A summary of the effects elicited by CA on neuronal cells may be found in Table 1.
Carnosol and Neuronal Cells
Martin et al. reported that carnosol upregulated the expression of HO-1 through a mechanism associated to PI3K/Akt signaling pathway and activation of Nrf2 in PC12 cells [58]. Carnosol was tested at different concentrations (1–100 μM), and authors found that carnosol at 10 μM (for 16 h) induced a several fold (>200-fold) increase in HO-1 mRNA expression. Carnosol activated several protein kinases, as for instance ERK, JNK, p38, and PI3K/Akt, which participate in Nrf2 regulation. However, only inhibition of the PI3K/Akt and, to a lower extent, p38 pathways resulted in decreased levels of HO-1. The blockade of other signaling pathways did not alter HO-1 expression or ARE binding activity. Carnosol induced a concentration-dependent increase in the amounts of Nrf2 in the nucleus of PC12 cells. The stronger effect was seen in cells exposed to carnosol at 10 μM. The authors also examined whether carnosol would exert any protective effect against pro-oxidants in PC12 cells. Carnosol (1–30 μM) pretreatment (30 min) prevented the increase in ROS production elicited by H2O2 in a concentration-dependent manner. Interestingly, carnosol (10 μM for 30 min) blocked the increase in ROS production triggered by increasing H2O2 concentrations (50–500 μM) in PC12 cells. The protective effects elicited by carnosol on PC12 cells were time dependent, since short-term anti-oxidant effects (30-min incubation with carnosol) on PC12 cells were not blocked by HO-1 or PI3K inhibitors (ZnPPIX and LY294002, respectively). However, the long-term anti-oxidant effects of carnosol (6-h incubation) on PC12 cells were partially, but significantly, reduced by the inhibition of HO-1 or PI3K. Thus, the induction of HO-1 through activation of PI3K by carnosol is important for the long-term protective effects elicited by carnosol on neuronal cells. Therefore, carnosol activated the Nrf2/ARE signaling pathway leading to augmented HO-1 protein expression by a mechanism dependent on the PI3K/Akt pathway (Table 2). Accordingly, Tamaki et al. (2009) shown that carnosol (5 μM) increased ARE binding activity leading to increased GSH levels in HT22 neuronal cells [19]. Nevertheless, authors did not demonstrate the mechanism modulating GSH synthesis improvement in that work.
CA and Glial Cells
CA also modulates the production of neurotrophic factors in glial cells, as demonstrated by Kosaka and Yokoi, which first reported a role for CA (1–100 μM for 4 days) in inducing an increase in the production of NGF in T98G human glioblastoma cells [59]. Mimura et al. reported that CA modulated NGF through a Nrf2-dependent mechanism in T98G glioblastoma cells and normal human astrocytes [60]. CA (10 and 50 μM for 24 h) upregulated NGF expression without altering the levels of neurotrophin-3 (NT-3) or BDNF (CA at 50 μM). Additionally, 50 μM CA augmented Nrf2 immunocontent (2–24 h), causing an upregulation in both HO-1 and thioredoxin reductase 1 (TXNRD1) expression. The authors demonstrated that the effect of CA on NGF, HO-1, and TXNRD1 was suppressed by Nrf2 silencing with siRNA. Interestingly, the effect of CA on NGF was not affected by the addition of tumor necrosis factor-α (TNF-α) or IL-1β, which are pro-inflammatory molecules. Actually, NGF expression is regulated by some pro-inflammatory agents, as TNF-α, IL-1β, and lipopolysaccharide (LPS) [61–64]. Thus, CA activated NGF expression by a Nrf2-associated mechanism in activated glial cells. Such data may indicate an anti-oxidant and protective role for CA in cases of neuroinflammation and also during physiological events, as for instance neuronal plasticity. Really, some authors have attributed an anti-inflammatory role for Nrf2 during impairment of neuronal function, as observed in cases of depression, a pathology in which neuronal plasticity may be altered [65–67]. Future investigations would be necessary to better understand the link between Nrf2 and inflammation in human brain disorders.
Yoshida et al. investigated whether CA was capable to upregulate NGF expression during a condition of hypoxia/reoxygenation induced in a human astrocyte cell line (U373MG) [68]. CA (20 μM) increased NGF levels during normoxia (48 h). However, the effect elicited by CA was augmented by the addition of edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one, 1 mM), which did act synergistically with CA in upregulating NGF expression during normoxia. Edaravone is able to penetrate the blood-brain barrier and functions as an anti-oxidant/free radical scavenger in cases of postischemic neuronal dysfunction [69, 70]. The synergism of CA and edaravone regarding the expression of NGF was also observed when cells were reoxygenated (48 h) after either 3-h or 4.5-h period under hypoxia. The authors also identified a role for JNK signaling pathway in the induction of NGF expression. On the other hand, when CA was combined with edaravone, both JNK and MEK1/2 signaling pathways were activated, leading to an upregulation in NGF expression. CA and more edaravone also modulated NGF through a p38 kinase-dependent manner, since SB203580 (an inhibitor of p38 kinase) partially abolished the effects of such compounds on NGF expression. CA and edaravone induced a higher level of activation of Nrf2 when compared with edaravone or CA alone, and Nrf2 activation caused an increase in NGF levels. Then, authors indicate a more potent effect of CA in human astrocytes when administrated in the presence of edaravone during situations of hypoxia. Importantly, CA and edaravone together maintained astrocytes viability during reoxygenation after a 4.5-h period under hypoxia. Thus, CA could be applied as an interesting strategy as a protective agent by activating Nrf2 in cases of ischemia/reperfusion that causes drastic oxygenation and ROS production in the reperfusion stage. Furthermore, CA would be effective in modulating neuronal plasticity by increasing NGF through a hormetic way [71]. In spite of this, further research is necessary in order to examine whether the effects seen in that experimental model will repeat in others.
