
Abstract
Clinical evidences showing zinc (Zn) accumulation in the post-mortem brain of Parkinson’s disease (PD) patients and experimental studies on rodents chronically exposed to Zn suggested its role in PD. While oxidative stress is implicated in Zn-induced neurodegeneration, roles of inflammation and apoptosis in degeneration of the nigrostriatal dopaminergic neurons have yet been elusive. The present study investigated the contribution of the nuclear factor kappa B (NF-κB), tumor necrosis factor-alpha (TNF-α), interleukin-1β (IL-1β), and B-cell lymphoma 2 (Bcl-2) family proteins in Zn-induced Parkinsonism. Male Wistar rats were treated with/without zinc sulfate (Zn; 20 mg/kg, intraperitoneally), twice a week, for 2–12 weeks. In a few sets, animals were treated intraperitoneally with a NF-κB inhibitor, pyrrolidine dithiocarbamate (PDTC; 100 mg/kg), a TNF-α inhibitor, pentoxyfylline (PTX; 50 mg/kg), and an anti-inflammatory agent, dexamethasone (DEX; 5 mg/kg), prior to Zn exposure along with respective controls. Zn caused neurobehavioral impairments and reduction in dopamine and its metabolites, tyrosine hydroxylase (TH)-positive neurons, catalase activity, and expression of TH, Bcl-2, and NOXA. On the contrary, Zn augmented lipid peroxidation, activity of superoxide dismutase, expression of TNF-α, IL-1β, Bcl-xl, and p53-upregulated modulator of apoptosis (PUMA), and translocation of NF-κB and Bax from the cytosol to the nucleus and mitochondria, respectively, with concomitant increase in the mitochondrial cytochrome c release and activation of procaspase-3 and -9. Pre-treatment with PTX, DEX, or PDTC invariably ameliorated Zn-induced changes in behavioral and neurodegenerative indexes, inflammatory mediators, and apoptosis. Results demonstrate that inflammation regulates Bax expression that subsequently contributes to the nigrostriatal dopaminergic neurodegeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a widespread progressive degenerative movement disorder of the central nervous system. It has a multi-factorial etiology with age and genetic and environmental factors as the major putative risk factors, which lead to the selective and progressive death of dopamine producing neurons in the substantia nigra pars compacta (SNpc) region of the midbrain [1]. Pesticides and heavy metals comprise the main environmental factors associated with increased incidences of PD [2, 3]. The role of Zn in PD pathogenesis, suggested by the increased accumulation of Zn in the substantia nigra region of the brain of PD patients [4], was supported by the animal studies documenting dopaminergic neurodegeneration in rodents chronically exposed to Zn resulting in PD phenotype [5–7]. Oxidative stress is established as a key player in Zn-induced dopaminergic neurodegeneration; however, the complete molecular mechanism is not yet clearly understood. Studies investigating mechanism of PD pathogenesis have revealed that reactive oxygen/nitrogen species (RONS) play an imperative role in inflammation by induction of pro-inflammatory mediators viz., nuclear factor-kappa B (NF-κB), tumor necrosis factor-α (TNF-α), and interleukins (IL-1β, IL-6) [8].
Chronic inflammation is recognized as a wrongdoer in PD [9, 10], which is supported by the fact that non-steroidal anti-inflammatory drugs slow down the disease progression and rescue from dopaminergic neurodegeneration [11, 12]. The role of activated microglia is also attributed to oxidative stress and inflammation-induced PD [13, 14]. This is substantiated by an increased expression or display of pro-inflammatory cytokines viz., TNF-α and IL-1β found in the cerebrospinal fluid (CSF), SNpc, and peripheral blood mononuclear cells of PD patients [15–17]. Furthermore, infusion with inflammatory agents, lipopolysaccharide (LPS) and histamine, resulting in the death of dopaminergic neurons [18, 19] also point towards contribution of inflammation in PD progression.
Nuclear factor-kappa B (NF-κB), one of the major transcription factors, is involved in inflammation since it regulates the expression of pro-inflammatory cytokines [20]. Increased nuclear translocation of NF-κB is reported in the brain of PD patients and toxin-based Parkinsonian models [21–23] that further suggests its involvement in PD pathogenesis. It is also supported by the fact that NF-κB inhibitors offer protection in 1-methyl-4-phenyl-1,2,3,4-tetrahydropyridine (MPTP) and pesticides-induced PD phenotypes [22, 24]. Interplay of inflammation with apoptosis ultimately results in the death of dopaminergic neurons [25]. Inflammation-mediated apoptotic dopaminergic neuronal cell death is reported both in sporadic and chemical-induced PD [26, 27]. Apoptosis is controlled by the B-cell lymphoma 2 (Bcl-2) family of proteins, which can function either as pro-survival or pro-apoptotic molecules [28]. Pro-survival proteins mainly include Bcl-2 and Bcl-xl, while Bcl-2 associated X protein (Bax), Bcl-2-antagonist/killer (Bak), Noxa, and tumor suppressor protein p53-upregulated modulator of apoptosis (Puma) are the proteins with pro-apoptotic functions. Involvement of the aforementioned Bcl-2 family proteins is implicated in sporadic and chemical-induced PD [26, 29, 30]. Although previous studies have shown the involvement of mitochondria-dependent caspase-mediated neuronal apoptosis in Zn-induced Parkinsonism, the role of Bcl-2 family of proteins is not yet explored.
