
Abstract
Oxidative stress is recognized as one of the major wrongdoers in Parkinson’s disease (PD) while glutathione S-transferase (GST), an endogenous antioxidant, protects from oxidative stress-induced neurodegeneration. Despite GST-pi (GST-π) encounters the toxic manifestations in PD, its role in zinc (Zn)-induced nigrostriatal dopaminergic neurodegeneration remains elusive. The study aimed to explore the role of GST-π in Zn-induced Parkinsonism and its underlying molecular mechanism. Male Wistar rats were treated intraperitoneally with zinc (zinc sulfate), twice a week, for 2–12 weeks. GST-π inducer, benzyl isothiocyanate (BITC) was also administered in a few sets of experiments along with respective vehicle. Catalytic activity and expression of GST-π protein, total GST activity, neurobehavioral indexes, striatal dopamine and its metabolites, nigral tyrosine hydroxylase (TH)-positive neurons and expression of TH and B-cell lymphoma-2 (Bcl-2) proteins were reduced in Zn-treated rats. Conversely, oxidative stress indicators, c-jun N-terminal kinase (JNK) activation, c-jun phosphorylation, cytochrome c release, Bcl-2-associated X protein (Bax) translocation, and procaspase 3/9 to caspase 3/9 conversion were significantly increased in Zn-exposed rats. BITC ameliorated GST-π activity/expression and normalized Zn-induced changes in neurodegenerative indicators, oxidative stress, JNK activation, c-jun phosphorylation and apoptotic indexes. The results demonstrate that Zn inhibits GST-π expression leading to increased oxidative stress and JNK activation, which induce apoptosis thereby degeneration of the nigrostriatal dopaminergic neurons.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a prevalent, chronic neurological disorder resulting in motor dysfunction caused by the selective loss of the dopamine synthesizing neurons in the substantia nigra (SN) region of midbrain. A multi-factorial aetiology has been suggested with age, genetic and environmental factors as the putative perils for the onset and progression of PD [1, 2]. Meta analysis studies have revealed a strong positive correlation between the exposure to heavy metals and increased risk of PD [3, 4]. Clinical evidences documenting increased Zn levels in the substantia nigra region of brain of PD patients implicated its role in PD pathogenesis [5]. This is substantiated by experimental studies reporting Zn-induced dopaminergic neurodegeneration and PD-like features in rodents [6,7,8,9].
Increased vulnerability of dopaminergic neurons towards oxidative stress is attributed to the presence of high levels of iron (Fe3+), α-synuclein, oxidized dopamine, polyunsaturated fatty acid (PUFA), and impaired calcium signaling [10]. Epidemiological evidences have established a strong association between oxidative stress and increased incidences of PD. It is supported by animal models of PD developed through toxicants exposure, which cause oxidative stress. Toxicants used for this purpose include 1-methyl 4-phenyl 1,2,3,6-tetrahydropyridine (MPTP), 6-hydroxy dopamine (6-OHDA), paraquat, rotenone, iron, and zinc [9,10,11]. Furthermore, reduced glutathione (GSH) content, elevated lipid peroxides, reduced mitochondrial complex I activity, quinines along with DNA damage and augmented c-jun N-terminal kinases/JNK-mediated activation of c-jun collectively signify the role of oxidative stress as a prime culprit in the dopaminergic neuronal loss in PD [12, 13].
