
Abstract
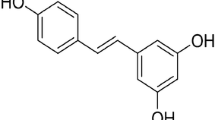
Resveratrol (3,4',5-trihydroxystilbene; C14H12O3) is a polyphenolic phytoalexin found in grapes, berries, peanuts, and wines. Resveratrol has been viewed as an antioxidant, anti-inflammatory, anti-apoptotic, and anticancer agent. Moreover, it has been reported that resveratrol modulates mitochondrial function, redox biology, and dynamics in both in vitro and in vivo experimental models. Resveratrol also attenuates mitochondrial impairment induced by certain stressors. Resveratrol upregulates, for example, mitochondria-located antioxidant enzymes, decreasing the production of reactive species by these organelles. Resveratrol also triggers mitochondrial biogenesis, ameliorating the mitochondria-related bioenergetics status in mammalian cells. In the present work, we discuss about the effects of resveratrol on brain mitochondria. Brain cells (both neuronal and glial) are susceptible to mitochondrial dysfunction due to their high demand for adenosine triphosphate (ATP). Additionally, brain cells consume oxygen (O2) at very high rates, leading to a proportionally high mitochondrial production of reactive species. Therefore, strategies focusing on the maintenance of mitochondrial function in these cell types are of pharmacological interest in the case of neurodegenerative diseases, which involve mitochondrial impairment and increased generation of reactive species, leading to neuroinflammation and cell death. The mechanism by which resveratrol protects mitochondrial function and dynamics is not completely understood, and further research would be necessary in order to investigate exactly how resveratrol affects mitochondria-related parameters. Furthermore, it is particularly important because resveratrol is able to induce cytotoxicity depending on its dosage.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Resveratrol (3,4',5-trihydroxystilbene; C14H12O3), a polyphenolic phytoalexin found in grapes, berries, peanuts, and wines, exhibits antioxidant, anti-inflammatory, anti-apoptotic, and anticancer capacities [1–6]. Resveratrol plays a central role in the “French paradox,” the inverse correlation between the intake of red wine and the incidence of cardiovascular disease [7, 8]. In addition to elicit beneficial effects in the cardiovascular system, resveratrol has been viewed as a neuroprotective agent. Resveratrol is effective in preventing redox impairment in brain cells in several in vitro and in vivo experimental models [9–13]. Additionally, resveratrol exerts anti-apoptotic actions in brain cells challenged with different types of stressors [14–18]. This polyphenol also modulates central nervous system function, as assessed in studies aiming to analyze the impacts of resveratrol on learning and memory abilities [19–23]. Resveratrol-induced antidepressive effects were also described experimentally [24–28]. Thus, resveratrol is able to cross the blood–brain barrier, exerting beneficial effects in both neuronal and glial cells [29]. Brain cells are very sensitive to energetic stress due to the high demand for adenosine triphosphate (ATP) in order to maintain neurotransmission, neuronal plasticity, synthesis of protein, cellular osmolarity, and cell division (depending on the cell type and the brain area), among other biological processes that consume ATP [30, 31]. Natural compounds that would be able to modulate bioenergetics in brain cells present potential to be used as neuroprotective agents [32]. In that context, resveratrol has been reported to be able to modulate mitochondrial function, redox biology, and dynamics in different cell types [12, 33–35].
Mitochondria are the major organelles involved in the synthesis of ATP in mammalian cells due to the presence of the components responsible for the maintenance of oxidative phosphorylation [36–38]. The electron flux between the complexes of the respiratory chain generates an electrochemical potential across the inner mitochondrial membrane that is further utilized by complex V (the so-called ATP synthase/ATPase) to produce ATP. Oxygen (O2) is the final acceptor of electrons and, in a reaction mediated by complex IV (also called cytochrome c oxidase), is reduced completely generating water [36]. The maintenance of the mitochondrial function is crucial to maintain the bioenergetic requirements of each cell type containing mitochondria. Perturbations in the mitochondrial function lead to several negative consequences, including impaired synthesis of biomolecules (lipids, proteins, nucleic acids, among others), disruption in the maintenance of cellular osmolarity, and cell death, to cite some examples [39, 40]. Actually, mitochondrial dysfunction has been associated with human diseases, such as neurodegeneration and cardiovascular diseases [41–48]. In that context, strategies aiming to prevent or to treat mitochondrial impairment attracted the attention of researchers mainly in the field of neurobiology, since the loss of neurons is irreversible in the majority of the brain areas [49, 50]. Additionally, mitochondria play a role in neuronal plasticity, which is a complex phenomenon necessary to maintain some central functions, such as learning and memory, among others [51, 52].
Several works have been published showing that dietary components may modulate mitochondrial function and dynamics, causing cytoprotection in different in vitro and in vivo experimental models involving animals and humans [12, 53–64]. In spite of this, there is a lack for information regarding the exact mechanism by which dietary factors alter mitochondria-related parameters causing benefits. Mitochondrial function may vary according to alterations in the redox environment and these organelles also contribute to change redox-related aspects in mammalian cells [65]. Number of mitochondria may be altered by the triggering of mitochondrial biogenesis (i.e., the synthesis of new mitochondria) or mitophagy (i.e., the degradation of mitochondria during autophagy) [66]. Additionally, mitochondrial fusion and fission alter mitochondrial number, size, and morphology [67]. Furthermore, mitochondria take a key role in mediating cell death through the intrinsic apoptotic pathway [68]. These mitochondria-related phenomena have been investigated in a research field that is called “mitochondrial medicine,” in which dietary factors are a major focus of interest [69].
Therefore, the aim of this work is to discuss how resveratrol would induce cytoprotective effects by modulating mitochondrial function, redox biology, and dynamics in several cell types. We focused on publications demonstrating the mechanisms by which resveratrol affects such parameters in mammalian cells. We also suggest here several experiments that would be useful to better understand how exposure to resveratrol impacts these organelles.
Mitochondrial Redox Biology: an Overview
The mitochondrial respiratory chain is a major site of reactive species production due to electron leakage [70]. The major sites of reactive species generation include complex I and complex III [71]. Superoxide anion radical (O2 −•) and hydrogen peroxide (H2O2) may be originated from alterations in the function of the respiratory chain due to variations in the energetic demand cells require [72]. Furthermore, changes in the O2 concentrations affect the respiratory chain activity and favor O2 −• production due to partial O2 reduction [70]. Several toxicants also cause mitochondrial dysfunction and increase the production of reactive species by the mitochondria [73, 74]. Mitochondria exhibit antioxidant defenses, such as the enzymatic and nonenzymatic. Among the enzymatic antioxidant defenses, Mn-superoxide dismutase (Mn-SOD) takes a crucial role in the conversion of O2 −• into H2O2, which reacts with glutathione peroxidase (GPx) or catalase (CAT) generating water [75–78].
The main nonenzymatic antioxidant defense is reduced glutathione (GSH) [75]. GSH is produced in the cytosol and its transport into mitochondria is necessary in order to attenuate redox impairment that may result from increased H2O2 production through the reactions of Mn-SOD [76] and/or from mitochondrial complexes [70]. Moreover, mitochondria-located monoamine oxidase (MAO) enzyme generates H2O2 during the reactions of neurotransmitter degradation [79]. H2O2 is a diffusible molecule that readily crosses biomembranes and may be viewed as both a manner to widespread oxidative stress and a signaling agent [80]. An imbalance in the activity of brain superoxide dismutases and CAT, for example, may render excessive H2O2 production and impaired redox homeostasis [81, 82]. Therefore, caution is needed when aiming to interfere with mitochondria-located SOD to avoid deregulation in the H2O2 concentrations in neuronal and glial cells, since the central nervous system (CNS) is rich in lipids and also presents high concentrations of iron and copper ions, as well as presents extremely high levels of reactive neurotransmitters [83]. The reaction of H2O2 with either iron or copper ions leads to the formation of hydroxyl (•OH), which is involved in neurodegenerative processes [84]. The reaction of H2O2 with O2 −• also generates •OH in such situations [83].
Mitochondria also contain an isoform of nitric oxide synthase (mtNOS), which produces nitric oxide (NO•), a free radical and signaling molecule [85, 86]. NO• may react with O2 −• from the respiratory chain yielding peroxynitrite (ONOO−), a reactive specie that reacts with tyrosine residues in proteins, leading to nitrosative stress [87, 88]. Moreover, peroxynitrous acid (ONOOH, which originated from ONOO−) generates nitryl cation (NO2 +), nitrogen diozide radical (·•NO2), and hydroxyl radical (·•OH) by homolytic fission [89, 90]. Nitrosative stress has been reported to affect mitochondrial function due to the damage ONOO− may cause in proteins present in the mitochondrial membranes [91, 92]. Additionally, nitrosative stress has been associated with several neurodegenerative diseases, such as Alzheimer’s disease [93], Parkinson’s disease [94–96], and Huntington’s disease [97, 98], among others [99–102].
The levels of antioxidant defenses in mitochondria are modulated by transcription factors, mainly nuclear factor erythroid 2-related factor 2 (Nrf2) and nuclear factor-κB (NF-κB) [103, 104]. Nrf2 has been viewed as a major redox modulator in mammalian cells [104]. Furthermore, Nrf2 participates in the regulation of mitochondria-related bioenergetics processes in different cell types [105–107]. Nrf2 is maintained in an inactive form in the cytosol through binding to Kelch-like ECH-associated protein 1 (Keap1) [108]. During exposure to pro-oxidant or electrophile agents, Nrf2 is released from Keap1 and translocates to the cell nucleus in order to modulate the expression of several antioxidant and detoxifying proteins [109]. The link between Nrf2 activation and maintenance of mitochondrial function has been examined as an important pharmacological target in cases of neurodegeneration, since mitochondrial impairment takes a crucial role in the pathophysiology of these conditions [104, 110]. The activation of NF-κB, a major immunomodulatory agent in mammalian cells, is regulated by the interaction with the inhibitor κB (IκB) proteins and has been linked to the maintenance of mitochondrial redox status, because NF-κB is involved in the control of Mn-SOD expression [103]. Additionally, it has been demonstrated that NF-κB upregulates the synthesis of glutathione-S-transferase (GST), which participates in phase II detoxification in several cell types [111]. GST increases the solubility of toxicants, favoring its excretion after conjugation with GSH [112]. Redox-active toxicants targeting mitochondria, for example, may be excreted more rapidly from cells, decreasing its effects on the organelles [111]. Both Nrf2 and NF-κB are modulated upstream by protein kinases, including the phosphoinositide-3-kinase (PI3K)/protein kinase B (PKB, also called Akt) signaling pathway, protein kinase C (PKC)-dependent signaling, and mitogen-activated protein kinases (MAPK), such as extracellular signal-regulated kinase (Erk), p38, and c-Jun N-terminal kinase (JNK), among others, as recently published in excellent reviews [105, 107, 113, 114].
Therefore, mitochondria are sources of reactive species and the maintenance of the function of these organelles is crucial to avoid redox impairment and cell death. Several works have been published demonstrating possible strategies to protect brain mitochondria by the modulation of antioxidant defenses located in the organelles [12, 53–62]. In spite of this, the exact mechanism underlying the chemically induced mitochondrial protection is not clear yet. Thereby, we have made several suggestions in this work that may be useful in further research involving modulation of mitochondrial redox biology in brain cells.
Mitochondrial Biogenesis: an Overview
Mitochondrial biogenesis (mitogenesis) is a complex process responsible for the synthesis of new mitochondria in mammalian cells [115–117]. Both nuclear and mitochondrial genomes are necessary to modulate mitochondrial biogenesis, which is an event needed to alter number of mitochondria according to the requirements of the cells [115]. Peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1α) is the major regulator in mitochondrial biogenesis by acting with the nuclear respiratory factors 1 and 2 (NRF-1 and NRF-2) [118]. PGC-1α upregulates NRF-1 and NRF-2, as well as the estrogen-related receptor α (ERRα) [119–121], leading to the expression of mitochondrial proteins encoded by nuclear DNA [122].