Pyrrolidine dithiocarbamate (PDTC), dexamethasone (DEX), and pentoxifylline (PTX) are anti-inflammatory agents, which are known to alleviate inflammation in varying pathological conditions [18, 31–33]. While PDTC is a selective inhibitor of NF-κB [34], pentoxifylline (PTX) is a methylxanthine derivative that is a phosphodiesterase inhibitor and potent inhibitor of TNF-α biosynthesis [35]. Dexamethasone (DEX), a synthetic glucocorticoid, is reported to reduce the expression of the pro-inflammatory cytokines in several pathological conditions [36]. Neuroprotective effects of PDTC, PTX, and DEX are known in PD and Zn is known to induce PD-like features [22, 24, 31, 32]; however, the role of inflammation and contribution of Bcl-2 family proteins in Zn-induced PD have not yet been elucidated. The present study was undertaken to investigate the contributions of inflammation and of Bcl-2 family proteins in Zn-induced dopaminergic neurodegeneration employing PTX, DEX, and PDTC.
Materials and Methods
Chemicals
Acetic acid, disodium hydrogen phosphate, dibutyl phthalate xylene, heptane sulfonic acid, nicotinamide adenine dinucleotide reduced form, nitroblue tetrazolium (NBT), phenazine methosulfate, potassium chloride, potassium dihydrogen phosphate, sodium dihydrogen phosphate, sodium fluoride, and xylene were purchased from Sisco Research Laboratories (SRL, Mumbai, India). Ethanol, Folin Ciocalteau reagent, nitric acid, hydrogen peroxide, methanol, n-butanol, potassium dichromate, sodium chloride, sodium hydroxide, and sucrose were purchased from Merck (Darmstadt, Germany). Agarose, acrylamide, bisacrylamide, pentoxyfylline (PTX), pyrrolidine dithiocarbamate (PDTC), mouse monoclonal anti-TNF-α antibody, biotinylated anti-mouse secondary antibody, bovine serum albumin, bromophenol blue, β-mercaptoethanol, dithiothreitol, ethylenediaminetetraacetic acid (EDTA), ethylene glycol tetraacetic acid (EGTA), ethidium bromide (EtBr), 2-hydroxyethyl-1-piperazine ethane sulfonic acid (HEPES), paraformaldehyde, phenylmethyl sulfonyl fluoride (PMSF), protease inhibitor cocktail, potassium hydroxide, sodium deoxycholate, sodium dodecyl sulfate (SDS), 3,3′-diaminobenzidine tetrahydrochloride (DAB) system, sodium orthovanadate, sodium pyrophosphate, thiobarbituric acid (TBA), Tris-base, triton X-100, Tween-20, xylene cyanol, and zinc sulfate (ZnSO4) were procured from Sigma-Aldrich (St. Louis, MO, USA). cDNA synthesis kits, dNTPs, magnesium chloride, Taq buffer, and Taq DNA polymerase were procured from MBI Fermentas (Amherst, NY, USA). Gene-specific primers were synthesized from Integrated DNA Technologies Ltd., Singapore. Neg-50 was purchased from Richard Allen Scientific (Kalamazoo, MI). Perchloric acid was purchased from Ranbaxy Private Limited (New Delhi, India). Mouse monoclonal anti-β-actin, anti-Bax, anti-Bcl-2, anti-caspase 3, anti-TH, anti-lamin A, anti-NF-κB, anti-Bcl-xl and anti-cytochrome c, goat polyclonal anti-IL-1β, and anti-Tim 44 and rabbit polyclonal anti-caspase 9 primary antibodies along with goat anti-mouse, rabbit anti-goat, and bovine anti-rabbit alkaline phosphatase (AP)-conjugated secondary antibodies were procured from Santa Cruz Biotechnology (Santa Cruz, CA, USA). 5-Bromo-4-chloro-3′-indolylphosphate/nitroblue tetrazolium (BCIP/NBT) system, normal goat serum, and streptavidin peroxidase were procured from Bangalore Genei India Pvt. Ltd. (Bangalore, India). Polyvinylidene difluoride (PVDF) membrane and mouse monoclonal anti-NeuN primary antibody were purchased from Millipore Corporation (MA, USA). Other chemicals required for this study were procured locally.
Animal Treatment
Male Wistar rats (150–180 g) were obtained from the animal house of CSIR–Indian Institute of Toxicology Research, Lucknow. The animals were housed under standard conditions of temperature and humidity with 12 h light/dark cycle and provided standard pellet diet and water ad libitum. Animals were treated with ZnSO4 [20 mg/kg body weight (b.w.)] intraperitoneally (i.p.) twice weekly for 2–12 weeks. In few subsets of 12 weeks of exposure, the animals were pre-treated with pentoxyfylline/PTX (50 mg/kg b.w.; i.p.), dexamethasone/DEX (5 mg/kg b.w.; i.p.), or pyrrolidine dithiocarbamate/PDTC (100 mg/kg b.w.; i.p.) 1 h prior to Zn along with respective controls.
Neurobehavioral Studies
To evaluate neurobehavioral changes following exposure to Zn and inflammatory modulators, spontaneous locomotor activity (SLA) and rotarod performance tests were performed using standard procedures as described elsewhere [14]. The results are expressed in terms of percent of control.
Extraction and Dissection of Brain
Animals were sacrificed by cervical dislocation and brains were dissected in ice-cold conditions to isolate the striatum and SN separately as described previously [7]. Striatum and SN were collectively used as nigrostriatal tissues in all the experiments except for estimation of neurotransmitters (dopamine, its metabolites, and serotonin) and immunohistochemical (IHC) studies where striatum and frozen brain sections were used respectively. A minimum of four animals were used per group for biochemical, protein/gene expression, and IHC analysis.