Indeed, CA exhibited an ability to increase HO-1 expression at 6 h (5, 10, and 20 μM) and at 24 h (5 μM) in BV2 mouse microglial cells, as demonstrated by Foresti et al., with an EC50 = 13.1 μM [72]. Additionally, CA (20 μM) increased bilirubin production in the presence (6 and 24 h) or absence (6 h) of hemin (10 μM), a substrate of HO-1. The effect of CA on HO-1 expression was dependent on thiol redox status, since N-acetylcysteine (NAC) co-treatment did suppress the upregulation of HO-1 expression elicited by CA. CA at 20 μM (but not at 5 and 10 μM) was able to decrease interferon-γ (INF-γ)-induced NO production in BV2 cells, as assessed through quantification of nitrite levels. On the other hand, CA at 5, 10, and 20 μM was effective in reducing NO levels in LPS-exposed BV2 cells. Thus, CA exhibited a partial effect on the production of NO, being more effective at high concentrations. Accordingly, Yanagitai et al. found that CA (1, 3, and 10 μM) co-treatment (24 h) decreased NO generation in a dose-dependent fashion in microglial MG6 cells exposed to LPS [8]. CA (10 μM) also was effective in reducing IL-6 immunocontent and the expression of IL-1β and nitric oxide synthase (NOS) in LPS-treated cells. Moreover, CA (10 μM) regulated HO-1, GCLM, GCLC, NQO-1, and xCT (Na+-independent cystine-glutamate exchanger) in MG6 cells that were exposed to LPS: CA induced a decrease in the expression of GCLC, NQO-1, and xCT when compared with MG6 cells that received LPS alone, demonstrating that CA inhibited microglial activation through both redox and immune mechanisms. LPS elicits oxidative and nitrosative stress in several experimental models and upregulates the expression of anti-oxidant enzymes [73, 74]. Therefore, CA prevented LPS-induced oxidative impairment in MG6 cells, as well as inhibited its activation. However, the link between redox protection and microglial activation was not addressed in that work.
Recently, Yoshida et al. found that CA suppressed the production of amyloid-β 1-42 and 1-43 (Aβ42 and Aβ43, respectively) peptides by upregulating the α-secretase TACE/ADAM17 in U373MG human astrocytoma cells [75]. CA (10–50 μM) decreased Aβ40 and Aβ42 production in normoxia condition (24 h). On the other hand, CA (30–50 μM, but not at 10 μM; 24-h treatment) was effective in reducing Aβ43 production by U373MG cells under the same situation. The protective effect of CA regarding the production of Aβ42 was maintained even during hypoxia (8 h) and reoxygenation (24 h) after an 8-h period under hypoxia. CA did not alter α-secretase a disintegrin and metalloproteinase 1 (ADAM1), β-secretase BACE, or γ-secretase PS1 expression levels. However, CA (50 μM for 8 h) increased the mRNA levels of the α-secretase TACE. CA increased sAPPα (which is a product of the non-amyloidogenic pathway; CA at 30 and 50 μM) and decreased β-cleaved soluble fragment of APP (sAPPβ; CA at 10, 30, and 50 μM) levels in U373MG cells, thus promoting α-cleavage in the place of β-cleavage. Interestingly, CA (2.3-fold increase) was more effective than resveratrol (1.5-fold increase) in inducing TACE expression. The authors demonstrated that the effect of CA on Aβ42 and on Aβ43 was partially associated to TACE expression, since TACE siRNA abolished the effect of such diterpene on the production of such Aβ peptides. Indeed, the effect of CA on increasing sAPPα levels was abolished by TACE expression inhibition. CA (50 μM for 3–24 h) also augmented SIRT1 mRNA levels in U373MG cells. SIRT1 silencing (through utilization of siRNA against SIRT1) did not affect the effect of CA on Aβ42 levels, i.e., CA inhibited Aβ42 production by a SIRT1-independent manner. Surprisingly, the mechanism by which CA downregulated the production and release of Aβ42 peptide did not involve enzymes that degrades Aβ, as for instance neprilysin (NEP), IDE, and endothelin-converting enzyme-1 (ECE1), because CA did not alter mRNA of such enzymes in this experimental model. CA did promote α-secretase TACE upregulation leading to increased sAPPα production, which would cause a competition between α- and β-secretases for APP cleavage, resulting in decreased Aβ production in astrocytes. Such evidences may play an important role during AD pathology, in which there is an overproduction of Aβ peptides that culminates in increased redox impairment, neuroinflammation mediates by glial cells, and cell death. A summary of the CA-triggered effects on glial cells may be found in Table 3.