Among these proteins, upregulation of the mitochondrial transcription factor A (TFAM) and mitochondrial transcription factors B1 and B2 (TFB1M and TFB2M) leads to alterations in the DNA located in mitochondria, causing the synthesis of mitochondrial RNA [123]. TFAM is involved with both transcription and replication of mitochondrial DNA (mtDNA) [124, 125]. Additionally, TFAM has been viewed as an important modulator of mtDNA homeostasis and repair [125–127]. The regulation of PGC-1α involves lysine acetylation by the NAD+-dependent deacetylase sirtuin 1 (SIRT1) [128, 129]. SIRT1, in turn, is modulated by AMP-activated protein kinase (AMPK), which regulates NAD+ levels, causing SIRT1 activation and upregulation of PGC-1α by a SIRT1-mediated deacetylation [130]. The AMPK/SIRT1/PGC-1α axis is a major signaling pathway orchestrating mitochondrial function and dynamics in mammalian cells. Mitochondrial biogenesis impairments have been reported to take a crucial role in several diseases [131–133] and may be a pharmacological target to prevent or to treat such disturbances [33, 35, 54–62, 134].
Resveratrol-Induced Mitochondria-Related Effects: Focus on Brain Cells
Resveratrol is an antioxidant, anti-inflammatory, and anticancer agent, as reported by several research groups [135–140]. Additionally, the effects of resveratrol regarding mitochondrial function and dynamics are focus of research due to the role these organelles present in both physiological and pathological conditions [12]. The impact of resveratrol exposure includes alterations in mitochondrial function, redox state, dynamics, and synthesis of new mitochondria. In the present review, we describe, discuss, and compare (whenever possible due to availability of reported data) the effects of resveratrol focusing on brain mitochondria. Mitochondrial dysfunction in neuronal and glial cells has been viewed as an important component of neurodegeneration [141], and modulation of mitochondria-related factors by dietary agents represents a very interesting strategy to prevent or to treat neurodegenerative disorders.
Redox Effects of Resveratrol on Mitochondria of Brain Cells
Resveratrol modulates mitochondria-related redox aspects by an indirect way through the induction of the expression of mitochondria-located antioxidant enzymes, as will be discussed in this section. It is noteworthy that some of the concentrations of resveratrol utilized in in vitro studies are considered high when analyzing the levels resveratrol reaches after in vivo administration, as observed in works aiming to obtain data regarding resveratrol bioavailability [142–144]. Furthermore, resveratrol cytotoxicity has been studied in vitro, indicating that increased delivery of resveratrol to normal cells may be risky to mitochondrial function [145]. Thus, caution is needed when considering resveratrol to improve mitochondrial function and/or dynamics therapeutically.
In Vitro Experimental Models
Resveratrol may exert both antioxidant and pro-oxidant effects in mammalian cells depending on its concentration. The pro-oxidant actions elicited by resveratrol have been utilized to induce, for example, apoptosis in cancer cells related to brain tissue [146]. In spite of this, we focused here only on the mitochondria-related antioxidant effects elicited by resveratrol due to the potential this polyphenol presents as a neuroprotective agent in cases of neurodegenerative disorders in which mitochondrial impairment takes a central role (Table 1).
Kairisalo et al. [147] evidenced that resveratrol may modulate the mitochondria-related redox biology by a mechanism associated with the upregulation of Mn-SOD. Resveratrol (50 μM for 24 h) upregulated the expression of Mn-SOD and thioredoxin-2 (Trx2), as well as increased the content of X-linked inhibitor of apoptosis protein (XIAP), an anti-apoptotic protein, in neuron-like PC6-3 cells (a pheochromocytoma cell line). The authors also found that resveratrol increased the levels of NF-κB in that experimental model, a transcription factor that is modulated, at least in part, by XIAP and is involved in the regulation of the expression of both Mn-SOD and Trx2 [163]. Therefore, it is very likely that resveratrol ameliorated mitochondrial redox parameters through a XIAP/NF-κB signaling pathway in neuron-like PC6-3 cells. The utilization of specific inhibitors would be useful to understand exactly how resveratrol enhanced the levels of antioxidant proteins located in mitochondria. Thus, further studies are needed focusing in the mechanism by which resveratrol elicited such effects. Additionally, it would be interesting to investigate the effects of resveratrol on the transcription factor NF-κB in brain cells in different experimental models. Actually, Jang and Surh [164] reported that resveratrol co-treatment at lower concentrations (25 μM for 36 h) protected PC12 pheochromocytoma cells against amyloid-β peptide cytotoxicity by inhibiting NF-κB and suppressing mitochondria-mediated cell death. Therefore, this is an important point to be addressed in future research.
Sheu et al. [148] reported that resveratrol protected human adult retinal pigment epithelial (ARPE) cells against acrolein-induced oxidative stress and cell death by a mechanism involving mitochondrial protection. Resveratrol (20 μM for 48 h prior chemical challenge) abrogated the acrolein-induced upregulation in Mn-SOD immunocontent without altering the levels of Cu/Zn-SOD in ARPE cells. Resveratrol also ameliorated the respiratory capacity and the rates of oxidative phosphorylation in the cells exposed to acrolein. Interestingly, resveratrol did not induce mitochondrial biogenesis in that experimental model. Thus, resveratrol probably protected mitochondrial function by an indirect antioxidant effect without involving the synthesis of new organelles. The exact mechanism underlying the resveratrol-elicited Mn-SOD upregulation deserves attention because several transcription factors are involved in the modulation of Mn-SOD expression, as reviewed elsewhere [165]. Additionally, the PGC-1α-dependent signaling pathway, which is a specific target of resveratrol in mammalian cells, also coordinates the expression of antioxidant enzymes [166, 167]. In this view, mechanistic studies are welcome in order to reveal how resveratrol modulated Mn-SOD expression causing mitochondrial protection. Furthermore, some research groups have demonstrated that resveratrol suppressed the loss of mitochondrial membrane potential (MMP) induced by different challenges in several experimental models [168, 169]. Disruption of MMP leads to increased production of reactive species due to the leakage of electrons from the respiratory chain [170]. In addition, loss of MMP affects ATP production, causing bioenergetics deficits and, in some cases, cell death [68]. Thus, rescuing of MMP in cases of deleterious events potentially affecting mitochondria causes benefits regarding mitochondrial function and cell fate.
Resveratrol (5–10 μM for 48 h) co-treatment elicited mitochondria-related antioxidant effects by a mechanism potentially involving the activation of SIRT1/PGC-1α signaling pathway in mouse dopaminergic cell line SN4741 exposed to 1-methyl-4-phenylpyridinium ion (MPP+; an in vitro experimental model of Parkinson’s disease), as reported by Mudò et al. [149]. Resveratrol upregulated the levels of SIRT1, PGC-1α, Trx2, and Mn-SOD, without affecting the expression of CAT, NRF1, heme oxygenase-1 (HO-1), and XIAP. Consequently, resveratrol decreased reactive species generation and abrogated the loss of viability in MPP+-treated SN4741 cells. Similar data regarding Mn-SOD enzyme were obtained in an in vivo experimental model performed by the authors using resveratrol at 20 mg/kg (i.p.) 30 min before and 3 days after MPP+ administration. It would be important to confirm these effects of resveratrol in clinical trials, for example, using blood cells as the sample.
Narayanan et al. [150] have shown that resveratrol (25 μM for 2 h in an experimental model of resveratrol preconditioning) upregulated the levels of uncoupling protein 2 (UCP2) in astrocytes obtained from both wild-type and Nrf2−/− mice. Resveratrol also enhanced the mitochondrial production of H2O2 by complexes I and III. However, resveratrol failed to alter Mn-SOD levels in both wild-type and Nrf2−/− astrocytes. Increased UCP2 levels may lead to enhanced mitochondrial uncoupling-dependent reactive species production, causing cytoprotection by mild redox impairment. Nonetheless, Della-Morte et al. [171] previously reported that resveratrol preconditioning (10 mg/kg) decreased UCP2 levels in the mitochondria of CA1 region of rat hippocampus and enhanced the ADP/O ratio (an index of mitochondrial efficiency) by a SIRT1-dependent manner. Those discrepancies may be due to different experimental models utilized by those research groups, and further research is needed in order to better examine the mechanisms induced by resveratrol.
Bellaver et al. [151] recently demonstrated that resveratrol (100 μM) pretreatment (for 1 h) protected primary hippocampal astrocyte cultures against azide, a mitochondrial toxicant. The authors found that resveratrol prevented downregulation of cytochrome c oxidase (complex IV) expression, as well as suppressed loss of MMP and mitochondrial generation of reactive species in azide-treated cells. Additionally, resveratrol upregulated Mn-SOD activity in that experimental model. Inhibition of HO-1 abrogated the mitochondrial effects induced by resveratrol, clearly indicating a link between HO-1 modulation and mitochondrial protection. Actually, it has been reported that HO-1, which is also an enzyme presenting anti-inflammatory actions [172, 173], may take a role in the maintenance of mitochondrial function and dynamics [60, 174, 175]. Even though the authors did not investigate whether resveratrol would be able to activate Nrf2 (the major regulator of HO-1 expression during exposure to xenobiotics and/or pro-oxidant conditions), it is very likely that this transcription factor is involved with the regulation of both redox environment and mitochondrial function [104, 176].
Overall, in vitro studies demonstrate that resveratrol presents a promising role as a mitochondria-targeting agent. Nevertheless, it is necessary to investigate the involvement of other signaling pathways in mediating the mitochondrial protection elicited by resveratrol, as well as other factors that may take a role in the mechanism underlying such resveratrol-induced effects, as suggested above.
In Vivo Experimental Models
The resveratrol-induced mitochondria-related redox effects were also tested in in vivo experimental models. The benefits natural compounds elicit on mitochondria of brain cells in vivo may take a role in the maintenance of cognitive functions, since mitochondria play a crucial role not only in the production of ATP in these cells but also modulate cell fate according to several intrinsic and extrinsic factors (Table 2).
Robb et al. [152] reported that resveratrol (200 mg/kg day−1 for 4 weeks) added to high fat diet strongly enhanced the immunocontents and activity of Mn-SOD in mice brain. Resveratrol intake also induced a slight increase in citrate synthase activity, an index of mitochondria abundance. Thus, the increase in Mn-SOD content and activity is very likely to be due to specific regulation of resveratrol regarding gene expression of Mn-SOD and not a consequence, at least in part, of mitochondrial biogenesis. Interestingly, subcutaneous administration of resveratrol (100 mg/kg day−1) by using a mini-pump did not modulate Mn-SOD levels in mice brain, showing that the way by which resveratrol is delivered to the organism takes an important role in the regulation of gene expression. It would be also necessary to examine whether resveratrol would be able to modulate the expression of other mitochondria-located antioxidant enzymes, such as catalase and glutathione peroxidase. The specific modulation of mitochondrial antioxidant enzymes by natural compounds would lead to decreased levels of oxidative and nitrosative stress markers in the organelles, as previously demonstrated by some research groups using other natural compounds [61, 177–179]. Resveratrol (30 mg/kg day−1 for 7 days prior induction of lesion) induced antioxidant and mitochondria-related anti-apoptotic effects in an animal model of cerebral ischemia using rats. However, the redox state of mitochondrial components, such as mitochondrial proteins and mitochondrial membranes, was not analyzed by the authors.
Interestingly, Zhao et al. [153] have shown that resveratrol at 20 mg/kg (for 12 weeks, intragastric administration), but no higher doses of resveratrol (40–80 mg/kg for the same period), increased Mn-SOD enzyme activity in the hippocampus of ovariectomized rats exposed to d-galactose, causing structural and cognitive ameliorations. The mechanism underlying the effect of resveratrol in hippocampal mitochondria was not addressed by the authors, needing further research mainly by a possible link this effect may present with hippocampal-related cognition.