Striatal Dopamine, its Metabolites, and Serotonin Content
Monoamine(s) and its metabolites (3,4-dihydroxy phenyl acetic acid/DOPAC and homovanillic acid/HVA) were estimated as described elsewhere [7]. Final results are expressed as percent of control.
Immunohistochemistry
Tyrosine hydroxylase (TH)/NeuN immunoreactivity was performed as described previously [6]. The number of TH-positive neurons was calculated bilaterally. A minimum of four animals were used per group for counting TH-positive neurons. The results are expressed as percent of controls.
Oxidative Stress Indexes
Lipid Peroxidation (LPO), Superoxide Dismutase (SOD), and Catalase
LPO levels in control and treated groups were determined using thiobarbituric acid (TBA) method as described previously [7]. LPO levels are expressed as nanomoles of malondialdehyde (MDA)/mg tissue.
SOD activity was estimated using the standard NBT method as described earlier [6]. The results are expressed as units per milliliter per minute. Catalase activity was determined as described elsewhere [7]. The enzymatic activity was calculated in terms of micromoles per minute per milligram of protein.
Protein Estimation
Protein content was determined in different sub-cellular fractions by using Lowry’s method as described elsewhere [7]. Protein concentration was calculated using bovine serum albumin as a standard.
Western Blotting
The protein expressions of TH, TNF-α, IL-1β, Bcl-xl, pro-caspase-9, and pro-caspase-3 were analyzed in the cytosolic fraction of the nigrostriatal tissue, protein expression of Bcl-2 was analyzed in the mitochondrial fraction, and Bax translocation and mitochondrial cytochrome c (cyt c) release were assessed by analyzing their relative protein levels in the cytosolic and mitochondrial fractions. The translocation of NF-κB was monitored by analyzing its relative protein levels in the nuclear and cytosolic fractions. The nuclear, mitochondrial, and cytosolic fractions were prepared using standard procedure described elsewhere [37]. The denatured proteins were resolved on 10–15 % SDS-polyacrylamide gel and electroblotted onto PVDF membrane. Blots were blocked overnight with Tris-buffered saline containing 0.05 % Tween-20 (TBS-T) and 5 % non-fat dry milk and subsequently incubated with specific primary antibodies for 2 h followed by incubation in respective AP-conjugated secondary antibodies for 1 h. Blots were developed using BCIP/NBT as the substrate. Relative band density was calculated with β-actin, lamin A, and Tim-44 as the reference for cytosolic, nuclear, and mitochondrial fractions, respectively. The data are expressed as mean ± standard error of means (SEM) of band density ratio of at least four individual experiments.
Gene Expression
Total RNA was isolated from nigrostriatal tissues of control and treated groups using Trizol reagent as described previously [7]. Revert Aid H-minus Mul M reverse transcriptase kit was used to synthesize c-DNA from total RNA as per given protocol. Amplification of BAX, BCL-2, BCL-XL, NOXA, PUMA, and β-ACTIN was carried out employing gene-specific primers designed using DNA star software under optimal conditions. Primer sequences were as follows—BAX: forward: 5′-GGAGCAGCCGCCCCAGGATG-3′, reverse: 5′- CACGCGGCCCCAGTTGAAGTTG-3′; BCL-2: forward: 5′-GCCGGGACGCGAAGTGCTATTG-3′, reverse: 5′-GCGGGCGTTCGGTTGCTCTC-3′; BCL-XL: forward: 5′-TAATCCCCATGGCAGCAGTGAAGC-3′, reverse: 5′-TGGGGGCAAGGTGGGAGGTG-3′; NOXA: forward: 5′-CGCGAAAGAGCACGATGAGA-3′, reverse: 5′-TTGAAGAGCTTGGAAATAAAAT-3′; PUMA: forward: 5′-CTCCCCCAGTCCCCATCCATCC-3′, reverse: 5′-TCCCTCCCCGCTCCCAGACTCC-3′; and β-ACTIN: forward: 5′-CTGGGACGATATGGAGAAGATTTG-3′, reverse: 5′-CATGGCTGGGGTGTTGAAGG-3′. Densitometric analysis was performed employing computerized software with β-ACTIN as the reference.
Statistical Analysis
Statistical analysis was performed by using two-way or one-way analysis of variance (ANOVA), and Bonferroni post hoc test and Newman-Keul’s post-test were used for multiple comparisons, respectively. The results are expressed as mean ± SEM. The differences were considered statistically significant when p values were less than 0.05.
Results
Gene Expression of BAX, BCL-2, BCL-XL, NOXA, and PUMA
Zn augmented the mRNA levels of BAX, BCL-XL, and PUMA in rats following 2, 4, 8, and 12 weeks of exposure in a time-dependent manner. However, the mRNA levels of NOXA and BCL-2 were found to be decreased in a time-dependent manner in Zn-exposed groups (Fig. 1a–c).

Effect of Zn on mRNA expression of BCL-2/BCL-XL (a), NOXA/PUMA (b), and BAX (c) along with β-ACTIN in rats following 2, 4, 8, and 12 weeks of exposure. Upper panel shows the representative gel picture and the lower panel shows the densitometric analysis of the same. Data are expressed as mean ± SEM (n = 4). ***p < 0.001 and **p < 0.01 as compared with controls
Neurobehavioral Analyses
Zn exposure exhibited a marked decrease in the SLA and rotarod performance in the exposed groups. Pre-treatment with PTX, DEX, or PDTC significantly attenuated the Zn-induced neurobehavioral impairments (Fig. 2a, b). PDTC was most effective in rescuing the animals from Zn-induced modulations in neurobehavioral activities followed by DEX and PTX (Fig. 2a, b). PTX, DEX, or PDTC per se did not alter the motor activities in the exposed groups.