Carnosol and Glial Cells
Kim et al. found that carnosol protected rat C6 glial cells against sodium nitroprusside (SNP)-induced toxicity [77]. C6 cell line presents astrocytic markers, e.g., glial fibrillary acidic protein (GFAP) and S-100 protein, and may be utilized as a model to investigate astrocytic diseases [76]. Carnosol pretreatment (1–10 μM for 1 h) prevented the increase in NO production triggered by SNP in a dose-dependent manner. Carnosol also alleviated the effect of SNP on cell viability. Additionally, carnosol pretreatment (10 μM for 1 h) inhibited SNP-induced apoptosis in C6 cells. Such effects were mediated by carnosol-induced inhibition of JNK1/2 phosphorylation and of caspase-3 activation. Carnosol at 10 μM also restored GSH levels in SNP-treated C6 cells. Carnosol also upregulated the expression of HO-1 through activation of the PI3K/Akt/Nrf2 signaling pathway in C6 cells. The effect of carnosol on Nrf2 activation and translocation were not blocked by ZnPP (inhibitor of HO-1). Thus, authors concluded that the activation of Nrf2 preceded the increase of HO-1 expression. On the other hand, ZnPP blocked the protective effects of carnosol on cell viability in the presence of SNP, demonstrating that HO-1 plays an important role in mediating the beneficial effects of carnosol on an experimental model of nitrosative stress. NO overproduction has been observed in cases of neurodegenerative diseases and may be a result of neuroinflammation, as occurs in the case of Parkinson’s disease (PD) and Alzheimer’s disease (AD) [78, 79]. Therefore, carnosol may be viewed as an agent able to alleviate the redox impairment induced in microglial cells, consequently leading to decreased levels of neuroinflammation. Indeed, Foresti et al. reported that carnosol elicited anti-oxidant and anti-inflammatory effects on BV2 mouse microglial cells [72]. The authors investigated the ability of 56 compounds that are known activators of Nrf2 signaling pathway to induce HO-1 expression. Among the molecules analyzed, authors found that carnosol (5, 10, and 20 μM) upregulated HO-1 expression with an EC50 = 9.4 μM (6 h). Additionally, carnosol (20 μM for 24 h) increased the generation of a HO-1 product, bilirubin. The increase in HO-1 activity elicited by carnosol was alleviated by a co-treatment with N-acetylcysteine (NAC), an anti-oxidant, showing that thiol redox status may be involved in the mechanism of HO-1 induction by carnosol. Interestingly, carnosol, did not change the levels of Δ-aminolevulinate synthase 1 (ALAS1) and biliverdin reductase (BVR), enzymes that participate in heme metabolism (ALAS1 is the rate-limiting enzyme in heme synthesis and BVR converts biliverdin to bilirubin by a reduction reaction) [80, 81]. Thus, carnosol increased bilirubin levels due to a direct action of HO-1 expression, and not by acting on other enzymes responsible for the synthesis of bilirubin. Carnosol (10 and 20 μM) also elicited a decrease in INF-γ-induced NO synthesis, as assessed through the quantification of nitrite. Carnosol was more effective against LPS-induced NO production, since the lower carnosol concentration tested (5 μM) was able to reduce NO production by 50 %. Carnosol also exhibited an anti-inflammatory effect by decreasing both prostaglandin E2 (PGE2) and TNF-α levels in INF-γ and LPS-treated BV2 cells. The effects of carnosol in decreasing the levels of pro-inflammatory molecules were not dependent, at least in part, on HO-1 and Nrf2. However, carnosol decreased INF-γ and LPS-induced NO production by a mechanism that depended on the Nrf2/HO-1 axis, since silencing HO-1 or Nrf2 expression led to an increase in NO levels in cells that were exposed to INF-γ and LPS in the presence of carnosol. The authors also observed that the Nrf2/HO-1 axis may play a role in the production of PGE2 and TNF-α. However, the link between Nrf2 and/or HO-1 and the production of pro-inflammatory molecules was not investigated in that work. Some authors have demonstrated that a refined cross-talk between Nrf2 and NF-κB may lead to decreased inflammation [82–84]. However, the full mechanism was not still clarified (Table 4).
Therefore, CA and carnosol mediate signaling pathways involved in both control of the redox environment and immune functions in both neuronal and glial cells. There is a lack for the complete way by which CA and carnosol elicited such effects, but evidences point to a cross-talk between the anti-oxidant and the immune-related signaling pathways as an important part of the mechanism stimulated by such diterpenes in mediating its protective effects on neuronal and glial cells. Importantly, it is necessary to examine whether the concentrations of CA and carnosol utilized in in vitro experimental models would be reached in brain cells in in vivo experimental designs, since the metabolism of CA and carnosol (including possible modifications that may be performed by gut bacteria in CA and carnosol) may affect its availability to brain tissues.
The In Vivo Effects of CA and Carnosol on Neuronal and Glial Cells
Carnosic Acid
Satoh et al. reported that CA (3 mg/25 g body weight orally administrated; catechol-type form) accumulated in C57BL/6 mouse brain within 1 h [22]. CA induced an increase in both GSH and GSSG (the oxidized form of GSH), as well as on the GSH/GSSG ratio. Additionally, CA upregulated HO-1 and γ-GCL expression in mouse brain. Interestingly, CA treatment (1 mg/kg i.p.) 1 h prior induction of middle cerebral artery occlusion (MCAO)/reperfusion (a model of focal cerebral ischemia) prevented the increase in infarct volume caused by MCAO in C57BL/6 mice. Taken together, such data demonstrate that CA penetrates the blood-brain barrier and exerts a protective effect on mammalian brain in vivo in an experimental model that induces redox disturbances and neuroinflammation. CA elicited such effects probably by activating the Keap1/Nrf2 pathway in a quinone-type form, since the catechol-type form does not activate Keap1/Nrf2 pathway, as demonstrated by the authors.
In the work by Azad et al., CA (10 mg/kg, i.p. 1 h before surgery, followed by CA at 3 mg/kg, i.p. 3–4 h after surgery with repetitions once a day during 12 days) protected rat hippocampus from amyloid-β-induced lesions in an experimental model of Alzheimer’s disease [85]. CA treatment was able to preserve the number of neurons in the cornu ammonis 1 (CA1) region by inhibiting apoptosis in such rat brain area. Even thought the mechanism by which CA exerted such protective effect was not investigated in that work, data presented by the authors showed that CA would protect neuronal cells from a peptide that may act through different ways in inducing cell dysfunction. More analyses would be useful to examine the exact mechanism by which CA elicits neuronal protection in vivo.