Effects of Resveratrol on Signaling Pathways Modulating Mitochondrial Fusion and Fission and Mitochondrial Biogenesis in Brain Cells
There is evidence that resveratrol triggers mitochondrial biogenesis in different cell types [12]. Additionally, resveratrol regulates mitochondrial fusion and fission in mammalian cells [158]. Mitochondrial fusion and fission are important to maintain mitochondrial function and morphology and have been implicated also in the maintenance of cell viability and in the complex regulation of cell death, as previously reviewed [180, 181]. Due to limited data reporting the effects of resveratrol on mitochondrial fusion and fission, we decided not to detail these events in the present work. The induction of mitochondrial biogenesis by natural or pharmacological agents attracted the attention of researchers due to the impact this strategy would present in human diseases in which mitochondrial dysfunction takes a role [33]. In this section, we describe, discuss, and compare (whenever possible) the effects induced by resveratrol regarding the synthesis of new mitochondria and/or mitochondrial dynamics in brain cells. We have focused on specific works that described the mechanisms underlying the mitochondrial biogenesis- and/or mitochondrial dynamics-related actions of resveratrol in brain cells (Table 3).
Alzheimer’s Disease
Manczak et al. [154] compared resveratrol with SS31 and MitoQ (two molecules designed to target mitochondria) regarding several parameters, including mitochondrial fusion and fission and mitochondrial biogenesis. The authors found that resveratrol (5 μM) pretreatment (for 48 h) protected mitochondria of N2a cells against exposure to amyloid-β (an experimental model mimicking Alzheimer’s disease). Resveratrol prevented amyloid-β-induced disruption of mitochondrial fusion and fission by maintaining the expression of genes such as mitofusin 2 (Mfn2), optic atrophy 1 (Opa1), dynamin-related protein 1 (Drp1), and mitochondrial fission 1 protein (Fis1). Resveratrol alone did not alter PGC-1α protein levels and failed to decrease the amyloid-β-elicited increase in the production of H2O2 by mitochondria. In that context, resveratrol did not trigger mitochondrial biogenesis. Interestingly, resveratrol alone inhibited cytochrome c oxidase enzyme activity in that experimental model. In spite of this, resveratrol prevented loss of MPP and inhibited swelling and fragmentation of mitochondria in amyloid-β-treated cells. On the other hand, comparisons made by the authors revealed that molecules specifically targeting mitochondria exhibited better effects than resveratrol. Therefore, it is very likely that strategies aiming to increase the delivery of resveratrol to mitochondria would exert more pronounced effects regarding the function and dynamics of these organelles. It would be especially useful in vivo, since amyloid-β peptides have been linked to mitochondrial dysfunction in experimental models using animals [40, 44].
Monocular Deprivation
Yu and Yang [155] have demonstrated that resveratrol (20 μM for 1 h) stimulated AMPK and acetyl-CoA carboxylase (ACC) phosphorylation in rat primary visual cortical neurons. Resveratrol also upregulated PGC-1α and NRF-1 expression by a dose-dependent manner. The authors also tested the effects of resveratrol on mitochondrial biogenesis-related signaling in the visual cortex of animals subjected to an in vivo experimental model of monocular deprivation (MD). Resveratrol (injected i.p. at 20 mg/kg body weight 1 day prior MD) prevented the MD-induced downregulation in the levels of phospho-AMPK, PGC-1α, and NRF-1 in the rat visual cortex. Importantly, the authors also analyzed whether resveratrol would be able to alter the number of mitochondria in vivo. Resveratrol enhanced the number of mitochondria per cell in the visual cortex of animals submitted to MD. Furthermore, resveratrol increased the levels of ATP in that brain area. In that context, resveratrol upregulated the signaling pathway associated with the control of mitochondrial biogenesis (AMPK-PGC-1α-NRF-1) and increased the number of mitochondria, resulting in the enhancement of mitochondrial quality and function. The authors also observed that resveratrol mimicked the effects of neuronal activity, which was induced by potassium chloride (KCl) experimentally. Further studies are necessary in order to better analyze the complete signaling pathway regulated by resveratrol and involved in the control of this biological event. The use of specific inhibitors and/or gene silencing/overexpression would be interesting strategies in order to reveal the exact mechanism by which resveratrol modulated the synthesis of new mitochondria. Additionally, it is recommended to investigate this issue in other brain areas due to the important role mitochondrial biogenesis would present in neurodegenerative processes in which mitochondrial dysfunction is involved.
Glaucomatous Neurodegeneration
Chen et al. [156] recently reported that resveratrol (40 μM for 12–24 h) co-treatment inhibited apoptosis in retinal ganglion cell line RGC-5 exposed to serum deprivation. Resveratrol suppressed the serum deprivation-induced loss of MMP and cytochrome c release, consequently blocking caspase-3 activation in RGC-5 cells by a mechanism dependent on SIRT1, since nicotinamide (a SIRT1 inhibitor) blocked the effects elicited by resveratrol. Resveratrol also maintained the normal mtDNA copy number and the number of mitochondria in serum-deprived cells by the same mechanism. Importantly, resveratrol rescued mitochondrial architecture by inhibiting serum deprivation-induced swelling of the organelles. The authors demonstrated that resveratrol enhanced SIRT1, NRF1, and TFAM levels in a time-dependent manner, affecting PGC-1α with little impact. Nonetheless, resveratrol favored PGC-1α entry into the cell nucleus, probably assisting with the translocation of this regulator to the nucleus, where it exerts its effect on modulating mitochondrial biogenesis. This is an important point to be addressed in further research: the exact role of resveratrol in mediating PGC-1α into the nucleus. The proteins translocase of the inner membrane (TIM) and translocase of the outer membrane (TOM) play a crucial role in the transport of proteins from the cytosol to the mitochondria, and whether resveratrol would modulate the mitochondrial transport remains to be investigated. Indirect evidence that resveratrol would modulate mitochondria-located SIRT have been reported by Morris-Blanco et al. [182]. This research group found that resveratrol (25 μM for 48 h) upregulated nicotinamide phosphoribosyltransferase (Nampt) in neuronal–glial cortical cultured cells. Nampt participates in the generation of NAD+, which regulates mitochondrial SIRTs, poly(ADP-ribose) polymerase (PARP), and the tricarboxylic acid (TCA) cycle. Resveratrol enhanced Nampt levels by a mechanism dependent on the activation of PKCε. Therefore, in addition to exhibiting the ability to modulate the cytoplasmic isoforms of SIRTs, resveratrol is very likely to upregulate mitochondria-located SIRT by a PKCε-Nampt axis. This modulation involving cytoplasmic–nucleus communication also deserves attention due to the impact alterations in the NAD+/NADH ratio would cause in mammalian cells, including regulation of bioenergetics parameters and cell fate.
Prenatal Stress
Cao et al. [157] have explored the effects of resveratrol on mitochondrial biogenesis in vitro and in a prenatal restraint stress (PRS) animal model. The authors have shown that resveratrol (100 μM for 24 h) stimulated the phosphorylation of AMPK and upregulated the expression of PGC-1α, complex I, and complex III in SH-SY5Y cells. Inhibition of AMPK by compound C revealed that the resveratrol-induced upregulation of complex I and complex III expression occurred by an AMPK-dependent manner. Compound C also blocked the in vitro effects elicited by resveratrol on Nrf2 and HO-1 expression. Interestingly, resveratrol (100 mg/kg day−1 during gestation, orally administrated) suppressed the PRS-induced downregulation in the levels of phospho-AMPK, PGC-1α, and mitochondrial complexes (I, III, IV, and V) in the rat hippocampus of both male and female offspring. Resveratrol also restored mtDNA in that experimental model. Redox effects elicited by resveratrol involved upregulation of Nrf2, NAD(P)H:quinone dehydrogenase 1 (NQO1), HO-1, and of the regulatory (GCLm) and catalytic (GCLc) subunits of glutamate-cysteine ligase in the hippocampus of the PRS offspring. Moreover, resveratrol decreased carbonyl and oxidized glutathione (GSSG) levels in that rat brain region. Cognitive dysfunction (deficits in exploration and success rate of exploration in T maze and escape latency in Morris water maze) was also ameliorated by resveratrol in the PRS group. Data obtained by the authors clearly demonstrate that resveratrol upregulated mitochondrial dynamics-related parameters in both in vitro and in vivo experimental models. However, redox aspects were not analyzed directly on mitochondria. Furthermore, it would be interesting to investigate whether the mitochondrial biogenesis-associated effects elicited by resveratrol would be related to the behavioral effects observed in that experimental model. Even though the authors have demonstrated that resveratrol enhanced the levels of neurotrophic factors in the hippocampus of the PRS group, it would be interesting to examine whether there is a causal link between upregulation of mitochondrial biogenesis and neuronal plasticity, for example. In another work, resveratrol (10 μM for 24 h) upregulated PGC-1α by an AMPK-dependent manner in an experimental model of insulin resistance using SH-SY5Y cells exposed to high glucose concentration [183]. Nonetheless, resveratrol enhanced the levels of complex I by an AMPK-independent mechanism in that work, contrasting with the data published by Cao et al. [157]. However, it is noteworthy that these research groups utilized very different experimental models to study the effects of resveratrol on mitochondria. Thus, data obtained by the authors have shown the complexity involved in the triggering of signaling pathways by resveratrol in mammalian cells. This should be considered when using this molecule pharmacologically.
Parkinson’s Disease
Pen et al. [158] have reported that resveratrol modulated mitochondrial biogenesis in PC12 cells exposed to rotenone (an experimental model mimicking Parkinson’s disease). Resveratrol (60 μM) pretreatment (24 h) enhanced mitochondrial mass (mtDNA) and prevented the rotenone-induced downregulation of proteins involved in the control of mitochondrial fusion and fission, such as Drp1, Fis1, OPA1, and MFN2. Resveratrol triggered mitochondrial biogenesis through a PGC-1α/mtTFA-dependent signaling pathway in rotenone-treated PC12 cells, resulting in increased levels of ATP. Resveratrol also elicited antioxidant effects by decreasing the formation of reactive species in that cell line. Data obtained by the authors were also confirmed in the brain of rats that were injected (i.p.) with resveratrol (50 mg/kg day−1 for 30 days) before administration of rotenone. Dysfunctional mitochondrial dynamics has been linked to neurodegeneration, such as Parkinson’s disease [12]. In that context, the use of resveratrol as a preventive agent in the case of impaired mitochondrial dynamics is of pharmacological interest.
Down Syndrome
Recently, Valenti et al. [159] have studied the effects of resveratrol (10 μM for 24 h) in neural progenitor cells (NPCs) obtained from the hippocampus of the Ts65Dn mouse, which has been utilized as an experimental model of Down syndrome. Ts65Dn NPCs exhibit mitochondria-related bioenergetics deficits, such as reduced activity of complexes I and V, as well as decreased ATP synthesis. Resveratrol increased complex I and complex V enzyme activities, consequently enhancing ATP production. Resveratrol also upregulated the immunocontents of PGC-1α, NRF-1, and TFAM in Ts65Dn NPCs. The content of mtDNA and the levels of complex I and complex V were augmented by resveratrol, strongly suggesting that this natural compound induced mitochondrial biogenesis. Resveratrol did not alter the levels of SIRT1 but increased its activity, since the amounts of acetylated histone 3 (a major target of SIRT1) were decreased by resveratrol treatment. Resveratrol also upregulated the levels of phospho-AMPK, which seems to be involved in the increments induced by resveratrol regarding the mitochondrial production of ATP in Ts65Dn NPCs, since the inhibition of AMPK abrogated the beneficial effects of resveratrol on mitochondrial function. Therefore, it is very likely that resveratrol activated mitochondrial biogenesis through the triggering of the AMPK-SIRT1-PGC-1α signaling pathway in Ts65Dn NPCs. The findings obtained by that research group are very interesting because it may be useful in the treatment of patients affected by Down syndrome, which present mitochondrial abnormalities, as previously reported [184, 185].
Amyotrophic Lateral Sclerosis
Interesting data demonstrating the in vivo ability of resveratrol in triggering mitochondrial biogenesis during energetic stress was published by Mancuso et al. [161]. Resveratrol-enriched diet (leading to a daily ingestion of 160 mg/kg resveratrol for 4 weeks) suppressed the increase in the levels of complexes I–V observed in the ventral part of the lumbar spinal cord in SOD1G93A mice, an experimental model of amyotrophic lateral sclerosis. Resveratrol also upregulated Fis-1 expression without affecting the levels of Mfn2, indicating that this polyphenol triggered mitochondrial fission but not mitochondrial fusion in SOD1G93A mice. The levels of phospho-AMPK were enhanced by resveratrol in those animals, indicating mitochondrial biogenesis. Resveratrol also normalized the autophagic flux in SOD1G93A mice by restoring the levels of LC3II and Beclin 1. Mitochondrial dysfunction has been associated with the neurodegeneration observed in amyotrophic lateral sclerosis [186–189], and the use of mitochondriotropic agents would be an interesting strategy in order to restore mitochondrial function and dynamics, leading to ameliorations in the redox and bioenergetic aspects altered in the cells of patients suffering from this disease.