Effect of Zn on SLA (a) and rotarod performance (b) in rats following 12 weeks of exposure in presence and absence of PTX, DEX, or PDTC. Data are expressed as mean ± SEM. ***p < 0.001 as compared with control; ### p < 0.001 and ## p < 0.01 as compared to Zn-treated group
Dopamine, its Metabolites, and Serotonin Levels
Zn significantly attenuated the striatal dopamine content and its metabolites (DOPAC and HVA) in the exposed groups, which were noticeably alleviated in PTX, DEX, or PDTC pre-treated Zn-exposed groups (Fig. 3a–c). Maximum protection was observed in PDTC pre-treated groups followed by DEX and PTX pre-treated groups (Fig. 3a–c). No change was obtained in the dopamine and its metabolite levels in the groups treated with PTX, DEX, or PDTC alone. Striatal serotonin levels were unaltered in any of the groups as compared with controls (data not shown).

Effect of Zn on striatal dopamine (a), DOPAC (b), and HVA (c) levels in rats following 12 weeks of exposure in presence and absence of PTX, DEX, or PDTC. Data are expressed as mean ± SEM (n = 4). ***p < 0.001 as compared with control; ### p < 0.001 and ## p < 0.01 as compared to Zn-treated group
Immunohistochemical Analysis
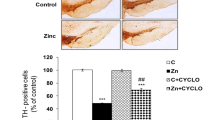
There was a significant decrease in the number of TH-positive cells in the Zn-exposed groups as compared with controls. Pre-treatment with PTX, DEX, or PDTC significantly mitigated the Zn-induced changes in the number of TH-positive neurons. Maximum protection was observed in PDTC pre-treated groups from Zn-induced dopaminergic neuronal loss followed by DEX and PTX pre-treated groups (Fig. 4a). PTX, DEX, or PDTC per se did not alter the number of TH-positive cells in the exposed groups as compared with controls (Fig. 4a).

Effect of Zn on TH/NeuN-positive neurons (a) and TH protein expression (b) in rats following 12 weeks of exposure in presence and absence of PTX, DEX, or PDTC. a Bar diagram showing percent change in TH/NeuN-positive cells from control groups. b Upper panel shows representative western blot and lower panel depicts the densitometric analysis of the same. Data are expressed as mean ± SEM (n = 4). ***p < 0.001 and **p < 0.01 as compared with control; ### p < 0.001 and ## p < 0.01 as compared to Zn-treated group
TH Protein Expression
Zn markedly attenuated the level of TH protein in the nigrostriatal tissues of exposed animals. Pre-treatment with PTX, DEX, or PDTC significantly prevented Zn-induced alterations in the TH protein expression (Fig. 4b). PDTC was more effective as compared to DEX or PTX in protecting against Zn-induced decrease in the TH protein level of exposed groups (Fig. 4b). PTX, DEX, or PDTC alone did not affect the expression of TH protein in the exposed groups.
Oxidative Stress Indexes
Zn elevated LPO levels and SOD activity in the nigrostriatal tissues of the exposed groups (Fig. 5a, b). Conversely, a significant reduction was observed in the catalase activity in the Zn-exposed groups (Fig. 5c). Pre-treatment with PTX, DEX, or PDTC significantly alleviated Zn-induced modulations in the LPO levels, SOD, and catalase activities. PDTC was the most effective as compared with DEX or PTX in preventing Zn-mediated modulations in the oxidative stress indices (Fig. 5a–c). PTX, DEX, or PDTC alone did not alter the LPO level, SOD, and catalase activities in the exposed groups.

Effect of Zn on LPO (a), SOD (b), and catalase (c) activities in rats after 12 weeks of treatment and their modulation by PTX, DEX, or PDTC. Data are expressed as mean ± SEM (n = 4). ***p < 0.001 and **p < 0.01 compared with control; ### p < 0.001, ## p < 0.01, and # p < 0.05 as compared to Zn-treated group
Translocation of NF-κB
Translocation of NF-κB from cytosol into nucleus indicates its activation, which in turn regulates the expression of several pro-inflammatory genes. Zn exposure resulted in increased translocation of NF-κB from the cytosol to the nucleus as denoted by decreased NF-κB protein level in the cytosolic fraction with a concurrent increase in the protein expression of the NF-κB in the nuclear fraction (Fig. 6a). Pre-treatment with PTX, DEX, or PDTC significantly prevented Zn-induced NF-κB translocation in exposed groups. Since PDTC is the known inhibitor of NF-κB activation, it showed maximum protection against Zn-induced NF-κB activation as compared with DEX followed by PTX (Fig. 6a). PTX, DEX, or PDTC alone did not modulate the translocation of NF-κB from cytosol to the nucleus (Fig. 6a).