Free radicals produced via oxidative stress initiate chain reaction in phospholipid bilayer of the cell membranes resulting in the formation of lipid peroxides and reactive aldehydes viz., malondialdehyde MDA, 4-hydroxy nonenal (4-HNE), etc. and the process is termed as lipid peroxidation. Glutathione S-transferases (GSTs) facilitate the removal of free radicals and neutralization of lipid peroxides and reactive aldehydes via conjugation with GSH thereby protecting from oxidative stress-mediated neuronal cell death [14]. Out of three forms of the cytosolic GSTs, GST alpha (GST-α), GST-mu (GST-µ), and GST-pi (GST-π), GST-π is predominantly expressed in the substantia nigra implicating its contribution in the detoxification of reactive oxygen species (ROS) [14, 15]. Reduced GST-π expression owing to single-nucleotide polymorphisms is associated with the increased incidences of PD [16, 17]. Moreover, genetic and pharmacological inhibitions of GST-π are known to increase MPTP- and rotenone-induced neurodegeneration. GST-π over-expression, on the other hand, protects from dopaminergic neuronal cell death substantiating its ameliorative effect in PD [15, 18, 19]. GST-π is recognized as the negative regulator of JNK pathway, which in turn directs c-jun-mediated apoptosis [20, 21]. The protection afforded by GST-π against MPTP and 6-OHDA-induced PD models via inhibition of JNK-activation further corroborates its involvement in JNK signaling [19, 22].
Previous studies have shown that oxidative stress contributes to the apoptotic neuronal cell death in Zn-induced Parkinsonism in rodent models [6, 9]. Although reduced glutathione (GSH) content and GST activity are reported in the brain following Zn exposure, the role of GST-π in Zn-induced dopaminergic neurodegeneration is not yet explored. The study was undertaken to decipher the involvement GST-π in Zn-induced nigrostriatal dopaminergic neurodegeneration. Besides, an association of GST-π with JNK-dependent programmed cell death is also elucidated by investigating the effect of benzyl isothiocyanate (BITC), a GST-π inducer [23] on the neurodegenerative indexes and oxidative stress along with apoptotic cell death markers in Zn-induced Parkinsonism.
Materials
Acetic acid, disodium hydrogen phosphate, dibutyl phthalate xylene (DPX), heptane sulfonic acid, nicotinamide adenine dinucleotide reduced form (NADH), nitroblue tetrazolium (NBT), phenazine methosulfate (PMS), 1-chloro-2,4-dinitrobenzene (CDNB), potassium chloride, glutathione reduced (GSH), potassium dihydrogen phosphate, sodium dihydrogen phosphate, sodium fluoride (NaF), and xylene were purchased from Sisco Research Laboratories Pvt. Ltd., Mumbai, India. Mouse monoclonal anti-β-actin, anti-Bax, anti-Bcl-2, anti-caspase 3, anti-TH, anti-cytochrome c, anti-JNK, anti-p-JNK, anti-p–c-jun, goat polyclonal anti-Tim 44 and rabbit polyclonal anti-caspase 9 primary antibodies along with goat anti-mouse, rabbit anti-goat, and bovine anti-rabbit alkaline phosphatase (AP)-conjugated secondary antibodies were procured from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Goat polyclonal anti-GST-π antibody was purchased from Abcam Technology, Cambridge, MA (USA). Polyvinylidene difluoride (PVDF) membrane and mouse monoclonal anti-NeuN primary antibody were purchased from Millipore Corporation (MA, USA). Ethanol, Folin Ciocalteau reagent, nitric acid, hydrogen peroxide, methanol, n-butanol, potassium dichromate, sodium chloride, sodium hydroxide, and sucrose were obtained from Merck (Darmstadt, Germany). Acrylamide, bis-acrylamide, biotinylated anti-mouse secondary antibody, bovine serum albumin (BSA), 5-Bromo-4-chloro-3′-indolyl phosphate/nitroblue tetrazolium (BCIP/NBT) system, bromophenol blue, benzyl isothiocyanate (BITC), β-mercaptoethanol, ethacrynic acid (EA), 3,3′-diaminobenzidine tetrahydrochloride (DAB) system, dithiothreitol (DTT), ethylene diamine tetraacetic acid (EDTA), ethylene glycol tetraacetic acid (EGTA), 2-hydroxyethyl-1-piperazine ethane sulfonic acid (HEPES), magnesium chloride (MgCl2), normal goat serum, paraformaldehyde, phenylmethyl sulfonyl fluoride (PMSF), protease inhibitor (PI) cocktail, potassium hydroxide, sodium deoxycholate, sodium dodecyl sulfate (SDS), sodium orthovanadate, sodium pyrophosphate, thiobarbituric acid (TBA), streptavidin peroxidase, Tris-base, triton X-100, tween-20 and zinc sulfate (ZnSO4) were procured from Sigma–Aldrich (St. Louis, MO, USA). Neg-50 was purchased from Richard Allen Scientific (Kalamazoo, MI). Perchloric acid was obtained from Ranbaxy Private Limited, New Delhi, India. Other chemicals required for this study were procured locally.