Huntington’s Disease
Naia et al. [160] have revealed that resveratrol ameliorated mitochondrial function in in vitro (striatal and cortical neurons obtained from YAC128 transgenic mice embryos) and in in vivo experimental models mimicking Huntington’s disease. Resveratrol was tested at the concentrations of 1 and 5 μM for 96 h, and the effects have varied according to the cell type studied. For example, only resveratrol at 5 μM restored MMP in YAC128 cortical neurons, whereas resveratrol at 1 or 5 μM ameliorated MMP in YAC128 striatal neurons. Resveratrol did not activate SIRT1 in vitro. Moreover, resveratrol (1 mg/kg day−1 administrated subcutaneously through a mini-pump for 28 days) upregulated the mRNA levels of MT-ND5 (complex I subunit NADH dehydrogenase 5) and MT-CO1 (complex IV subunit cytochrome c oxidase I), which are encoded by mitochondria and take a role in the respiratory chain, in the YAC128 mice cortex. Interestingly, resveratrol did not affect the expression of cytochrome c (CYTC, which is encoded by the cell nucleus) in that mice brain region. Resveratrol upregulated SIRT1 in the striatum of both wild-type and YAC128 mice, whereas it did not affect SIRT1 in the cortex of YAC128 mice. Therefore, differential effects were elicited by resveratrol according to the mice brain region, among other factors. Further studies would be useful in order to reveal whether resveratrol would be able to increase the number of mitochondria in such brain regions.
Aging
Similarly, Palomera-Avalos et al. [162] have shown that resveratrol administration (through diet at 1 g/kg w/w for 8 weeks, rendering 160 mg/kg day−1 to each animal) improved cognition and mitochondria-related parameters in senescence-accelerated prone mouse (SAMP8) subjected to high-fat diet (HFD). Resveratrol induced an increase in the size of hippocampal mitochondria (mitochondrial fusion) through a mechanism involving upregulation of OPA1 and MFN2 proteins. Moreover, resveratrol ingestion caused an increase in the levels of the components of the respiratory chain (complexes I–IV) and of complex V (ATP synthase/ATPase). The authors also found that resveratrol increased the levels of phospho-AMPK without altering SIRT1 or PGC-1α. SAMP8 mice present spontaneously reduced learning and memory capacities and have been utilized in studies focusing aging [190]. The SAMP8 animals exhibit mitochondrial impairment regarding both bioenergetics and redox-related aspects [190–192]. In that context, resveratrol would be an interesting pharmacological agent presenting cytoprotective effects during aging. More studies are necessary in order to evaluate whether resveratrol would be able to modulate mitochondrial activity (through measuring the activity of complexes, for example). Moreover, we recommend testing the effects of resveratrol on SAMP8-mitochondrial susceptibility, i.e., the ability of mitochondria to resist against chemical challenges in ex vivo experiments after in vivo administration of the polyphenol.
Effects of Resveratrol on Autophagy in Brain Cells: Is There a Link with Mitochondria and the Maintenance of Mitochondrial Function and Dynamics?
Resveratrol is able to induce autophagy in brain cells depending on its concentration, among other factors (e.g., duration of the exposure, nutritional state of the cells) [16, 193, 194]. However, it is not still clear whether there is a link between the mitochondria-related effects elicited by resveratrol and the triggering of autophagy. Since autophagy is utilized by cells as an adaptive strategy during several forms of stress, and mitochondrial also play a role during stress response, it would be interesting to examine exactly how autophagy and mitochondrial biogenesis and dynamics interact in these conditions in the presence of resveratrol.
Wu et al. [195] reported that resveratrol (50 μM for 24–72 h) induced autophagy in rotenone-treated human neuroblastoma SH-SY5Y cells by a mechanism associated with the AMPK/SIRT1 signaling pathway. The activation of autophagy by resveratrol inhibited the rotenone-induced cell death in SH-SY5Y cells. Additionally, the authors have found that resveratrol increased α-synuclein clearance in PC12 cells expressing wild-type α-synuclein, A30P, or A53T α-synuclein mutants. α-Synuclein accumulation has been viewed in dopaminergic neurons obtained from patients suffering from Parkinson’s disease [196, 197]. Autophagy is involved in the clearance of several proteins, including α-synuclein, in neuronal cells, avoiding protein accumulation and widespread damage [198, 199]. The authors did not investigate mitochondria-related parameters in that work. However, autophagy is linked to mitochondrial function due to its role in controlling the levels of damage organelles and proteins [51]. In this view, autophagy is necessary to maintain mitochondrial function. Additionally, accumulation of α-synuclein may occur in cytosol and mitochondria, limiting mitochondrial function and increasing the production of reactive species [200–204].
A direct link between resveratrol-induced autophagy and the mitochondria-related apoptosis was reported by Jeong et al. [205], who demonstrated that resveratrol (2 μM, pretreatment for 12 h) blocked the effects of prion protein on MMP, Bax translocation to mitochondria, and cytochrome c release from the organelles by an autophagy-dependent manner, since the autophagy inhibitor 3-methyladenine (3-MA) abrogated the effects of cytoprotective effects of resveratrol in prion protein-treated SH-SY5Y cells. It is important to note that very low concentrations of resveratrol efficiently protected SH-SY5Y cells in that experimental model. This is a very interesting finding, since several studies demonstrate that the bioavailability of resveratrol is limited.
The relationship between resveratrol and autophagy has been also studied by Lin et al. [206] using the dopaminergic cell line SH-SY5Y exposed to rotenone. The authors found a link between resveratrol-dependent HO-1 expression and autophagy and cell death in experimental model. Resveratrol (20 μM) pretreatment (for 24 h) protected SH-SY5Y cells from the toxicity induced by rotenone through a mechanism associated with HO-1, which activated the autophagy-related signaling pathway and inhibited the mitochondria-associated intrinsic apoptotic pathway. In fact, HO-1 has been studied regarding its role in modulating autophagy and cell death in several experimental models [207–209]. However, it remains to be fully understood how HO-1 interacts with mitochondria during autophagy causing cytoprotection in mammalian cells by inhibiting mitochondria-related apoptosis.
The mitochondria–autophagy link needs more studies in order to be fully understood. This research area is complex because the response to stress, for example, may vary according to the cell type analyzed. Additionally, the effects elicited in vitro may be different from those observed in vivo due to resveratrol bioavailability. This is particularly important in the case of investigating strategies to induce apoptosis in cancer cells, which initiate autophagy as a response to chemical stressors, for example, decreasing the efficacy of the treatment in some cases [210, 211].
Conclusion
Resveratrol modulates mitochondrial function and dynamics by diverse mechanisms, causing cytoprotective effects in both in vitro and in vivo experimental models involving brain cells. It would be important to investigate whether resveratrol would cause mitochondria-related cytotoxic effects in brain cells in order to avoid triggering of cell death in healthy cells by the intrinsic apoptotic pathway, which is dependent on the release of pro-apoptotic factors by mitochondria, or by necrosis due to energetic collapse. Additionally, studying how the resveratrol-induced effects on mitochondria would be associated with neuronal plasticity would be useful in the treatment or prevention of several neurodegenerative diseases. Based on the fact that the resveratrol concentrations utilized in in vitro studies to induce mitochondrial effects may be considered high due to decreased bioavailability of this polyphenol in vivo, it may be recommended also to investigate the impact of nanotechnology-related strategies to improve the delivery of resveratrol to mammalian cells according to the particularities of each tissue, causing better effects in mitochondria. Overall, the mitochondria-resveratrol field is gradually expanding in research areas associated with neurobiology, pharmacology, nutrition, and biotechnology. Therefore, it is expected that the number of publications in this area will be increased, improving our knowledge about the use of resveratrol as a modulator of mitochondrial function, redox biology, and dynamics.
3-MA, 3-methyladenine; ACC, acetyl-CoA carboxylase; AMPK, AMP-activated protein kinase; CAT, catalase; CNS, central nervous system; Drp1, dynamin-related protein 1; Erk, extracellular signal-regulated kinase; ERRα, estrogen-related receptor α; Fis1, mitochondrial fission 1 protein; GCLc, catalytic subunit of glutamate-cysteine ligase; GCLm, regulatory subunit of glutamate-cysteine ligase; GPx, glutathione peroxidase; GSH, reduced glutathione; GSSG, oxidized glutathione; GST, glutathione-S-transferase; HFD, high fat diet; HO-1, heme oxygenase-1; IκB, inhibitor κB; JNK, c-Jun N-terminal kinase; Keap1, Kelch-like ECH-associated protein 1; MAPK, mitogen-activated protein kinases; MD, monocular deprivation; Mfn2, mitofusin 2; MMP, mitochondrial membrane potential; Mn-SOD, Mn-superoxide dismutase; MPP +, 1-methyl-4-phenylpyridinium ion; MT-CO1, complex IV subunit cytochrome c oxidase I; MT-ND5, complex I subunit NADH dehydrogenase 5; mtDNA, mitochondrial DNA; mtNOS, mitochondrial nitric oxide synthase; Nampt, nicotinamide phosphoribosyltransferase; NF-κB, nuclear factor-κB; NPCs, neural progenitor cells; NQO1, NAD(P)H:quinone dehydrogenase 1; Nrf2, nuclear factor erythroid 2-related factor 2; NRF-1, nuclear respiratory factor 1; NRF-2, nuclear respiratory factor 2; Opa1, optic atrophy 1; PARP, poly(ADP-ribose) polymerase; PI3K, phosphoinositide-3-kinase; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1-α; PKB, protein kinase B; PKC, protein kinase C; PRS, prenatal restraint stress; SIRT1, NAD+-dependent deacetylase sirtuin 1; TCA, tricarboxylic acid; TFAM, mitochondrial transcription factor A; TFB1M, mitochondrial transcription factor B1; TFB2M, mitochondrial transcription factor B2; TIM, translocase of the inner membrane; TOM, translocase of the outer membrane; Trx2, thioredoxin-2; UCP2, uncoupling protein 2; XIAP, X-linked inhibitor of apoptosis protein.