a Effect of Zn on translocation NF-κB from cytosol to nucleus by analyzing its protein expression in cytosolic and nuclear fractions in the nigrostriatal tissues of rats after 12 weeks of exposure in presence and absence of PTX, DEX, or PDTC. b Effect of Zn on TNF-α and IL-1β protein expression in rats following 12 weeks of exposure and their modulation by PTX, DEX, or PDTC. Upper panels of both (a) and (b) illustrate the representative blot pictures and lower panels depict the densitometric analysis of the same. Data are expressed as mean ± SEM (n = 4). ***p < 0.001, **p < 0.01, and *p < 0.05 as compared with control; ### p < 0.001, ## p < 0.01, and # p < 0.05 as compared to Zn-treated group
Expression of TNF-α and IL-1β
Zn significantly elevated the protein expression of TNF-α and IL-1β in the nigrostriatal tissues of exposed groups, which was markedly mitigated by pre-treatment with PTX, DEX, or PDTC (Fig. 6b). PDTC and PTX pre-treatment exhibited greater rescue in the levels of TNF-α protein in Zn-exposed groups as compared with DEX pre-treated groups. Similar results were obtained in case of IL-1β protein, i.e., all the three agents alleviated Zn-induced increase in the levels of IL-1β protein with PDTC being the most effective followed by DEX and PTX (Fig. 6b). PTX, DEX, or PDTC treatment per se did not change the protein levels of TNF-α and IL-1β in the exposed animals.
Protein Expression of Bcl-2 and Bcl-xl
The protein expression analysis for anti-apoptotic proteins Bcl-2 and Bcl-xl was also performed to validate the results observed in gene analysis of Zn-exposed animals. Zn exposure resulted in a decline of Bcl-2 protein expression in the mitochondrial fraction of the nigrostriatal tissues of the exposed groups showing results similar to that obtained in gene expression experiments, while Bcl-xl protein levels were elevated in the cytosolic fraction of the Zn-exposed groups, further affirming the gene expression results (Fig. 7a, b). Pre-treatment with PTX, DEX, or PDTC significantly attenuated Zn-mediated modulations in the protein expression of Bcl-2 and Bcl-xl (Fig. 7a, b). The effect was more pronounced in PDTC pre-exposed groups as compared with DEX and PTX (Fig. 7a, b). PTX, DEX, or PDTC treatment alone exhibited no significant change in the protein expression of Bcl-2 and Bcl-xl as compared with controls.

a Effect of Zn exposure on Bax and Bcl-2 levels associated with mitochondrial fraction and their modulation by PTX, DEX, or PDTC. Upper panel exhibits representative blot picture and lower panel shows the densitometric analysis of the same with Tim-44 as the reference. b Effect of Zn on cytosolic Bax and Bcl-xl protein levels in rats following 12 weeks of exposure and their modulation by PTX, DEX, or PDTC. Upper panel exhibits representative western blot and lower panel shows the densitometric analysis of the same with β-actin as the reference. Data are expressed as mean ± SEM (n = 4). ***p < 0.001 and **p < 0.01 as compared with control; ### p < 0.001 and ## p < 0.01 as compared to Zn-treated groups
Translocation of Bax
The translocation of Bax from cytosol to mitochondrial membrane is an indicator for mitochondrial outer membrane permeabilization (MOMP) leading to mitochondria-dependent apoptotic pathway. The protein expression of Bax was attenuated in the cytosolic fraction with a concurrent increase in the level of mitochondria-associated Bax protein of Zn-exposed groups indicating increased translocation of Bax from the cytosol to the mitochondria in Zn-treated groups (Fig. 7a, b). Pre-treatment with PTX, DEX, or PDTC significantly abated Zn-induced translocation of Bax from cytosol to mitochondria. Mitigation of Zn-induced Bax translocation was more pronounced in PDTC pre-exposed groups as compared with DEX or PTX (Fig. 7a, b). PDTC, DEX, or PTX per se did not influence the Bax translocation in the exposed groups.
Mitochondrial cyt c Release
Zn exposure increased mitochondrial cyt c release as evident by reduced cyt c levels in the mitochondrial fraction and concomitant increase in the cyt c levels of cytosolic fraction of Zn-exposed groups (Fig. 8a). Pre-treatment with PTX, DEX, or PDTC markedly abated Zn-induced mitochondrial cyt c release (Fig. 8a). PDTC was more effective in alleviating Zn-induced cyt c release as compared with DEX and PTX (Fig. 8a). PTX, DEX, or PDTC alone did not affect the cyt c release in the exposed animals.

Effect of Zn on mitochondrial cyt c release into cytosol (a) and protein levels of pro-caspase-3 and pro-caspase-9 (b) in the nigrostriatal tissues of rats in presence and absence of PTX, DEX, or PDTC. Upper panels of both (a) and (b) exhibit representative blot pictures and lower panels show the densitometric analysis of the same. Data are expressed as mean ± SEM (n = 4). ***p < 0.001 and **p < 0.01 as compared with control; ### p < 0.001 and ## p < 0.01 as compared to Zn-treated group
Caspase Activation
Zn exposure decreased the protein levels of pro-caspase 3 and pro-caspase 9 suggesting their activation in Zn-exposed groups (Fig. 8b). Pre-treatment with PTX, DEX, or PDTC noticeably mitigated this Zn-induced activation of pro-caspase 3 and pro-caspase 9 (Fig. 8b). The maximum protective effect was observed in PDTC pre-treated groups followed by DEX and PTX pre-treated groups (Fig. 8b). PTX, DEX, or PDTC treatment alone did not alter the levels of pro-caspase 3 and pro-caspase 9 in the exposed groups.