Wu et al. demonstrated that a pretreatment with CA (20 mg/kg body weight orally administrated for 3 weeks) was able to protect the striatum of Wistar rats against oxidative damage induced by 6-OHDA exposure [43]. CA decreased lipid peroxidation and oxidation of GSH elicited by 6-OHDA. Moreover, CA improved rat behavior regarding locomotory activity and distance travelled when compared with the 6-OHDA treated group. CA also decreased both the number and time of rotation of 6-OHDA treated rats in a behavioral task. CA exerted such protective effects probably by increasing the immunocontent of anti-oxidant enzymes, as for instance GCLC, GCLM, GR, and SOD. The exact mechanism by which CA modulated the expression of the anti-oxidant enzymes was not addressed in such work, but a role for Nrf2/ARE pathway is very plausible. CA also decreased the rates of apoptosis in rat striatum by increasing the levels of B cell CLL/lymphoma 2 (Bcl-2) and decreasing the amounts of cleaved caspase-3 (active form), BCL2-associated X protein (Bax), and cleaved PARP (a target of activated caspase-3), through downregulation of JNK1/2 and p38 mitogen-activated protein kinase (p38 MAPK), leading to an increased number of tyrosine hydroxylase (TH)-positive cells in such brain region. Thus, CA was able to inhibit apoptosis triggered by 6-OHDA, clearly demonstrating its role as a neuroprotective agent against chemically induced stress and cell death by a pro-oxidant drug. Please see Table 5 for a summary regarding the effects of CA on mammalian brain cells.
Carnosol
Carnosol (0.01–10 mg/kg, oral route 1 h before the execution of behavioral tasks) elicited anti-depressant effects in mice, as reported by Machado et al. [87]. Animals that received carnosol (0.01 and 0.1 mg/kg through oral route) spent less time immobilized in the tail suspension test (TST) when compared with the control group (which ingested vehicle before each task) (Table 6). Interestingly, the authors did not find any psychostimulant effect associated to carnosol treatment in the open field test, leading to the conclusion that the anti-depressant effects induced by carnosol and observed in the TST were specific. The same research group previously demonstrated that an extract of R. officinalis (rosemary; 10–100 mg/kg, oral route for 14 days) exerted anti-depressant effects in an experimental model utilizing mice [86, 88]. The authors attributed such effects to the ability of this extract to modulate the monoaminergic system in mice brain, since specific antagonists of dopaminergic receptors blocked the beneficial effects elicited by the extract. However, authors did not analyze the specific component of the extract that would be the responsible for the anti-depressant effects seen in that work. Sasaki et al. observed that mice that received rosemary extract (50–100 mg/kg in drinking water for 7 days) presented decreased immobility time in TST [89]. Rosemary extract (100 mg/kg) increased the levels of norepinephrine (NE), dopamine (DA), serotonin (5-HT), choline (Ch), and acetylcholine (ACh) in the mice brain cortex. Additionally, rosemary extract (50 and 100 mg/kg) upregulated the expression of tyrosine hydroxylase (TH) and pyruvate carboxylase (PC) in mice brain. On the other hand, rosemary extract (100 mg/kg) decreased mitogen-activated protein kinase (MKP-1) expression in mice brain. Inhibition of MKP-1 may play a role in the anti-depressant effects elicited by plant components, as previously published [89]. In another experimental model, Zanella et al. reported that rosemary extract (150–300 mg/kg oral route, 30 min–4 h before each behavioral task) improved mice memory in social recognition and in step-down inhibitory avoidance tasks [90]. Therefore, rosemary extract or its components are able to positively modulate brain functions and behavior of mammals. More analyses are necessary to investigate the mechanism underlying such effects of R. officinalis extract or carnosol on animal behavior.
Future Directions
CA and carnosol are the major diterpenes found in rosemary and exert 90 % of the anti-oxidant capacity of that plant. However, there is a lack of studies demonstrating the protective effects and the underlying mechanisms elicited by these compounds regarding mammalian brain cells. Therefore, it would be very interesting:
-
To examine alternative signaling pathways that may be modulated by CA and carnosol and that are involved in its protective effects in brain cells in vitro and in vivo;
-
To verify the role of CA and carnosol in modulating signaling pathways associated to apoptosis and/or autophagy in neuronal and glial cells, as well as to investigate whether there is a interplay between modulation of such pathways and other involved with the maintenance of redox and immune-related functions in brain cells in vitro and in vivo;
-
To analyze the role of CA and carnosol in other experimental models of neurodegeneration;
-
To investigate whether there is a role for CA and carnosol in preventing, as well treating, brain tumors;
-
To evaluate whether CA and carnosol elicit neurotoxicity in vitro and in vivo;
-
To perform clinical trials considering the anti-oxidant and anti-inflammatory potentials of CA and carnosol in different human populations including those subjects suffering from neurological disturbances. A search in the Website clinicaltrials.gov (20 August 2015) resulted in zero (0) studies investigating the effects of CA or carnosol on human health. Some results were obtained when “rosemary” was utilized in the searching machine of the system. However, the therapeutical potential of CA and carnosol would deserve more attention pharmacologically.
Conclusion
CA and carnosol are neuroprotective agents exerting anti-oxidant and anti-inflammatory effects through specific modulation of signaling pathways involved in cytoprotection. More studies are necessary to analyze additional mechanisms by which CA and carnosol would protect neuronal and glial cells.