References
Shakibaei M, Harikumar KB, Aggarwal BB (2009) Resveratrol addiction: to die or not to die. Mol Nutr Food Res 53:115–128. doi:10.1002/mnfr.200800148
Hsieh TC, Wu JM (2010) Resveratrol: biological and pharmaceutical properties as anticancer molecule. Biofactors 36:360–369
Kalantari H, Das DK (2010) Physiological effects of resveratrol. Biofactors 36:401–406
Schnekenburger M, Dicato M, Diederich M (2014) Plant-derived epigenetic modulators for cancer treatment and prevention. Biotechnol Adv 32:1123–1132. doi:10.1016/j.biotechadv.2014.03.009
Chan S, Kantham S, Rao VM, Palanivelu MK, Pham HL, Shaw PN, McGeary RP, Ross BP (2016) Metal chelation, radical scavenging and inhibition of Aβ42 fibrillation by food constituents in relation to Alzheimer's disease. Food Chem 199:185–194. doi:10.1016/j.foodchem.2015.11.118
Blanquer-Rosselló MD, Hernández-López R, Roca P, Oliver J, Valle A (2016) Resveratrol induces mitochondrial respiration and apoptosis in SW620 colon cancer cells. Biochim Biophys Acta. doi:10.1016/j.bbagen.2016.10.009
Kopp P (1998) Resveratrol, a phytoestrogen found in red wine. A possible explanation for the conundrum of the ʻFrench paradoxʼ? Eur J Endocrinol 138:619–620
Yap S, Qin C, Woodman OL (2010) Effects of resveratrol and flavonols on cardiovascular function: physiological mechanisms. Biofactors 36:350–359
Albani D, Polito L, Signorini A, Forloni G (2010) Neuroprotective properties of resveratrol in different neurodegenerative disorders. Biofactors 36:370–376
Anandhan A, Tamilselvam K, Vijayraja D, Ashokkumar N, Rajasankar S, Manivasagam T (2010) Resveratrol attenuates oxidative stress and improves behaviour in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) challenged mice. Ann Neurosci 17:113–119. doi:10.5214/ans.0972-7531.1017304
Sun AY, Wang Q, Simonyi A, Sun GY (2010) Resveratrol as a therapeutic agent for neurodegenerative diseases. Mol Neurobiol 41:375–383. doi:10.1007/s12035-010-8111-y
de Oliveira MR, Nabavi SF, Manayi A, Daglia M, Hajheydari Z, Nabavi SM (2016) Resveratrol and the mitochondria: from triggering the intrinsic apoptotic pathway to inducing mitochondrial biogenesis, a mechanistic view. Biochim Biophys Acta 1860:727–745. doi:10.1016/j.bbagen.2016.01.017
Ahmed T, Javed S, Javed S, Tariq A, Šamec D, Tejada S, Nabavi SF, Braidy N et al (2016) Resveratrol and Alzheimer's disease: mechanistic insights. Mol Neurobiol. doi:10.1007/s12035-016-9839-9
Simonyi A, Wang Q, Miller RL, Yusof M, Shelat PB, Sun AY, Sun GY (2005) Polyphenols in cerebral ischemia: novel targets for neuroprotection. Mol Neurobiol 31:135–147
Lin CJ, Chen TH, Yang LY, Shih CM (2014) Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Dis 5:e1147. doi:10.1038/cddis.2014.123
Deng H, Mi MT (2016) Resveratrol attenuates Aβ25-35 caused neurotoxicity by inducing autophagy through the TyrRS-PARP1-SIRT1 signaling pathway. Neurochem Res 41:2367–2379. doi:10.1007/s11064-016-1950-9
Venigalla M, Sonego S, Gyengesi E, Sharman MJ, Münch G (2016) Novel promising therapeutics against chronic neuroinflammation and neurodegeneration in Alzheimer's disease. Neurochem Int 95:63–74. doi:10.1016/j.neuint.2015.10.011
Zeng W, Zhang W, Lu F, Gao L, Gao G (2016) Resveratrol attenuates MPP+-induced mitochondrial dysfunction and cell apoptosis via AKT/GSK-3β pathway in SN4741 cells. Neurosci Lett. doi:10.1016/j.neulet.2016.11.054
Sharma M, Gupta YK (2002) Chronic treatment with trans-resveratrol prevents intracerebroventricular streptozotocin induced cognitive impairment and oxidative stress in rats. Life Sci 71:2489–2498
Schmatz R, Mazzanti CM, Spanevello R, Stefanello N, Gutierres J, Corrêa M, da Rosa MM, Rubin MA et al (2009) Resveratrol prevents memory deficits and the increase in acetylcholinesterase activity in streptozotocin-induced diabetic rats. Eur J Pharmacol 610:42–48. doi:10.1016/j.ejphar.2009.03.032
Bagatini PB, Xavier LL, Bertoldi K, Moysés F, Lovatel G, Neves LT, Barbosa S, Saur L et al (2017) An evaluation of aversive memory and hippocampal oxidative status in streptozotocin-induced diabetic rats treated with resveratrol. Neurosci Lett 636:184–189. doi:10.1016/j.neulet.2016.10.059
Gocmez SS, Gacar N, Utkan T, Gacar G, Scarpace PJ, Tumer N (2016) Protective effects of resveratrol on aging-induced cognitive impairment in rats. Neurobiol Learn Mem 131:131–136. doi:10.1016/j.nlm.2016.03.022
Wang R, Zhang Y, Li J, Zhang C (2016) Resveratrol ameliorates spatial learning memory impairment induced by Aβ1-42 in rats. Neuroscience. doi:10.1016/j.neuroscience.2016.08.051
Hurley LL, Tizabi Y (2013) Neuroinflammation, neurodegeneration, and depression. Neurotox Res 23:131–144. doi:10.1007/s12640-012-9348-1
Wang X, Xie Y, Zhang T, Bo S, Bai X, Liu H, Li T, Liu S et al (2016) Resveratrol reverses chronic restraint stress-induced depression-like behaviour: involvement of BDNF level, ERK phosphorylation and expression of Bcl-2 and Bax in rats. Brain Res Bull 125:134–143. doi:10.1016/j.brainresbull.2016.06.014
Ge JF, Xu YY, Qin G, Cheng JQ, Chen FH (2016) Resveratrol ameliorates the anxiety- and depression-like behavior of subclinical hypothyroidism rat: possible involvement of the HPT axis, HPA axis, and Wnt/β-catenin pathway. Front Endocrinol (Lausanne) 7:44. doi:10.3389/fendo.2016.00044
Liu S, Li T, Liu H, Wang X, Bo S, Xie Y, Bai X, Wu L et al (2016) Resveratrol exerts antidepressant properties in the chronic unpredictable mild stress model through the regulation of oxidative stress and mTOR pathway in the rat hippocampus and prefrontal cortex. Behav Brain Res 302:191–199. doi:10.1016/j.bbr.2016.01.037
Tizabi Y (2016) Duality of antidepressants and neuroprotectants. Neurotox Res 30:1–13. doi:10.1007/s12640-015-9577-1
Wang Q, Xu J, Rottinghaus GE, Simonyi A, Lubahn D, Sun GY, Sun AY (2002) Resveratrol protects against global cerebral ischemic injury in gerbils. Brain Res 958:439–447
Dinuzzo M, Mangia S, Maraviglia B, Giove F (2012) The role of astrocytic glycogen in supporting the energetics of neuronal activity. Neurochem Res 37:2432–2438. doi:10.1007/s11064-012-0802-5
Falkowska A, Gutowska I, Goschorska M, Nowacki P, Chlubek D, Baranowska-Bosiacka I (2015) Energy metabolism of the brain, including the cooperation between astrocytes and neurons, especially in the context of glycogen metabolism. Int J Mol Sci 16:25959–25981. doi:10.3390/ijms161125939
de Oliveira MR (2016) The dietary components carnosic acid and carnosol as neuroprotective agents: a mechanistic view. Mol Neurobiol 53:6155–6168
Valero T (2014) Mitochondrial biogenesis: pharmacological approaches. Curr Pharm Des 20:5507–5509
Gibellini L, Bianchini E, De Biasi S, Nasi M, Cossarizza A, Pinti M (2015) Natural compounds modulating mitochondrial functions. Evid Based Complement Alternat Med 2015:527209. doi:10.1155/2015/527209
Tellone E, Galtieri A, Russo A, Giardina B, Ficarra S (2015) Resveratrol: a focus on several neurodegenerative diseases. Oxidative Med Cell Longev 2015:392169. doi:10.1155/2015/392169
Papa S, Martino PL, Capitanio G, Gaballo A, De Rasmo D, Signorile A, Petruzzella V (2012) The oxidative phosphorylation system in mammalian mitochondria. Adv Exp Med Biol 942:3–37. doi:10.1007/978-94-007-2869-1_1
Chaban Y, Boekema EJ, Dudkina NV (2014) Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim Biophys Acta 1837:418–426. doi:10.1016/j.bbabio.2013.10.004
Russell AP, Foletta VC, Snow RJ, Wadley GD (2014) Skeletal muscle mitochondria: a major player in exercise, health and disease. Biochim Biophys Acta 1840:1276–1284. doi:10.1016/j.bbagen.2013.11.016
Zhou L, Chen P, Peng Y, Ouyang R (2016) Role of oxidative stress in the neurocognitive dysfunction of obstructive sleep apnea syndrome. Oxidative Med Cell Longev 2016:9626831
Islam MT (2017) Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res 39:73–82
Brown GC, Bal-Price A (2003) Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol Neurobiol 27:325–355
Naoi M, Maruyama W, Shamoto-Nagai M, Yi H, Akao Y, Tanaka M (2005) Oxidative stress in mitochondria: decision to survival and death of neurons in neurodegenerative disorders. Mol Neurobiol 31:81–93
Kasote DM, Hegde MV, Katyare SS (2013) Mitochondrial dysfunction in psychiatric and neurological diseases: cause(s), consequence(s), and implications of antioxidant therapy. Biofactors 39:392–406
Cadonic C, Sabbir MG, Albensi BC (2016) Mechanisms of mitochondrial dysfunction in Alzheimer's disease. Mol Neurobiol 53:6078–6090. doi:10.1007/s12035-015-9515-5
Hu H, Tan CC, Tan L, Yu JT (2016) A mitocentric view of Alzheimer's disease. Mol Neurobiol. doi:10.1007/s12035-016-0117-7
Balog J, Mehta SL, Vemuganti R (2016) Mitochondrial fission and fusion in secondary brain damage after CNS insults. J Cereb Blood Flow Metab 36:2022–2033
Bergman O, Ben-Shachar D (2016) Mitochondrial oxidative phosphorylation system (OXPHOS) deficits in schizophrenia: possible interactions with cellular processes. Can J Psychiatr 61:457–469. doi:10.1177/0706743716648290
Zulian A, Schiavone M, Giorgio V, Bernardi P (2016) Forty years later: mitochondria as therapeutic targets in muscle diseases. Pharmacol Res 113(Pt A):563–573. doi:10.1016/j.phrs.2016.09.043
Luo C, Ikegaya Y, Koyama R (2016) Microglia and neurogenesis in the epileptic dentate gyrus. Neurogenesis (Austin) 3:e1235525
Gonçalves JT, Schafer ST, Gage FH (2016) Adult neurogenesis in the hippocampus: from stem cells to behavior. Cell 167:897–914. doi:10.1016/j.cell.2016.10.021
Raefsky SM, Mattson MP (2016) Adaptive responses of neuronal mitochondria to bioenergetic challenges: roles in neuroplasticity and disease resistance. Free Radic Biol Med 102:203–216. doi:10.1016/j.freeradbiomed.2016.11.045
Mattson MP (2007) Mitochondrial regulation of neuronal plasticity. Neurochem Res 32:707–715
Atamna H, Mackey J, Dhahbi JM (2012) Mitochondrial pharmacology: electron transport chain bypass as strategies to treat mitochondrial dysfunction. Biofactors 38:158–166
Gruber J, Fong S, Chen CB, Yoong S, Pastorin G, Schaffer S, Cheah I, Halliwell B (2013) Mitochondria-targeted antioxidants and metabolic modulators as pharmacological interventions to slow ageing. Biotechnol Adv 31:563–592. doi:10.1016/j.biotechadv.2012.09.005
de Oliveira MR, Ferreira GC, Schuck PF, Dal Bosco SM (2015) Role for the PI3K/Akt/Nrf2 signaling pathway in the protective effects of carnosic acid against methylglyoxal-induced neurotoxicity in SH-SY5Y neuroblastoma cells. Chem Biol Interact 242:396–406. doi:10.1016/j.cbi.2015.11.003
de Oliveira MR, Nabavi SF, Habtemariam S, Erdogan Orhan I, Daglia M, Nabavi SM (2015) The effects of baicalein and baicalin on mitochondrial function and dynamics: a review. Pharmacol Res 100:296–308. doi:10.1016/j.phrs.2015.08.021