Discussion
Inflammation in conjunction with apoptosis plays a critical role in the death of dopaminergic neurons [25]. The present study was conducted to investigate the link among NF-κB, inflammatory molecules, oxidative stress, and apoptosis leading to Zn-induced PD. Neurobehavioral impairments including reduced SLA and rotarod performance and decline in dopamine content and its metabolites observed in Zn exposed animals were in concurrence with the previous reports documenting that Zn exposure results in PD phenotype in rodents [6, 7, 14]. Unaltered serotonin levels in exposed groups (data not shown) further confirmed the preferential loss of TH-positive neurons and not the serotoninergic neurons as reported earlier [6, 7]. Loss of dopaminergic neurons and decreased TH expression observed in the SNPc following Zn exposure further established neurotoxic potential of Zn as also illustrated in the previous reports [6, 14]. Mitigation of Zn-induced changes in neurobehavioral indexes and levels of dopamine and its metabolites in PDTC, DEX, and PTX pre-treated groups indicated that NF-κB and pro-inflammatory mediators (TNF-α, IL-1β, etc.) might be associated with Zn-induced Parkinsonism. These results are supported by the studies, which have shown the protective effect of anti-inflammatory agents in pesticides- and LPS-induced Parkinsonism [18, 22, 31]. PDTC, DEX, and PTX conferred neuroprotection against Zn-induced loss of TH-positive neurons and also implicated the role of inflammation in Zn-mediated neurotoxicity as observed in pesticides and other toxicant models of the nigrostriatal dopaminergic neurodegeneration [22, 38, 39].
Increased LPO and SOD activity along with reduced catalase activity in Zn-treated animals reaffirmed the role of oxidative stress in Zn-induced Parkinsonism [5–7]. Amelioration of Zn-induced changes in oxidative stress indexes by PTX, DEX, or PDTC observed in the current study implicated the participation of inflammatory mediators in Zn-induced oxidative stress. The results are consistent with the previous investigations that have illustrated the involvement of inflammatory mediators in PD phenotypes and ameliorative potentials of PDTC, DEX, etc. [18, 31, 41].
NF-κB regulates the expression of TNF-α, IL-1β, IL-6, and other inflammatory mediators, thereby contributing to toxic manifestation of PD involving oxidative stress and inflammation [21, 24, 25, 38–40]. Increased levels of NF-κB in the nuclear fraction with a concomitant decrease in the cytosolic fraction, i.e., increased nuclear translocation of NF-κB, indicated Zn-induced activation of NF-κB. Additionally, elevated expression of TNF-α and IL-1β observed in Zn-exposed animals suggested the involvement of pro-inflammatory cytokines in Zn-induced Parkinsonism. Mitigation of Zn-induced expression of TNF-α, IL-1β, and translocation of NF-κB by PDTC indicated that NF-κB is involved in the regulation of TNF-α and IL-1β [34, 41]. Alleviation in Zn-induced expression of TNF-α by PTX, along with reduction of NF-κB translocation and IL-1β protein expression, could be due to either its anti-inflammatory efficacy or due to TNF-α-mediated regulation of NF-κB and IL-1β expression [35, 42]. Amelioration of Zn-induced NF-κB activation and expression of TNF-α and IL-1β by DEX affirmed a crosstalk between NF-κB and pro-inflammatory cytokines [43, 44]. Results also suggested an association of inflammation with Zn-induced neurodegeneration akin to other models of PD [8].
Apoptosis is the major pathway involved in neuronal cell death leading to PD and is regulated by Bcl-2 family of proteins [26, 28]. Therefore, the effects of Zn on Bcl-2 family proteins were examined in the study. Bcl-2 family proteins regulate the structural integrity of the mitochondrial membrane by controlling mitochondrial outer membrane permeabilization (MOMP) through Bax and Bak [45, 46]. Pro-apoptotic proteins, Noxa and Puma, facilitate mitochondrial permeabilization via Bak and Bax oligomerization. Noxa indirectly activates Bak oligomerization while Puma facilitates Bax translocation to the outer mitochondrial membrane [47]. While decreased gene expression of NOXA and increased gene expression of BCL-XL suggested negative regulation of Bak-mediated pathway, attenuation in BCL-2 gene expression with concurrent increase in PUMA after Zn exposure implicated participation of Bax in neuronal cell apoptosis [48, 49]. A significant augmentation in the mitochondrial Bax and decline in the cytosolic Bax reaffirmed its contribution in Zn-induced apoptosis. Bax is known to induce mitochondrial cyt c release [50], and increased cyt c content in the cytosolic fraction with concurrent reduction in the mitochondrial fraction observed in Zn-exposed animals indicated that Bax-mediated mitochondria-dependent apoptosis could contribute in Zn-induced neurodegeneration as observed in the previous studies [29, 30, 51]. Mitigation of Zn-induced changes in the expression of Bcl-2 and Bcl-xl and Bax translocation by PDTC, PTX, and DEX implied that inflammation guided the Bax-mediated neuronal apoptosis in Zn-induced Parkinsonism. Attenuated levels of pro-caspase 9 and pro-caspase 3 observed in Zn-exposed animals clearly indicated the cyt c-mediated activation of the caspase cascade [6, 14]. Marked alleviation of Zn-induced mitochondrial cyt c release and pro-caspase activation by DEX, PTX, or PDTC showed the contribution of inflammation in Zn-induced dopaminergic neuronal apoptosis. The results showed that inflammation led to the activation of Bax-mediated and mitochondria-dependent neuronal apoptosis and an existence of a crosstalk between oxidative stress and inflammation, which collectively lead to the nigrostriatal dopaminergic neurodegeneration in Zn-induced Parkinsonism.
Conclusions
The results demonstrate that inflammation regulates Bax-mediated mitochondria-dependent apoptosis of the nigrostriatal dopaminergic neurons leading to Zn-induced Parkinsonism.