Reference
Birtić S, Dussort P, Pierre FX, Bily AC, Roller M (2015) Carnosic acid. Phytochemistry 115:9–19. doi:10.1016/j.phytochem.2014.12.026
Aruoma OI, Halliwell B, Aeschbach R, Löligers J (1992) Antioxidant and pro-oxidant properties of active rosemary constituents: carnosol and carnosic acid. Xenobiotica 22(2):257–268
Kuzmenko AI, Morozova RP, Nikolenko IA, Donchenko GV, Richheimer SL, Bailey DT (1999) Chemiluminescence determination of the in vivo and in vitro antioxidant activity of RoseOx and carnosic acid. J Photochem Photobiol, B 48(1):63–67. doi:10.1016/S1011-1344(99)00011-1
Erkan N, Ayranci G, Ayranci E (2008) Antioxidant activities of rosemary (Rosmarinus officinalis L.) extract, blackseed (Nigella sativa L.) essential oil, carnosic acid, rosmarinic acid and sesamol. Food Chem 110(1):76–82. doi:10.1016/j.foodchem.2008.01.058
Park JA, Kim S, Lee SY, Kim CS, Kim do K, Kim SJ, Chun HS (2008) Beneficial effects of carnosic acid on dieldrin-induced dopaminergic neuronal cell death. Neuroreport 19(13):1301–1304. doi:10.1097/WNR.0b013e32830abc1f
Lian KC, Chuang JJ, Hsieh CW, Wung BS, Huang GD, Jian TY, Sun YW (2010) Toxicol Appl Pharmacol 245(1):21–35. doi:10.1016/j.taap.2010.01.003
Mengoni ES, Vichera G, Rigano LA, Rodriguez-Puebla ML, Galliano SR, Cafferata EE, Pivetta OH, Moreno S, Vojnov AA (2011) Suppression of COX-2, IL-1β and TNF-α expression and leukocyte infiltration in inflamed skin by bioactive compounds from Rosmarinus officinalis L. Fitoterapia 82(3):414–421. doi:10.1016/j.fitote.2010.11.023
Yanagitai M, Itoh S, Kitagawa T, Takenouchi T, Kitani H, Satoh T (2012) Carnosic acid, a pro-electrophilic compound, inhibits LPS-induced activation of microglia. Biochem Biophys Res Commun 418(1):22–26. doi:10.1016/j.bbrc.2011.12.087
Satoh T, McKercher SR, Lipton SA (2013) Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic Biol Med 65:645–657. doi:10.1016/j.freeradbiomed.2013.07.022
Pérez-Fons L, Garzón MT, Micol V (2010) Relationship between the antioxidant capacity and effect of rosemary (Rosmarinus officinalis L.) polyphenols on membrane phospholipid order. J Agric Food Chem 58(1):161–171. doi:10.1021/jf9026487
Maruoka H, Sasaya H, Sugihara K, Shimoke K, Ikeuchi T (2011) Low-molecular-weight compounds having neurotrophic activity in cultured PC12 cells and neurons. J Biochem 150(5):473–475. doi:10.1093/jb/mvr113
Wijeratne SS, Cuppett SL (2007) Potential of rosemary (Rosemarinus officinalis L.) diterpenes in preventing lipid hydroperoxide-mediated oxidative stress in Caco-2 cells. J Agric Food Chem 55(4):1193–1199. doi:10.1021/jf063089m
Haraguchi H, Saito T, Okamura N, Yagi A (1995) Inhibition of lipid peroxidation and superoxide generation by diterpenoids from Rosmarinus officinalis. Planta Med 61(4):333–336. doi:10.1055/s-2006-958094
Johnson JJ (2011) Carnosol: a promising anti-cancer and anti-inflammatory agent. Cancer Lett 305(1):1–7. doi:10.1016/j.canlet.2011.02.005
Visanji JM, Thompson DG, Padfield PJ (2006) Induction of G2/M phase cell cycle arrest by carnosol and carnosic acid is associated with alteration of cyclin A and cyclin B1 levels. Cancer Lett 237(1):130–136. doi:10.1016/j.canlet.2005.05.045
Poeckel D, Greiner C, Verhoff M, Rau O, Tausch L, Hörnig C, Steinhilber D, Schubert-Zsilavecz M, Werz O (2008) Carnosic acid and carnosol potently inhibit human 5-lipoxygenase and suppress pro-inflammatory responses of stimulated human polymorphonuclear leukocytes. Biochem Pharmacol 76(1):91–97. doi:10.1016/j.bcp.2008.04.013
Johnson JJ, Syed DN, Suh Y, Heren CR, Saleem M, Siddiqui IA, Mukhtar H (2010) Disruption of androgen and estrogen receptor activity in prostate cancer by a novel dietary diterpene carnosol: implications for chemoprevention. Cancer Prev Res (Phila) 3(9):1112–1123. doi:10.1158/1940-6207
Fawcett JR, Bordayo EZ, Jackson K, Liu H, Peterson J, Svitak A, Frey WH 2nd (2002) Inactivation of the human brain muscarinic acetylcholine receptor by oxidative damage catalyzed by a low molecular weight endogenous inhibitor from Alzheimer’s brain is prevented by pyrophosphate analogs, bioflavonoids and other antioxidants. Brain Res 950(1-2):10–20. doi:10.1016/S0006-8993(02)02981-5
Tamaki Y, Tabuchi T, Takahashi T, Kosaka K, Satoh T (2010) Activated glutathione metabolism participates in protective effects of carnosic acid against oxidative stress in neuronal HT22 cells. Planta Med 76(7):683–688. doi:10.1055/s-0029-1240622
del Baño MJ, Lorente J, Castillo J, Benavente-García O, del Río JA, Ortuño A, Quirin KW, Gerard D (2003) Phenolic diterpenes, flavones, and rosmarinic acid distribution during the development of leaves, flowers, stems, and roots of Rosmarinus officinalis. Antioxidant activity. J Agric Food Chem 51(15):4247–4253. doi:10.1021/jf0300745
Luis JC, Johnson CB (2005) Seasonal variations of rosmarinic and carnosic acids in rosemary extracts. Analysis of their in vitro antiradical activity. Span J Agric Res 3(1):106–112