de Oliveira MR, Nabavi SM, Braidy N, Setzer WN, Ahmed T, Nabavi SF (2016) Quercetin and the mitochondria: a mechanistic view. Biotechnol Adv 34:532–549. doi:10.1016/j.biotechadv.2015.12.014
de Oliveira MR, Jardim FR, Setzer WN, Nabavi SM, Nabavi SF (2016) Curcumin, mitochondrial biogenesis, and mitophagy: exploring recent data and indicating future needs. Biotechnol Adv 34:813–826. doi:10.1016/j.biotechadv.2016.04.004
Oliveira MR, Nabavi SF, Daglia M, Rastrelli L, Nabavi SM (2016) Epigallocatechin gallate and mitochondria—a story of life and death. Pharmacol Res 104:70–85. doi:10.1016/j.phrs.2015.12.027
de Oliveira MR, Peres A, Ferreira GC, Schuck PF, Gama CS, Bosco SM (2016) Carnosic acid protects mitochondria of human neuroblastoma SH-SY5Y cells exposed to paraquat through activation of the Nrf2/HO-1Axis. Mol Neurobiol. doi:10.1007/s12035-016-0100-3
de Oliveira MR, Peres A, Ferreira GC, Schuck PF, Bosco SM (2016) Carnosic acid affords mitochondrial protection in chlorpyrifos-treated Sh-Sy5y cells. Neurotox Res 30:367–379. doi:10.1007/s12640-016-9620-x
de Oliveira MR, Ferreira GC, Schuck PF (2016) Protective effect of carnosic acid against paraquat-induced redox impairment and mitochondrial dysfunction in SH-SY5Y cells: role for PI3K/Akt/Nrf2 pathway. Toxicol in Vitro 32:41–54. doi:10.1016/j.tiv.2015.12.005
Ueta CB, Gomes KS, Ribeiro MA, Mochly-Rosen D, Ferreira JC (2016) Disruption of mitochondrial quality control in peripheral artery disease: new therapeutic opportunities. Pharmacol Res 115:96–106. doi:10.1016/j.phrs.2016.11.016
Sandoval-Acuña C, Ferreira J, Speisky H (2014) Polyphenols and mitochondria: an update on their increasingly emerging ROS-scavenging independent actions. Arch Biochem Biophys 559:75–90. doi:10.1016/j.abb.2014.05.017
Jiang T, Sun Q, Chen S (2016) Oxidative stress: a major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson's disease and Alzheimer's disease. Prog Neurobiol 147:1–19. doi:10.1016/j.pneurobio.2016.07.005
Zhang Y, Xu H (2016) Translational regulation of mitochondrial biogenesis. Biochem Soc Trans 44:1717–1724. doi:10.1042/BST20160071C
Lee H, Yoon Y (2016) Mitochondrial fission and fusion. Biochem Soc Trans 44:1725–1735. doi:10.1042/BST20160129
Green DR, Galluzzi L, Kroemer G (2014) Metabolic control of cell death. Science 345:1250256. doi:10.1126/science.1250256
Picard M, Wallace DC, Burelle Y (2016) The rise of mitochondria in medicine. Mitochondrion 30:105–116. doi:10.1016/j.mito.2016.07.003
Zorov DB, Juhaszova M, Sollott SJ (2014) Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94:909–950. doi:10.1152/physrev.00026.2013
Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, Uchida K, Arimura K et al (1999) Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res 85:357–363
Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J Physiol 552:335–344
de Oliveira MR, Jardim FR (2016) Cocaine and mitochondria-related signaling in the brain: a mechanistic view and future directions. Neurochem Int 92:58–66. doi:10.1016/j.neuint.2015.12.006
de Oliveira MR (2015) Vitamin A and retinoids as mitochondrial toxicants. Oxidative Med Cell Longev 2015:140267. doi:10.1155/2015/140267
Lu SC (2013) Glutathione synthesis. Biochim Biophys Acta 1830:3143–3153. doi:10.1016/j.bbagen.2012.09.008
Flynn JM, Melov S (2013) SOD2 in mitochondrial dysfunction and neurodegeneration. Free Radic Biol Med 62:4–12. doi:10.1016/j.freeradbiomed.2013.05.027
Morris G, Anderson G, Dean O, Berk M, Galecki P, Martin-Subero M, Maes M (2014) The glutathione system: a new drug target in neuroimmune disorders. Mol Neurobiol 50:1059–1084. doi:10.1007/s12035-014-8705-x
Barrera G, Gentile F, Pizzimenti S, Canuto RA, Daga M, Arcaro A, Cetrangolo GP, Lepore A et al (2016) Mitochondrial dysfunction in cancer and neurodegenerative diseases: spotlight on fatty acid oxidation and lipoperoxidation products. Antioxidants (Basel) 5:7. doi:10.3390/antiox5010007
Edmondson DE (2014) Hydrogen peroxide produced by mitochondrial monoamine oxidase catalysis: biological implications. Curr Pharm Des 20:155–160
Veal EA, Day AM, Morgan BA (2007) Hydrogen peroxide sensing and signaling. Mol Cell 26:1–14
Pinho RA, Andrades ME, Oliveira MR, Pirola AC, Zago MS, Silveira PC, Dal-Pizzol F, Moreira JC (2006) Imbalance in SOD/CAT activities in rat skeletal muscles submitted to treadmill training exercise. Cell Biol Int 30:848–853
Andrades M, Ritter C, de Oliveira MR, Streck EL, Fonseca Moreira JC, Dal-Pizzol F (2011) Antioxidant treatment reverses organ failure in rat model of sepsis: role of antioxidant enzymes imbalance, neutrophil infiltration, and oxidative stress. J Surg Res 167:e307–e313. doi:10.1016/j.jss.2009.08.005
Halliwell B (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97:1634–1658
Gutteridge JM (1994) Hydroxyl radicals, iron, oxidative stress, and neurodegeneration. Ann N Y Acad Sci 738:201–213
Ghafourifar P, Cadenas E (2005) Mitochondrial nitric oxide synthase. Trends Pharmacol Sci 26:190–195
Finocchietto PV, Franco MC, Holod S, Gonzalez AS, Converso DP, Antico Arciuch VG, Serra MP, Poderoso JJ et al (2009) Mitochondrial nitric oxide synthase: a masterpiece of metabolic adaptation, cell growth, transformation, and death. Exp Biol Med (Maywood) 234:1020–1028. doi:10.3181/0902-MR-81
Valez V, Cassina A, Batinic-Haberle I, Kalyanaraman B, Ferrer-Sueta G, Radi R (2013) Peroxynitrite formation in nitric oxide-exposed submitochondrial particles: detection, oxidative damage and catalytic removal by Mn-porphyrins. Arch Biochem Biophys 529:45–54. doi:10.1016/j.abb.2012.10.012
Campolo N, Bartesaghi S, Radi R (2014) Metal-catalyzed protein tyrosine nitration in biological systems. Redox Rep 19:221–231. doi:10.1179/1351000214Y.0000000099
Alvarez B, Radi R (2003) Peroxynitrite reactivity with amino acids and proteins. Amino Acids 25:295–311
Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM (2007) Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci 8:766–775
De Oliveira MR, Oliveira MW, Da Rocha RF, Moreira JC (2009) Vitamin A supplementation at pharmacological doses induces nitrosative stress on the hypothalamus of adult Wistar rats. Chem Biol Interact 180:407–413. doi:10.1016/j.cbi.2009.02.006
de Oliveira MR, da Rocha RF, Stertz L, Fries GR, de Oliveira DL, Kapczinski F, Moreira JC (2011) Total and mitochondrial nitrosative stress, decreased brain-derived neurotrophic factor (BDNF) levels and glutamate uptake, and evidence of endoplasmic reticulum stress in the hippocampus of vitamin A-treated rats. Neurochem Res 36:506–517. doi:10.1007/s11064-010-0372-3
Swomley AM, Butterfield DA (2015) Oxidative stress in Alzheimer disease and mild cognitive impairment: evidence from human data provided by redox proteomics. Arch Toxicol 89:1669–1680. doi:10.1007/s00204-015-1556-z
Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM et al (2004) Nitrosative stress linked to sporadic Parkinson's disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci U S A 101:10810–10814
Gu Z, Nakamura T, Yao D, Shi ZQ, Lipton SA (2005) Nitrosative and oxidative stress links dysfunctional ubiquitination to Parkinson's disease. Cell Death Differ 12:1202–1204
Pal R, Miranda M, Narayan M (2011) Nitrosative stress-induced Parkinsonian Lewy-like aggregates prevented through polyphenolic phytochemical analog intervention. Biochem Biophys Res Commun 404:324–329. doi:10.1016/j.bbrc.2010.11.117
Sorolla MA, Rodríguez-Colman MJ, Vall-llaura N, Tamarit J, Ros J, Cabiscol E (2012) Protein oxidation in Huntington disease. Biofactors 38:173–185
Tasset I, Sánchez-López F, Agüera E, Fernández-Bolaños R, Sánchez FM, Cruz-Guerrero A, Gascón-Luna F, Túnez I (2012) NGF and nitrosative stress in patients with Huntington's disease. J Neurol Sci 315:133–136. doi:10.1016/j.jns.2011.12.014
Bossy-Wetzel E, Schwarzenbacher R, Lipton SA (2004) Molecular pathways to neurodegeneration. Nat Med 10(Suppl):S2–S9. doi:10.1038/nm1067
Anderson G, Maes M, Berk M (2013) Schizophrenia is primed for an increased expression of depression through activation of immuno-inflammatory, oxidative and nitrosative stress, and tryptophan catabolite pathways. Prog Neuro-Psychopharmacol Biol Psychiatry 42:101–114. doi:10.1016/j.pnpbp.2012.07.016
Moylan S, Berk M, Dean OM, Samuni Y, Williams LJ, O'Neil A, Hayley AC, Pasco JA et al (2014) Oxidative & nitrosative stress in depression: why so much stress? Neurosci Biobehav Rev 45:46–62. doi:10.1016/j.neubiorev.2014.05.007
Morris G, Walder K, Puri BK, Berk M, Maes M (2016) The deleterious effects of oxidative and nitrosative stress on palmitoylation, membrane lipid rafts and lipid-based cellular signalling: new drug targets in neuroimmune disorders. Mol Neurobiol 53:4638–4658. doi:10.1007/s12035-015-9392-y
Delhalle S, Deregowski V, Benoit V, Merville MP, Bours V (2002) NF-kappaB-dependent MnSOD expression protects adenocarcinoma cells from TNF-alpha-induced apoptosis. Oncogene 21:3917–3924
Esteras N, Dinkova-Kostova AT, Abramov AY (2016) Nrf2 activation in the treatment of neurodegenerative diseases: a focus on its role in mitochondrial bioenergetics and function. Biol Chem 397:383–400. doi:10.1515/hsz-2015-0295
Hayes JD, Dinkova-Kostova AT (2014) The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 39:199–218. doi:10.1016/j.tibs.2014.02.002
Piantadosi CA, Suliman HB (2012) Transcriptional control of mitochondrial biogenesis and its interface with inflammatory processes. Biochim Biophys Acta 1820:532–541. doi:10.1016/j.bbagen.2012.01.003
Denzer I, Münch G, Friedland K (2016) Modulation of mitochondrial dysfunction in neurodegenerative diseases via activation of nuclear factor erythroid-2-related factor 2 by food-derived compounds. Pharmacol Res 103:80–94. doi:10.1016/j.phrs.2015.11.019
Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL (2013) The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol 1:45–49. doi:10.1016/j.redox.2012.10.001
Zhang M, An C, Gao Y, Leak RK, Chen J, Zhang F (2013) Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog Neurobiol 100:30–47. doi:10.1016/j.pneurobio.2012.09.003
Gilmore TD (2006) Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 25:6680–6684
Lin CY, Chen JH, Fu RH, Tsai CW (2014) Induction of Pi form of glutathione S-transferase by carnosic acid is mediated through PI3K/Akt/NF-κB pathway and protects against neurotoxicity. Chem Res Toxicol 27:1958–1966. doi:10.1021/tx5003063
Sheehan D, Meade G, Foley VM, Dowd CA (2001) Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem J 360:1–16
Nguyen T, Nioi P, Pickett CB (2009) The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 284:13291–13295. doi:10.1074/jbc.R900010200