References
Dauer W, Przedborski S (2003) Parkinson’s disease: mechanisms and models. Neuron 39(6):889–909
Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ (1997) Occupational exposures to metals as risk factors for Parkinson’s disease. Neurology 48:650–658
Singh C, Ahmad I, Kumar A (2007) Pesticides and metals induced Parkinson’s disease: involvement of free radicals and oxidative stress. Cell Mol Biol 53:19–28
Dexter DT, Carayon A, Javoy-Agid F, Agid Y, Wells FR, Daniel SE (1991) Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 114:1953–1975
Kumar A, Ahmad I, Shukla S, Singh BK, Patel DK, Pandey HP, Singh C (2010) Effect of zinc and paraquat co-exposure on neurodegeneration: modulation of oxidative stress and expression of metallothioneins, toxicant responsive and transporter genes in rats. Free Radic Res 44:950–965
Kumar A, Singh BK, Ahmad I, Shukla S, Patel DK, Srivastava G, Kumar V, Pandey HP et al (2012) Involvement of NADPH oxidase and glutathione in zinc-induced dopaminergic neurodegeneration in rats: similarity with paraquat neurotoxicity. Brain Res 1438:48–64
Singh BK, Kumar A, Ahmad I, Kumar V, Patel DK, Jain SK, Singh C (2011) Oxidative stress in Zn-induced dopaminergic neurodegeneration: implications of superoxide dismutase and heme oxygenase-1. Free Radic Res 45:1207–1222
Mosley RL, Benner EJ, Kadiu I, Thomas M, Boska MD, Hasan K, Gendelamn HE (2006) Neuroinflammation, oxidative stress and pathogenesis of Parkinson’s disease. Clin Neurosci Res 6:261–281
Bartels AL, Leenders KL (2007) Neuro-inflammation in the pathophysiology of Parkinson’s disease: evidence from animal models to human in vivo studies with [11C]-PK11195 PET. Mov Disord 22:1852–1856
Nolan YM, Sullivan AM, Toulouse A (2013) Parkinson’s disease in the nuclear age of neuroinflammation. Trends Mol Med 19:187–196
Chen H, Zhang SM, Hernan MA, Schwarzschild MA, Willett WC, Colditz GA, Speizer FE, Ascherio A (2003) Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch Neurol 60:1059–1064
Wahner AD, Bronstein JM, Bordelon YM, Ritz B (2007) Non-steroidal anti-inflammatory drugs may protect against Parkinson disease. Neurology 69:1836–1842
Teismann P, Schulz JB (2004) Cellular pathology of Parkinson’s disease: astrocytes, microglia and inflammation. Cell Tissue Res 318:149–161
Kumar V, Singh BK, Chauhan AK, Singh D, Patel DK, Singh C (2015) Minocycline rescues from zinc-induced nigrostriatal dopaminergic neurodegeneration: biochemical and molecular interventions. Mol Neurobiol. doi:10.1007/s12035-015-9137-y
Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T (1994) Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett 165:208–210
Nagatsu T, Sawada M (2005) Inflammatory process in Parkinson’s disease: role for cytokines. Curr Pharm Des 11:999–1016
Bessler H, Djaldetti R, Salman H, Bergman M, Djaldetti M (1999) IL-1β, IL-2, IL-6 and TNF-α production by peripheral blood mononuclear cells from patients with Parkinson’s disease. Biomed Pharmacother 53:141–145
Castano A, Herrera AJ, Cano J, Machado A (2002) The degenerative effect of a single intranigral injection of LPS on the dopaminergic system is prevented by dexamethasone, and not mimicked by rh-TNF-α IL-1β IFN-γ. J Neurochem 81:150–157
Vizuete ML, Merino M, Venero JL, Santiago M, Cano J, Machado A (2000) Histamine infusion induces a selective dopaminergic neuronal death along with an inflammatory reaction in rat substantia nigra. J Neurochem 75:540–552
Peterson LJ, Flood PM (2012) Oxidative stress and microglial cells in Parkinson’s disease. Mediat Inflamm 2012:401264
Hunot S, Brugg B, Ricard D, Michel PP, Muriel MP, Ruberg M, Faucheux BA, Agid Y et al (1997) Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with Parkinson disease. Proc Natl Acad Sci U S A 94:7531–7536
Gupta SP, Patel S, Yadav S, Singh AK, Singh S, Singh MP (2010) Involvement of nitric oxide in maneb- and paraquat-induced Parkinson’s disease. Neurochem Res 35:1206–1213
Yuan YH, Sun JD, Wu MM, Hu JF, Peng SY, Chen NH (2013) Rotenone could activate microglia through NF-kB pathway. Neurochem Res 38:1553–1560
Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ, Hartley DM, Ghosh S, Mosley RL et al (2007) Selective inhibition of NF-κB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A 104:18754–18759
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140:918–934
Venderova K, Park DS (2012) Programmed cell death in Parkinson’s disease. Cold Spring Harb Perspect Med 2:a009365
Litteljohn D, Mangano E, Clarke M, Bobyn J, Moloney K, Hayley S (2011) Inflammatory mechanisms of neurodegeneration in toxin-based models of Parkinson’s disease. Parkinson’s Disease. 2011:Article ID 713517, 18 pages
Kuwana T, Newmeyer DD (2003) Bcl-2 family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol 15:691–699
Singhal NK, Srivastava G, Patel DK, Jain SK, Singh MP (2011) Melatonin or silymarin reduces maneb- and paraquat-induced Parkinson’s disease phenotype in the mouse. J Pineal Res 50:97–109
Agrawal S, Singh A, Tripathi P, Mishra M, Singh PK, Singh MP (2015) Cypermethrin-induced nigrostriatal dopaminergic neurodegeneration alters the mitochondrial function: a proteomics study. Mol Neurobiol 51(2):448–465