Satoh T, Kosaka K, Itoh K, Kobayashi A, Yamamoto M, Shimojo Y, Kitajima C, Cui J, Kamins J, Okamoto S, Izumi M, Shirasawa T, Lipton AS (2008) Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J Neurochem 104(4):1116–1131. doi:10.1111/j.1471-4159.2007.05039.x
Romo Vaquero M, García Villalba R, Larrosa M, Yáñez-Gascón MJ, Fromentin E, Flanagan J, Roller M, Tomás-Barberán FA, Espín JC, García-Conesa MT (2013) Bioavailability of the major bioactive diterpenoids in a rosemary extract: metabolic profile in the intestine, liver, plasma, and brain of Zucker rats. Mol Nutr Food Res 57(10):1834–1846. doi:10.1002/mnfr.201300052
Doolaege EH, Raes K, De Vos F, Verhé R, De Smet S (2011) Absorption, distribution and elimination of carnosic acid, a natural antioxidant from Rosmarinus officinalis, in rats. Plant Foods Hum Nutr 66(2):196–202. doi:10.1007/s11130-011-0233-5
Satoh T, Izumi M, Inukai Y, Tsutsumi Y, Nakayama N, Kosaka K, Shimojo Y, Kitajima C, Itoh K, Yokoi T, Shirasawa T (2008) Carnosic acid protects neuronal HT22 Cells through activation of the antioxidant-responsive element in free carboxylic acid- and catechol hydroxyl moieties-dependent manners. Neurosci Lett 434(3):260–265. doi:10.1016/j.neulet.2008.01.079
Talalay P (2000) Chemoprotection against cancer by induction of phase 2 enzymes. Biofactors 12(1-4):5–11
Itoh K, Tong KI, Yamamoto M (2004) Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med 36(10):1208–1213. doi:10.1016/j.freeradbiomed.2004.02.075
Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, Kang MI, Kobayashi A, Yokoyama S, Yamamoto M (2006) Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell 21(5):689–700. doi:10.1016/j.molcel.2006.01.013
Kraft AD, Johnson DA, Johnson JA (2004) Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci 24(5):1101–1112. doi:10.1523/JNEUROSCI.3817-03.2004
Hong F, Freeman ML, Liebler DC (2005) Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem Res Toxicol 18(12):1917–1926. doi:10.1021/tx0502138
Chen JH, Ou HP, Lin CY, Lin FJ, Wu CR, Chang SW, Tsai CW (2012) Carnosic acid prevents 6-hydroxydopamine-induced cell death in SH-SY5Y cells via mediation of glutathione synthesis. Chem Res Toxicol 25(9):1893–1901. doi:10.1021/tx300171u
Lu SC (2013) Glutathione synthesis. Biochim Biophys Acta 1830(5):3143–3153. doi:10.1016/j.bbagen.2012.09.008
Lu SC (2009) Regulation of glutathione synthesis. Mol Aspects Med 30(1-2):42–59. doi:10.1016/j.mam.2008.05.005
Lin CY, Chen JH, Fu RH, Tsai CW (2014) Induction of Pi form of glutathione S-transferase by carnosic acid is mediated through PI3K/Akt/NF-κB pathway and protects against neurotoxicity. Chem Res Toxicol 27(11):1958–1966. doi:10.1021/tx5003063
Salminen A, Hyttinen JM, Kaarniranta K (2011) AMP-activated protein kinase inhibits NF-κB signaling and inflammation: impact on healthspan and lifespan. J Mol Med (Berl) 89(7):667–676. doi:10.1007/s00109-011-0748-0
Castro-Caldas M, Carvalho AN, Rodrigues E, Henderson C, Wolf CR, Gama MJ (2012) Glutathione S-transferase pi mediates MPTP-induced c-Jun N-terminal kinase activation in the nigrostriatal pathway. Mol Neurobiol 45(3):466–477. doi:10.1007/s12035-012-8266-9
Carvalho AN, Marques C, Rodrigues E, Henderson CJ, Wolf CR, Pereira P, Gama MJ (2013) Ubiquitin-proteasome system impairment and MPTP-induced oxidative stress in the brain of C57BL/6 wild-type and GSTP knockout mice. Mol Neurobiol 47(2):662–672. doi:10.1007/s12035-012-8368-4
Hou CW, Lin YT, Chen YL, Wang YH, Chou JL, Ping LY, Jeng KC (2012) Neuroprotective effects of carnosic acid on neuronal cells under ischemic and hypoxic stress. Nutr Neurosci 15(6):257–263. doi:10.1179/1476830512Y.0000000021
Jiang J, Borisenko GG, Osipov A, Martin I, Chen R, Shvedova AA, Sorokin A, Tyurina YY, Potapovich A, Tyurin VA, Graham SH, Kagan VE (2004) Arachidonic acid-induced carbon-centered radicals and phospholipid peroxidation in cyclo-oxygenase-2-transfected PC12 cells. J Neurochem 90(5):1036–1049. doi:10.1111/j.1471-4159.2004.02577.x
Nakanishi M, Rosenberg DW (2013) Multifaceted roles of PGE2 in inflammation and cancer. Semin Immunopathol 35(2):123–137. doi:10.1007/s00281-012-0342-8
Trebino CE, Stock JL, Gibbons CP et al (2003) Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc Natl Acad Sci U S A 100(15):9044–9049. doi:10.1073/pnas.1332766100
Kalinski P (2012) Regulation of immune responses by prostaglandin E2. J Immunol 188(1):21–28. doi:10.4049/jimmunol.1101029
Wu CR, Tsai CW, Chang SW, Lin CY, Huang LC, Tsai CW (2015) Carnosic acid protects against 6-hydroxydopamine-induced neurotoxicity in in vivo and in vitro model of Parkinson’s disease: involvement of antioxidative enzymes induction. Chem Biol Interact 225:40–46. doi:10.1016/j.cbi.2014.11.011
Halliwell B (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97(6):1634–1658. doi:10.1111/j.1471-4159.2006.03907.x
Itoh K, Ye P, Matsumiya T, Tanji K, Ozaki T (2015) Emerging functional cross-talk between the Keap1-Nrf2 system and mitochondria. J Clin Biochem Nutr 56(2):91–97. doi:10.3164/jcbn.14-134