Ghosh S, Dass JF (2016) Study of pathway cross-talk interactions with NF-κB leading to its activation via ubiquitination or phosphorylation: a brief review. Gene 584:97–109. doi:10.1016/j.gene.2016.03.008
Hock MB, Kralli A (2009) Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol 71:177–203. doi:10.1146/annurev.physiol.010908.163119
Jornayvaz FR, Shulman GI (2010) Regulation of mitochondrial biogenesis. Essays Biochem 47:69–84. doi:10.1042/bse0470069
Ungvari Z, Sonntag WE, de Cabo R, Baur JA, Csiszar A (2011) Mitochondrial protection by resveratrol. Exerc Sport Sci Rev 39:128–132. doi:10.1097/JES.0b013e3182141f80
Scarpulla RC (2011) Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 1813:1269–1278. doi:10.1016/j.bbamcr.2010.09.019
Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, Oakeley EJ, Kralli A (2004) The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc Natl Acad Sci U S A 101:6472–6477
Virbasius CA, Virbasius JV, Scarpulla RC (1993) NRF-1, an activator involved in nuclear-mitochondrial interactions, utilizes a new DNA-binding domain conserved in a family of developmental regulators. Genes Dev 7:2431–2445
Schreiber SN, Knutti D, Brogli K, Uhlmann T, Kralli A (2003) The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha). J Biol Chem 278:9013–9018
Anderson R, Prolla T (2009) PGC-1alpha in aging and anti-aging interventions. Biochim Biophys Acta 1790:1059–1066. doi:10.1016/j.bbagen.2009.04.005
Gleyzer N, Vercauteren K, Scarpulla RC (2005) Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol Cell Biol 25:1354–1366
Fisher RP, Clayton DA (1988) Purification and characterization of human mitochondrial transcription factor 1. Mol Cell Biol 8:3496–3509
Canugovi C, Maynard S, Bayne AC, Sykora P, Tian J, de Souza-Pinto NC, Croteau DL, Bohr VA (2010) The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair (Amst) 9:1080–1089. doi:10.1016/j.dnarep.2010.07.009
Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM et al (2004) Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet 13:935–944
Picca A, Lezza AM (2015) Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: useful insights from aging and calorie restriction studies. Mitochondrion 25:67–75. doi:10.1016/j.mito.2015.10.001
Nemoto S, Fergusson MM, Finkel T (2005) SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J Biol Chem 280:16456–16460
Sugden MC, Caton PW, Holness MJ (2010) PPAR control: it's SIRTainly as easy as PGC. J Endocrinol 204:93–104. doi:10.1677/JOE-09-0359
Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P et al (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458:1056–1060. doi:10.1038/nature07813
Paoli A, Bianco A, Damiani E, Bosco G (2014) Ketogenic diet in neuromuscular and neurodegenerative diseases. Biomed Res Int 2014:474296. doi:10.1155/2014/474296
Wang KZ, Zhu J, Dagda RK, Uechi G, Cherra SJ 3rd, Gusdon AM, Balasubramani M, Chu CT (2014) ERK-mediated phosphorylation of TFAM downregulates mitochondrial transcription: implications for Parkinson's disease. Mitochondrion 17:132–140. doi:10.1016/j.mito.2014.04.008
Banerjee K, Munshi S, Frank DE, Gibson GE (2015) Abnormal glucose metabolism in Alzheimer's disease: relation to autophagy/mitophagy and therapeutic approaches. Neurochem Res 40:2557–2569. doi:10.1007/s11064-015-1631-0
de Olivera MR (2016) Evidence for genistein as a mitochondriotropic molecule. Mitochondrion 29:35–44. doi:10.1016/j.mito.2016.05.005
Schmitt CA, Heiss EH, Dirsch VM (2010) Effect of resveratrol on endothelial cell function: molecular mechanisms. Biofactors 36:342–349
Dragone T, Cianciulli A, Calvello R, Porro C, Trotta T, Panaro MA (2014) Resveratrol counteracts lipopolysaccharide-mediated microglial inflammation by modulating a SOCS-1 dependent signaling pathway. Toxicol in Vitro 28:1126–1135. doi:10.1016/j.tiv.2014.05.005
Sun D, Yue Q, Guo W, Li T, Zhang J, Li G, Liu Z, Sun J (2015) Neuroprotection of resveratrol against neurotoxicity induced by methamphetamine in mouse mesencephalic dopaminergic neurons. Biofactors 41:252–260
Lopez MS, Dempsey RJ, Vemuganti R (2015) Resveratrol neuroprotection in stroke and traumatic CNS injury. Neurochem Int 89:75–82. doi:10.1016/j.neuint.2015.08.009
Wang Y, Jiang Y, Fan X, Tan H, Zeng H, Wang Y, Chen P, Huang M et al (2015) Hepato-protective effect of resveratrol against acetaminophen-induced liver injury is associated with inhibition of CYP-mediated bioactivation and regulation of SIRT1-p53 signaling pathways. Toxicol Lett 236:82–89. doi:10.1016/j.toxlet.2015.05.001
Steiner N, Balez R, Karunaweera N, Lind JM, Münch G, Ooi L (2016) Neuroprotection of Neuro2a cells and the cytokine suppressive and anti-inflammatory mode of action of resveratrol in activated RAW264.7 macrophages and C8-B4 microglia. Neurochem Int 5:46–54. doi:10.1016/j.neuint.2015.10.013
Arun S, Liu L, Donmez G (2016) Mitochondrial biology and neurological diseases. Curr Neuropharmacol 14:143–154
Erdogan CS, Vang O (2016) Challenges in analyzing the biological effects of resveratrol. Nutrients 8:353. doi:10.3390/nu8060353
Morbidelli L (2016) Polyphenol-based nutraceuticals for the control of angiogenesis: analysis of the critical issues for human use. Pharmacol Res 111:384–393. doi:10.1016/j.phrs.2016.07.011
Weiskirchen S, Weiskirchen R (2016) Resveratrol: how much wine do you have to drink to stay healthy? Adv Nutr 7:706–718. doi:10.3945/an.115.011627
Rowlands BD, Lau CL, Ryall JG, Thomas DS, Klugmann M, Beart PM, Rae CD (2015) Silent information regulator 1 modulator resveratrol increases brain lactate production and inhibits mitochondrial metabolism, whereas SRT1720 increases oxidative metabolism. J Neurosci Res 93:1147–1156. doi:10.1002/jnr.23570
Varoni EM, Lo Faro AF, Sharifi-Rad J, Iriti M (2016) Anticancer molecular mechanisms of resveratrol. Front Nutr 3:8. doi:10.3389/fnut.2016.00008
Kairisalo M, Bonomo A, Hyrskyluoto A, Mudò G, Belluardo N, Korhonen L, Lindholm D (2011) Resveratrol reduces oxidative stress and cell death and increases mitochondrial antioxidants and XIAP in PC6.3-cells. Neurosci Lett 488:263–266. doi:10.1016/j.neulet.2010.11.042
Sheu SJ, Liu NC, Ou CC, Bee YS, Chen SC, Lin HC, Chan JY (2013) Resveratrol stimulates mitochondrial bioenergetics to protect retinal pigment epithelial cells from oxidative damage. Invest Ophthalmol Vis Sci 54:6426–6438. doi:10.1167/iovs.13-12024
Mudò G, Mäkelä J, Di Liberto V, Tselykh TV, Olivieri M, Piepponen P, Eriksson O, Mälkiä A et al (2012) Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson's disease. Cell Mol Life Sci 69:1153–1165. doi:10.1007/s00018-011-0850-z
Narayanan SV, Dave KR, Saul I, Perez-Pinzon MA (2015) Resveratrol preconditioning protects against cerebral ischemic injury via nuclear erythroid 2-related factor 2. Stroke 46:1626–1632. doi:10.1161/STROKEAHA.115.008921
Bellaver B, Bobermin LD, Souza DG, Rodrigues MD, de Assis AM, Wajner M, Gonçalves CA, Souza DO et al (2016) Signaling mechanisms underlying the glioprotective effects of resveratrol against mitochondrial dysfunction. Biochim Biophys Acta 1862:1827–1838. doi:10.1016/j.bbadis.2016.06.018
Robb EL, Winkelmolen L, Visanji N, Brotchie J, Stuart JA (2008) Dietary resveratrol administration increases MnSOD expression and activity in mouse brain. Biochem Biophys Res Commun 372:254–259. doi:10.1016/j.bbrc.2008.05.028
Zhao H, Niu Q, Li X, Liu T, Xu Y, Han H, Wang W, Fan N et al (2012) Long-term resveratrol consumption protects ovariectomized rats chronically treated with D-galactose from developing memory decline without effects on the uterus. Brain Res 1467:67–80. doi:10.1016/j.brainres.2012.05.040
Manczak M, Mao P, Calkins MJ, Cornea A, Reddy AP, Murphy MP, Szeto HH, Park B et al (2010) Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer's disease neurons. J Alzheimers Dis 20:S609–S631. doi:10.3233/JAD-2010-100564
Yu L, Yang SJ (2010) AMP-activated protein kinase mediates activity-dependent regulation of peroxisome proliferator-activated receptor gamma coactivator-1alpha and nuclear respiratory factor 1 expression in rat visual cortical neurons. Neuroscience 169:23–38. doi:10.1016/j.neuroscience.2010.04.063
Chen S, Fan Q, Li A, Liao D, Ge J, Laties AM, Zhang X (2013) Dynamic mobilization of PGC-1α mediates mitochondrial biogenesis for the protection of RGC-5 cells by resveratrol during serum deprivation. Apoptosis 18:786–799. doi:10.1007/s10495-013-0837-3
Cao K, Zheng A, Xu J, Li H, Liu J, Peng Y, Long J, Zou X et al (2014) AMPK activation prevents prenatal stress-induced cognitive impairment: modulation of mitochondrial content and oxidative stress. Free Radic Biol Med 75:156–166. doi:10.1016/j.freeradbiomed.2014.07.029
Peng K, Tao Y, Zhang J, Wang J, Ye F, Dan G, Zhao Y, Cai Y et al (2016) Resveratrol regulates mitochondrial biogenesis and fission/fusion to attenuate rotenone-induced neurotoxicity. Oxidative Med Cell Longev 2016:6705621. doi:10.1155/2016/6705621
Valenti D, de Bari L, de Rasmo D, Signorile A, Henrion-Caude A, Contestabile A, Vacca RA (2016) The polyphenols resveratrol and epigallocatechin-3-gallate restore the severe impairment of mitochondria in hippocampal progenitor cells from a Down syndrome mouse model. Biochim Biophys Acta 1862:1093–1104. doi:10.1016/j.bbadis.2016.03.003
Naia L, Rosenstock TR, Oliveira AM, Oliveira-Sousa SI, Caldeira GL, Carmo C, Laço MN, Hayden MR et al (2016) Comparative mitochondrial-based protective effects of resveratrol and nicotinamide in Huntington's disease models. Mol Neurobiol. doi:10.1007/s12035-016-0048-3
Mancuso R, del Valle J, Modol L, Martinez A, Granado-Serrano AB, Ramirez-Núñez O, Pallás M, Portero-Otin M et al (2014) Resveratrol improves motoneuron function and extends survival in SOD1(G93A) ALS mice. Neurotherapeutics 11:419–432. doi:10.1007/s13311-013-0253-y
Palomera-Avalos V, Griñán-Ferré C, Puigoriol-Ilamola D, Camins A, Sanfeliu C, Canudas AM, Pallàs M (2016) Resveratrol protects SAMP8 brain under metabolic stress: focus on mitochondrial function and Wnt pathway. Mol Neurobiol. doi:10.1007/s12035-016-9770-0
Kairisalo M, Korhonen L, Blomgren K, Lindholm D (2007) X-linked inhibitor of apoptosis protein increases mitochondrial antioxidants through NF-kappaB activation. Biochem Biophys Res Commun 364:138–144
Jang JH, Surh YJ (2003) Protective effect of resveratrol on beta-amyloid-induced oxidative PC12 cell death. Free Radic Biol Med 34:1100–1110
Miao L, St Clair DK (2009) Regulation of superoxide dismutase genes: implications in disease. Free Radic Biol Med 47:344–356. doi:10.1016/j.freeradbiomed.2009.05.018