Methodology
Animal treatment
Male Wistar rats (150–180 g) were obtained from the animal house of CSIR-Indian Institute of Toxicology Research, Lucknow. The animals were housed under standard conditions of temperature and humidity with 12-h light/dark cycle and fed standard pellet diet and water ad libitum. Animals were treated with normal saline or ZnSO4 [20 mg/kg body weight (b.w.); intraperitoneally (i.p.)] twice a week for 2–12 weeks. In subsets, the animals were treated with Zn for 12 weeks in the presence or absence of BITC along with respective controls. BITC (10 mg/kg b.w.; i.p.) was administered, daily, 1 h prior to Zn treatment. The study was initiated after the approval from Institutional Animal Ethics Committee.
Behavioral studies
Motor performance was measured by spontaneous locomotor activity (SLA) and rotarod test. SLA was measured employing infrared beam-activated movement monitoring chamber (OptoVarimax-Mini A; Columbus Instruments, Columbus, OH) [24]. Rotarod performance was checked using Omni rotor (Omnitech Electronics Inc., Columbus, OH, USA). After offering the training to the animals for three consecutive days, the time spent by the animals on the rotating rod was recorded [24]. The results are expressed as % of control.
Decapitation and dissection of brain
The animals were sacrificed by the cervical dislocation and decapitated. The brain was dissected under ice cold conditions and the nigrostriatal tissue (striatum and SN) was collected as described previously [6]. Neurotransmitters were estimated in the striatum while TH/NeuN-immunoreactivity was performed in the frozen brain sections. All other experiments were performed in the nigrostriatal tissue.
Striatal dopamine, its metabolites, and serotonin content
Striatal dopamine, its metabolites, such as 3,4-dihydroxyphenyl acetic acid (DOPAC) and homovanillic acid (HVA) along with serotonin were measured as described elsewhere [6]. Values were calculated using known amount of respective standards. Results are expressed in the percent change with reference to control.
Immunohistochemistry
TH-positive dopaminergic neurons in NeuN positive cells were calculated in control and treated groups as described previously [24]. In brief, animals were anaesthetized; brain was perfused with ice-cold normal saline and paraformaldehyde. The brain was isolated, post fixed in paraformaldehyde and cryoprotected in sucrose gradients. The coronal sections were cut and processed for TH/NeuN-immunoreactivity [24]. Image of the coronal sections was captured with a bright field microscope (Leica Mikroskopie-GMBH; Wetzlar, Germany) at 10X magnification. TH-positive neurons were counted bilaterally using computerized analysis software (QWin Pro, Leica, Germany).
GST, GST-α, GST-µ, and GST-π activities
Total GST activity was determined using CDNB method as described previously [6]. Similarly, GST-α, GST-µ, and GST-π activities were estimated using enzyme-specific substrates in terms of nmoles/min/mg protein. For GST-α and GST-µ, cumene hydroperoxide/CHP (2.5 mM) and 1,2-dichloro-4-nitrobenzene/DCNB (1.0 mM), respectively, were used as substrates [25] while ethacrynic acid (0.2 mM) was used as substrate for GST-π [26]. Briefly, the reaction mixture contained GSH, tissue homogenate and substrate in phosphate buffer (pH 6.5). The absorbance was measured at 270 nm (GST-π) and at 340 nm (GST-α/GST-µ) for 3 min at the interval of 30 s and enzyme activity was calculated. Final results are expressed in terms of percent of control.
Lipid peroxidation (LPO), superoxide dismutase (SOD), and catalase
LPO levels were determined in terms of nmoles MDA/mg tissue by the standard TBA method [24]. SOD (units/ml/min) and catalase (µmoles/min/mg protein) were measured as described earlier [24]. The results are expressed in percentage of control.