Elomri A, Han J, Ben Abdrabbah M, Isoda H (2012) Down regulation effect of Rosmarinus officinalis polyphenols on cellular stress proteins in rat pheochromocytoma PC12 cells. Cytotechnology 64(3):231–240. doi:10.1007/s10616-011-9352-y
Luo W, Sun W, Taldone T, Rodina A, Chiosis G (2010) Heat shock protein 90 in neurodegenerative diseases. Mol Neurodegener 5:24. doi:10.1186/1750-1326-5-24
Zhao L, Rosales C, Seburn K, Ron D, Ackerman SL (2010) Alteration of the unfolded protein response modifies neurodegeneration in a mouse model of Marinesco-Sjögren syndrome. Hum Mol Genet 19(1):25–35. doi:10.1093/hmg/ddp464
Kimura N, Shimada N, Ishijima Y, Fukuda M, Takagi Y, Ishikawa N (2003) Nucleoside diphosphate kinases in mammalian signal transduction systems: recent development and perspective. J Bioenerg Biomembr 35(1):41–47
El Omri A, Han J, Yamada P, Kawada K, Ben Abdrabbah M, Isoda H (2010) Rosmarinus officinalis polyphenols activate cholinergic activities in PC12 cells through phosphorylation of ERK1/2. J Ethnopharmacol 131(2):451–458. doi:10.1016/j.jep.2010.07.006
Kosaka K, Mimura J, Itoh K, Satoh T, Shimojo Y, Kitajima C, Maruyama A, Yamamoto M, Shirasawa T (2010) Role of Nrf2 and p62/ZIP in the neurite outgrowth by carnosic acid in PC12h cells
Vaka SR, Murthy SN, Repka MA, Nagy T (2011) Upregulation of endogenous neurotrophin levels in the brain by intranasal administration of carnosic acid. J Pharm Sci 100(8):3139–3145. doi:10.1002/jps.22528
Davis JB (1996) Oxidative mechanisms in beta-amyloid cytotoxicity. Neurodegeneration 5(4):441–444
Markesbery WR (1997) Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med 23(1):134–147. doi:10.1016/S0891-5849(96)00629-6
Gupta A, Iadecola C (2015) Impaired Aβ clearance: a potential link between atherosclerosis and Alzheimer’s disease. Front Aging Neurosci 7:115. doi:10.3389/fnagi.2015.00115
Salminen A, Haapasalo A, Kauppinen A, Kaarniranta K, Soininen H, Hiltunen M (2015) Impaired mitochondrial energy metabolism in Alzheimer’s disease: Impact on pathogenesis via disturbed epigenetic regulation of chromatin landscape. Prog Neurobiol 131:1–20. doi:10.1016/j.pneurobio.2015.05.001
Meng P, Yoshida H, Matsumiya T (2013) Carnosic acid suppresses the production of amyloid-β 1-42 by inducing the metalloprotease gene TACE/ADAM17 in SH-SY5Y human neuroblastoma cells. Neurosci Res 75(2):94–102. doi:10.1016/j.neures.2012.11.007
Martin D, Rojo AI, Salinas M, Diaz R, Gallardo G, Alam J, De Galarreta CM, Cuadrado A (2004) Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J Biol Chem 279(10):8919–8929. doi:10.1074/jbc.M309660200
Kosaka K, Yokoi T (2003) Carnosic acid, a component of rosemary (Rosmarinus officinalis L.), promotes synthesis of nerve growth factor in T98G human glioblastoma cells. Biol Pharm Bull 26(11):1620–1622. doi:10.1248/bpb.26.1620
Mimura J, Kosaka K, Maruyama A, Satoh T, Harada N, Yoshida H, Satoh K, Yamamoto M, Itoh K (2011) Nrf2 regulates NGF mRNA induction by carnosic acid in T98G glioblastoma cells and normal human astrocytes. J Biochem 150(2):209–217. doi:10.1093/jb/mvr065
Vigé X, Costa E, Wise BC (1991) Mechanism of nerve growth factor mRNA regulation by interleukin-1 and basic fibroblast growth factor in primary cultures of rat astrocytes. Mol Pharmacol 40(2):186–192
Friedman WJ, Thakur S, Seidman L, Rabson AB (1996) Regulation of nerve growth factor mRNA by interleukin-1 in rat hippocampal astrocytes is mediated by NFkappaB. J Biol Chem 271(49):31115–31120. doi:10.1074/jbc.271.49.31115
Galve-Roperh I, Malpartida JM, Haro A, Brachet P, Díaz-Laviada I (1997) Regulation of nerve growth factor secretion and mRNA expression by bacterial lipopolysaccharide in primary cultures of rat astrocytes. J Neurosci Res 49(5):569–575
Sofroniew MV, Howe CL, Mobley WC (2001) Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci 24:1217–1281. doi:10.1146/annurev.neuro.24.1.1217
Maes M, Fišar Z, Medina M, Scapagnini G, Nowak G, Berk M (2012) New drug targets in depression: inflammatory, cell-mediated immune, oxidative and nitrosative stress, mitochondrial, antioxidant, and neuroprogressive pathways. And new drug candidates—Nrf2 activators and GSK-3 inhibitors. Inflammopharmacology 20(3):127–150. doi:10.1007/s10787-011-0111-7
Martín-de-Saavedra MD, Budni J, Cunha MP et al (2013) Nrf2 participates in depressive disorders through an anti-inflammatory mechanism. Psychoneuroendocrinology 38(10):2010–2022. doi:10.1016/j.psyneuen.2013.03.020
Bakunina N, Pariante CM, Zunszain PA (2015) Immune mechanisms linked to depression via oxidative stress and neuroprogression. Immunology 144(3):365–373. doi:10.1111/imm.12443
Yoshida H, Mimura J, Imaizumi T, Matsumiya T, Ishikawa A, Metoki N, Tanji K, Ota K, Hayakari R, Kosaka K, Itoh K, Satoh K (2011) Edaravone and carnosic acid synergistically enhance the expression of nerve growth factor in human astrocytes under hypoxia/reoxygenation. Neurosci Res 69(4):291–298. doi:10.1016/j.neures.2010.12.016
Yoshida H, Yanai H, Namiki Y, Fukatsu-Sasaki K, Furutani N, Tada N (2006) Neuroprotective effects of edaravone: a novel free radical scavenger in cerebrovascular injury. CNS Drug Rev 12(1):9–20. doi:10.1111/j.1527-3458.2006.00009.x