Lu Z, Xu X, Hu X, Fassett J, Zhu G, Tao Y, Li J, Huang Y et al (2010) PGC-1 alpha regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid Redox Signal 13:1011–1022. doi:10.1089/ars.2009.2940
Nijland PG, Witte ME, van het Hof B, van der Pol S, Bauer J, Lassmann H, van der Valk P, de Vries HE et al (2014) Astroglial PGC-1alpha increases mitochondrial antioxidant capacity and suppresses inflammation: implications for multiple sclerosis. Acta Neuropathol Commun 2:170. doi:10.1186/s40478-014-0170-2
Quincozes-Santos A, Bobermin LD, Tramontina AC, Wartchow KM, Tagliari B, Souza DO, Wyse AT, Gonçalves CA (2014) Oxidative stress mediated by NMDA, AMPA/KA channels in acute hippocampal slices: neuroprotective effect of resveratrol. Toxicol in Vitro 28:544–551. doi:10.1016/j.tiv.2013.12.021
Wang R, Liu YY, Liu XY, Jia SW, Zhao J, Cui D, Wang L (2014) Resveratrol protects neurons and the myocardium by reducing oxidative stress and ameliorating mitochondria damage in a cerebral ischemia rat model. Cell Physiol Biochem 34:854–864. doi:10.1159/000366304
Henry-Mowatt J, Dive C, Martinou JC, James D (2004) Role of mitochondrial membrane permeabilization in apoptosis and cancer. Oncogene 23:2850–2860
Della-Morte D, Dave KR, DeFazio RA, Bao YC, Raval AP, Perez-Pinzon MA (2009) Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway. Neuroscience 159:993–1002. doi:10.1016/j.neuroscience.2009.01.017
Guo C, Yang L, Wan CX, Xia YZ, Zhang C, Chen MH, Wang ZD, Li ZR et al (2016) Anti-neuroinflammatory effect of Sophoraflavanone G from Sophora alopecuroides in LPS-activated BV2 microglia by MAPK, JAK/STAT and Nrf2/HO-1 signaling pathways. Phytomedicine 23:1629–1637. doi:10.1016/j.phymed.2016.10.007
Kim CS, Choi HS, Joe Y, Chung HT, Yu R (2016) Induction of heme oxygenase-1 with dietary quercetin reduces obesity-induced hepatic inflammation through macrophage phenotype switching. Nutr Res Pract 10:623–628
Choi YK, Park JH, Baek YY, Won MH, Jeoung D, Lee H, Ha KS, Kwon YG et al (2016) Carbon monoxide stimulates astrocytic mitochondrial biogenesis via L-type Ca2+ channel-mediated PGC-1α/ERRα activation. Biochem Biophys Res Commun 479:297–304. doi:10.1016/j.bbrc.2016.09.063
Yu J, Shi J, Wang D, Dong S, Zhang Y, Wang M, Gong L, Fu Q et al (2016) Heme oxygenase-1/carbon monoxide-regulated mitochondrial dynamic equilibrium contributes to the attenuation of endotoxin-induced acute lung injury in rats and in lipopolysaccharide-activated macrophages. Anesthesiology 125:1190–1201
Holmström KM, Baird L, Zhang Y, Hargreaves I, Chalasani A, Land JM, Stanyer L, Yamamoto M et al (2013) Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol Open 2:761–770. doi:10.1242/bio.20134853
de Oliveira MR, Fürstenau CR, de Souza IC, da Costa Ferreira G (2016) Tanshinone I attenuates the effects of a challenge with H2O2 on the functions of tricarboxylic acid cycle and respiratory chain in SH-SY5Y cells. Mol Neurobiol doi. doi:10.1007/s12035-016-0267-7
de Oliveira MR, Schuck PF, Bosco SM (2016) Tanshinone I induces mitochondrial protection through an Nrf2-dependent mechanism in paraquat-treated human neuroblastoma SH-SY5Y cells. Mol Neurobiol. doi:10.1007/s12035-016-0009-x
de Oliveira MR, Peres A, Gama CS, Bosco SM (2016) Pinocembrin provides mitochondrial protection by the activation of the Erk1/2-Nrf2 signaling pathway in SH-SY5Y neuroblastoma cells exposed to paraquat. Mol Neurobiol. doi:10.1007/s12035-016-0135-5
Cerveny KL, Tamura Y, Zhang Z, Jensen RE, Sesaki H (2007) Regulation of mitochondrial fusion and division. Trends Cell Biol 17:563–569. doi:10.1016/j.tcb.2007.08.006
Westermann B (2010) Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11:872–884. doi:10.1038/nrm3013
Morris-Blanco KC, Cohan CH, Neumann JT, Sick TJ, Perez-Pinzon MA (2014) Protein kinase C epsilon regulates mitochondrial pools of Nampt and NAD following resveratrol and ischemic preconditioning in the rat cortex. J Cereb Blood Flow Metab 34:1024–1032. doi:10.1038/jcbfm.2014.51
Peng Y, Liu J, Shi L, Tang Y, Gao D, Long J, Liu J (2016) Mitochondrial dysfunction precedes depression of AMPK/AKT signaling in insulin resistance induced by high glucose in primary cortical neurons. J Neurochem 137:701–713. doi:10.1111/jnc.13563
Valenti D, Tullo A, Caratozzolo MF, Merafina RS, Scartezzini P, Marra E, Vacca RA (2010) Impairment of F1F0-ATPase, adenine nucleotide translocator and adenylate kinase causes mitochondrial energy deficit in human skin fibroblasts with chromosome 21 trisomy. Biochem J 431:299–310. doi:10.1042/BJ20100581
Valenti D, Manente GA, Moro L, Marra E, Vacca RA (2011) Deficit of complex I activity in human skin fibroblasts with chromosome 21 trisomy and overproduction of reactive oxygen species by mitochondria: involvement of the cAMP/PKA signalling pathway. Biochem J 435:679–688. doi:10.1042/BJ20101908
Bozzo F, Mirra A, Carrì MT (2017) Oxidative stress and mitochondrial damage in the pathogenesis of ALS: new perspectives. Neurosci Lett 636:3–8. doi:10.1016/j.neulet.2016.04.065
Carrì MT, D'Ambrosi N, Cozzolino M (2016) Pathways to mitochondrial dysfunction in ALS pathogenesis. Biochem Biophys Res Commun. doi:10.1016/j.bbrc.2016.07.055
Moreno-Ortega AJ, Al-Achbili LM, Alonso E, de Los Ríos C, García AG, Ruiz-Nuño A, Cano-Abad MF (2016) Neuroprotective effect of the novel compound ITH33/IQM9.21 against oxidative stress and Na(+) and Ca(2+) overload in motor neuron-like NSC-34 cells. Neurotox Res 30:380–391. doi:10.1007/s12640-016-9623-7
Zhou ZD, Saw WT, Tan EK (2016) Mitochondrial CHCHD-containing proteins: physiologic functions and link with neurodegenerative diseases. Mol Neurobiol. doi:10.1007/s12035-016-0099-5
Wang Q, Liu Y, Zou X, Wang Q, An M, Guan X, He J, Tong Y et al (2008) The hippocampal proteomic analysis of senescence-accelerated mouse: implications of Uchl3 and mitofilin in cognitive disorder and mitochondria dysfunction in SAMP8. Neurochem Res 33:1776–1782. doi:10.1007/s11064-008-9628-6
Eckert GP, Schiborr C, Hagl S, Abdel-Kader R, Müller WE, Rimbach G, Frank J (2013) Curcumin prevents mitochondrial dysfunction in the brain of the senescence-accelerated mouse-prone 8. Neurochem Int 62:595–602. doi:10.1016/j.neuint.2013.02.014
Wu FJ, Xue Y, Liu XF, Xue CH, Wang JF, Du L, Takahashi K, Wang YM (2014) The protective effect of eicosapentaenoic acid-enriched phospholipids from sea cucumber Cucumaria frondosa on oxidative stress in PC12 cells and SAMP8 mice. Neurochem Int 64:9–17. doi:10.1016/j.neuint.2013.10.015
Feng Y, Cui Y, Gao JL, Li R, Jiang XH, Tian YX, Wang KJ, Li MH et al (2016) Neuroprotective effects of resveratrol against traumatic brain injury in rats: involvement of synaptic proteins and neuronal autophagy. Mol Med Rep 13:5248–5254. doi:10.3892/mmr.2016.5201
Zhang Y, Cao X, Zhu W, Liu Z, Liu H, Zhou Y, Cao Y, Liu C et al (2016) Resveratrol enhances autophagic flux and promotes ox-LDL degradation in HUVECs via upregulation of SIRT1. Oxidative Med Cell Longev 2016:7589813. doi:10.1155/2016/7589813
Wu Y, Li X, Zhu JX, Xie W, Le W, Fan Z, Jankovic J, Pan T (2011) Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson's disease. Neurosignals 19:163–174. doi:10.1159/000328516
Lotharius J, Brundin P (2002) Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci 3:932–942
Olanow CW, Brundin P (2013) Parkinson's disease and alpha synuclein: is Parkinson's disease a prion-like disorder? Mov Disord 28:31–40. doi:10.1002/mds.25373
Zhang QS, Heng Y, Yuan YH, Chen NH (2016) Pathological α-synuclein exacerbates the progression of Parkinson's disease through microglial activation. Toxicol Lett 265:30–37. doi:10.1016/j.toxlet.2016.11.002
Mack JM, Schamne MG, Sampaio TB, Pértile RA, Fernandes PA, Markus RP, Prediger RD (2016) Melatoninergic system in Parkinson's disease: from neuroprotection to the management of motor and nonmotor symptoms. Oxidative Med Cell Longev 2016:3472032
Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK (2008) Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem 283:9089–9100. doi:10.1074/jbc.M710012200
Reeve AK, Park TK, Jaros E, Campbell GR, Lax NZ, Hepplewhite PD, Krishnan KJ, Elson JL et al (2012) Relationship between mitochondria and α-synuclein: a study of single substantia nigra neurons. Arch Neurol 69:385–393. doi:10.1001/archneurol.2011.2675
Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, Hu X, McCoy J et al (2016) α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson's disease. Sci Transl Med 8:342ra78. doi:10.1126/scitranslmed.aaf3634
Ingelsson M (2016) Alpha-synuclein oligomers-neurotoxic molecules in Parkinson's disease and other Lewy body disorders. Front Neurosci 10:408. doi:10.3389/fnins.2016.00408
Redmann M, Darley-Usmar V, Zhang J (2016) The role of autophagy, mitophagy and lysosomal functions in modulating bioenergetics and survival in the context of redox and Proteotoxic damage: implications for neurodegenerative diseases. Aging Dis 7:150–162
Jeong JK, Moon MH, Bae BC, Lee YJ, Seol JW, Kang HS, Kim JS, Kang SJ et al (2012) Autophagy induced by resveratrol prevents human prion protein-mediated neurotoxicity. Neurosci Res 73:99–105. doi:10.1016/j.neures.2012.03.005
Lin TK, Chen SD, Chuang YC, Lin HY, Huang CR, Chuang JH, Wang PW, Huang ST et al (2014) Resveratrol partially prevents rotenone-induced neurotoxicity in dopaminergic SH-SY5Y cells through induction of heme oxygenase-1 dependent autophagy. Int J Mol Sci 15:1625–1646. doi:10.3390/ijms15011625
Carchman EH, Rao J, Loughran PA, Rosengart MR, Zuckerbraun BS (2011) Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology 53:2053–2062. doi:10.1002/hep.24324
Dong C, Zheng H, Huang S, You N, Xu J, Ye X, Zhu Q, Feng Y et al (2015) Heme oxygenase-1 enhances autophagy in podocytes as a protective mechanism against high glucose-induced apoptosis. Exp Cell Res 337:146–159. doi:10.1016/j.yexcr.2015.04.005
Surolia R, Karki S, Kim H, Yu Z, Kulkarni T, Mirov SB, Carter AB, Rowe SM et al (2015) Heme oxygenase-1-mediated autophagy protects against pulmonary endothelial cell death and development of emphysema in cadmium-treated mice. Am J Physiol Lung Cell Mol Physiol 309:L280–L292. doi:10.1152/ajplung.00097.2015
Belounis A, Nyalendo C, Le Gall R, Imbriglio TV, Mahma M, Teira P, Beaunoyer M, Cournoyer S et al (2016) Autophagy is associated with chemoresistance in neuroblastoma. BMC Cancer 16:891
Noonan J, Zarrer J, Murphy BM (2016) Targeting autophagy in glioblastoma. Crit Rev Oncog 21:241–252
Acknowledgements
None to declare.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Jardim, F.R., de Rossi, F.T., Nascimento, M.X. et al. Resveratrol and Brain Mitochondria: a Review. Mol Neurobiol 55, 2085–2101 (2018). https://doi.org/10.1007/s12035-017-0448-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0448-z