Kurkowska-Jastrzebska I, Litwina T, Joniecb I, Ciesielskaa A, Przybylkowskib A, Czlonkowskia A, Czlonkowska A (2004) Dexamethasone protects against dopaminergic neurons damage in a mouse model of Parkinson’s disease. Int Immunopharmacol 4:1307–1318
Herrero MT, Estrada C, Maatouk L, Vyas S (2015) Inflammation in Parkinson’s disease: role of glucocorticoids. Front Neuroanat 9:article 32:1–12
Chao CC, Hu S, Close K, Choi CS, Molitor TW, Novick WJ, Peterson PK (1992) Cytokine release from microglia: differential inhibition by pentoxifylline and dexamethasone. J Infect Dis 166:847–853
Liu SF, Ye X, Malik AB (1999) Inhibition of NF-kappaB activation by pyrrolidine dithiocarbamate prevents in vivo expression of proinflammatory genes. Circulation 100:1330–1337
Coimbra R, Melbostad H, Loomis W, Tobar M, Hoyt DB (2005) Phosphodiesterase inhibition decreases nuclear factor-kappa B activation and shifts the cytokine response toward anti-inflammatory activity in acute endotoxemia. J Trauma 59:575–582
Kimberlin DW, Willis SA, Mccracken GH Jr, Nisen PD (1995) Protein synthesis-dependent induction of interleukin-1β by lipopolysaccharide is inhibited by dexamethasone via mRNA destabilization in human astroglial cells. J Clin Immunol 4:199–204
Dimauro I, Pearson T, Caporossi D, Jackson MJ (2012) A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue. BMC Res Notes 5:513
Wilms H, Rosenstiel P, Sievers J, Deusch G, Zecca L, Lucius R (2003) Activation of microglia by human neuromelanin is NF-κB dependent and involves p38 mitogen-activated protein kinase: implications for Parkinson’s disease. FASEB J 17:500–502
Yang HJ, Wanga L, Xia YY, Chang PN, Feng ZW (2010) NF-κB mediates MPP+-induced apoptotic cell death in neuroblastoma cells SH-EP1 through JNK and c-Jun/AP-1. Neurochem International 56:128–134
Ferger B, Leng A, Mura A, Hengerer B, Feldon J (2004) Genetic ablation of tumor necrosis factor-alpha (TNF-a) and pharmacological inhibition of TNF synthesis attenuates MPTP toxicity in mouse striatum. J Neurochem 89:822–833
Wilms H, Zecca L, Rosenstiel P, Sievers J, Deuschl G, Lucius R (2007) Inflammation in Parkinson’s diseases and other neurodegenerative diseases: cause and therapeutic implications. Curr Pharm Des 13:1925–1928
Ji Q, Zhang L, Jia H, Yang J, Xu J (2004) Pentoxyfylline inhibits endotoxin-induced NF-kappa B activation and associated production of proinflammatory cytokines. Ann Clin Lab Sci 34:427–436
Castro-Caldas M, Mendes AF, Carvalho AP, Duarte CB, Lopes MC (2003) Dexamethasone prevents interleukin-1β-induced nuclear factor-κB activation by upregulating IkB-α synthesis, in lymphoblastic cells. Med Inflamm 12:37–46
Fischer R, Maier O (2015) Interrelation of oxidative stress and inflammation in neurodegenerative disease: role of TNF. Oxid Med Cell Long 2015: article ID 610813,18 pages
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP et al (1997) Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275:1129–1132
Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB et al (1997) Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275:983–986
Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J et al (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4:321–328
Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC (2005) Pro-apoptotic Bak is sequestered by Mc1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 19:1294–1305
Hara H, Kamiya T, Adachi T (2009) Zinc induces expression of the BH3-only protein PUMA through p53 and ERK pathways in SH-SY5Y neuroblastoma cells. Neurochem Res 34:1498–1506
Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC (1998) Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci U S A 95:4997–5002
Vila M, Jackson-Lewis V, Vukosavic S, Djaldetti R, Liberatore G, Offen D, Korsmeyer SJ, Przedborski S (2001) Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl- 4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A 98:2837–2842
Acknowledgments
Council of Scientific and Industrial Research (CSIR), New Delhi, Department of Biotechnology, New Delhi, and Department of Science and Technology are gratefully acknowledged for rendering doctoral scholarships to Amit Kumar Chauhan, Vinod Kumar, and Namrata Mittra, respectively. Research grant made available to Chetna Singh through CSIR-network program “neurodegenerative diseases: causes and corrections” (miND; BSC0115) is sincerely acknowledged. The CSIR-IITR communication number of this article is 3335.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The guidelines of the committee for the purpose of control and supervision of experiments on animals (CPCSEA) were strictly followed. The study was initiated after the approval of the institutional animal ethics committee.
Conflicts of Interest
The authors declare that they have no competing interests.
Additional information
CSIR-IITR communication number: 3335
Rights and permissions
About this article

Cite this article
Chauhan, A.K., Mittra, N., Kumar, V. et al. Inflammation and B-cell Lymphoma-2 Associated X Protein Regulate Zinc-Induced Apoptotic Degeneration of Rat Nigrostriatal Dopaminergic Neurons. Mol Neurobiol 53, 5782–5795 (2016). https://doi.org/10.1007/s12035-015-9478-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9478-6