Higashi Y (2009) Edaravone for the treatment of acute cerebral infarction: role of endothelium-derived nitric oxide and oxidative stress. Expert Opin Pharmacother 10(2):323–331. doi:10.1517/14656560802636888
Mattson MP, Cheng A (2006) Neurohormetic phytochemicals: low-dose toxins that induce adaptive neuronal stress responses. Trends Neurosci 29(11):632–639. doi:10.1016/j.tins.2006.09.001
Foresti R, Bains SK, Pitchumony TS, de Castro Brás LE, Drago F, Dubois-Randé JL, Bucolo C, Motterlini R (2013) Small molecule activators of the Nrf2-HO-1 antioxidant axis modulate heme metabolism and inflammation in BV2 microglia cells. Pharmacol Res 76:132–148. doi:10.1016/j.phrs.2013.07.010
Sperner-Unterweger B, Kohl C, Fuchs D (2014) Immune changes and neurotransmitters: possible interactions in depression? Prog Neuropsychopharmacol Biol Psychiatry 48:268–276. doi:10.1016/j.pnpbp.2012.10.006
Cederbaum AI, Lu Y, Wang X, Wu D (2015) Synergistic toxic interactions between CYP2E1, LPS/TNFα, and JNK/p38 MAP kinase and their implications in alcohol-induced liver injury. Adv Exp Med Biol 815:145–172. doi:10.1007/978-3-319-09614-8_9
Yoshida H, Meng P, Matsumiya T et al (2014) Carnosic acid suppresses the production of amyloid-β 1-42 and 1-43 by inducing an α-secretase TACE/ADAM17 in U373MG human astrocytoma cells. Neurosci Res 79:83–93. doi:10.1016/j.neures.2013.11.004
Mangoura D, Sakellaridis N, Jones J, Vernadakis A (1989) Early and late passage C-6 glial cell growth: similarities with primary glial cells in culture. Neurochem Res 14(10):941–947
Kim SY, Park E, Park JA et al (2010) The plant phenolic diterpene carnosol suppresses sodium nitroprusside-induced toxicity in c6 glial cells. J Agric Food Chem 58(3):1543–1550. doi:10.1021/jf903294x
Vincent VA, Tilders FJ, Van Dam AM (1998) Production, regulation and role of nitric oxide in glial cells. Mediators Inflamm 7(4):239–255. doi:10.1080/09629359890929
Barres BA, Barde Y (2000) Neuronal and glial cell biology. Curr Opin Neurobiol 10(5):642–648. doi:10.1016/S0959-4388(00)00134-3
Jaronczyk K, Bui L, Soong JM, McLaughlin BE, Marks GS, Brien JF, Nakatsu K (2004) The source of heme for vascular heme oxygenase II: de novo heme biosynthesis in rat aorta. Can J Physiol Pharmacol 82(4):218–224
Wegiel B, Otterbein LE (2012) Go green: the anti-inflammatory effects of biliverdin reductase. Front Pharmacol 3:47. doi:10.3389/fphar.2012.00047
Cheng L, Li F, Ma R, Hu X (2015) Forsythiaside inhibits cigarette smoke-induced lung inflammation by activation of Nrf2 and inhibition of NF-κB. Int Immunopharmacol 28(1):494–499. doi:10.1016/j.intimp.2015.07.011
Choudhury S, Ghosh S, Gupta P, Mukherjee S, Chattopadhyay S (2015) Inflammation-induced ROS generation causes pancreatic cell death through modulation of Nrf2-NF-κB and SAPK/JNK pathway. Free Radic Res 20:1–41. doi:10.3109/10715762.2015.1075016
Jarrott B, Williams SJ (2015) Chronic brain inflammation: the neurochemical basis for drugs to reduce inflammation. Neurochem Res. doi:10.1007/s11064-015-1661-7
Azad N, Rasoolijazi H, Joghataie MT, Soleimani S (2011) Neuroprotective effects of carnosic acid in an experimental model of Alzheimer’s disease in rats. Cell J 13(1):39–44
Machado DG, Bettio LE, Cunha MP, Capra JC, Dalmarco JB, Pizzolatti MG, Rodrigues AL (2009) Antidepressant-like effect of the extract of Rosmarinus officinalis in mice: involvement of the monoaminergic system. Prog Neuropsychopharmacol Biol Psychiatry 33(4):642–650. doi:10.1016/j.pnpbp.2009.03.004
Machado DG, Cunha MP, Neis VB et al (2013) Antidepressant-like effects of fractions, essential oil, carnosol and betulinic acid isolated from Rosmarinus officinalis L. Food Chem 136(2):999–1005. doi:10.1016/j.foodchem.2012.09.028
Machado DG, Cunha MP, Neis VB et al (2012) Rosmarinus officinalis L. hydroalcoholic extract, similar to fluoxetine, reverses depressive-like behavior without altering learning deficit in olfactory bulbectomized mice. J Ethnopharmacol 143(1):158–169. doi:10.1016/j.jep.2012.06.017
Chen Y, Wang H, Zhang R, Wang H, Peng Z, Sun R, Tan Q (2012) Microinjection of sanguinarine into the ventrolateral orbital cortex inhibits Mkp-1 and exerts an antidepressant-like effect in rats. Neurosci Lett 506(2):327–331. doi:10.1016/j.neulet.2011.11.038
Zanella CA, Treichel H, Cansian RL, Roman SS (2012) The effects of acute administration of the hydroalcoholic extract of rosemary (Rosmarinus officinalis L.) (Lamiaceae) in animal models of memory. Braz J Pharm Sci 48(3):389–397. doi:10.1590/S1984-82502012000300005
Acknowledgments
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest Statement
None.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(XLSX 12 kb)
Rights and permissions
About this article

Cite this article
de Oliveira, M.R. The Dietary Components Carnosic Acid and Carnosol as Neuroprotective Agents: a Mechanistic View. Mol Neurobiol 53, 6155–6168 (2016). https://doi.org/10.1007/s12035-015-9519-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9519-1