Western blotting
The cytosolic and mitochondrial fractions were separated using standard procedure as described previously [9]. Protein content in tissue lysate or sub-cellular fractions was determined using Lowry’s method [27] using bovine serum albumin as a standard. The protein expression was analyzed by western blotting in the cytosolic/mitochondrial fraction of the nigrostriatal tissue. Translocation of Bax and cyt c release was assessed by analyzing their relative levels in the cytosolic and mitochondrial fractions. The denatured proteins were resolved on SDS–polyacrylamide gel (10–15%) and electroblotted onto PVDF membrane. Blots were blocked with tris-buffered saline containing 0.1% Tween-20 (TBS-T) and 5% non-fat dry milk or 3% BSA and subsequently incubated with primary antibodies against TH, JNK, p-JNK, p–c-jun, GST-π, pro-caspase-9, pro-caspase-3, Bax, Bcl-2, cyt c, β-actin, or Tim-44 for 2 h followed by incubation with AP-conjugated respective secondary antibody. Blots were visualized using BCIP/NBT as the substrate. Relative band density was calculated with β-actin and Tim-44 as the reference for cytosolic and mitochondrial fractions, respectively.
Statistical analysis
A minimum of four independent sets of experiments were performed for the study. The results are expressed as mean ± standard error of means (SEM). Statistical analysis was performed by using one/two-way analysis of variance (ANOVA) and the Newman–Keuls/Bonferroni post-test was used for multiple comparisons. The differences were considered statistically significant only when ‘p’ value was less than 0.05.
Results
Effect of Zn exposure on GST activity and protein expression
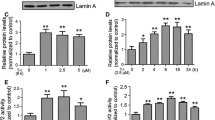
Zn exposure significantly reduced total GST (CDNB-related) and GST-π activities in a time-dependent manner while no noticeable change was observed in GST-α or GST-µ activity (Table 1). Furthermore, the western blot analysis of GST-π protein revealed the similar results i.e., Zn attenuated protein expression of GST-π in an exposure time-dependent manner as compared with control group (Fig. 1).

Effect of Zn on GST-π in the nigrostriatal tissue of rat brain following 2–12 weeks of exposure. The upper panel shows the representative western blot of GST-π and β-actin while the lower panel shows the densitometric analysis of the same. Data are expressed as mean ± SEM (n = 4) and the value *p < 0.05 and ***p < 0.001 are expressed as compared with control
Neurobehavioral analysis
Zn exposure decreased the locomotor activity and rotarod performance in the exposed groups (Fig. 2a). Pre-treatment with BITC significantly attenuated Zn-induced neurobehavioral anomalies (Fig. 2a). BITC per se did not alter the motor activities in the exposed groups.

a Effect of Zn on SLA and rotarod performance test in the presence or absence of BITC. b Effect of BITC on Zn-induced alterations in the striatal dopamine and its metabolites i.e., DOPAC and HVA along with serotonin. Data are expressed as mean ± SEM (n = 4). The value **p < 0.01 and ***p < 0.001 are expressed as compared with control and ##p < 0.01 and ###p < 0.001 as compared with Zn-treated groups
Measurement of the dopamine, its metabolites, and serotonin levels
Zn caused marked decline in the striatal dopamine and its metabolites (DOPAC and HVA) in the exposed groups. However, BITC noticeably restored the level of dopamine and its metabolites in Zn-exposed groups (Fig. 2b). BITC alone did not exhibit any change in the dopamine and its metabolites in the exposed groups. Striatal serotonin levels were unaffected in any of the exposed groups as compared with controls (Fig. 2b).
TH-immunoreactivity and protein expression
Zn reduced the number of TH-positive dopaminergic neurons as compared with control (Fig. 3a). BITC pre-treatment markedly rescued from Zn-induced TH-positive neuronal loss (Fig. 3a). No change was observed in the number of TH-positive neurons in the groups exposed to BITC alone as compared with control (Fig. 3a).

a Effect of BITC on Zn-induced modulations in the number of TH-positive neurons in the substantia nigra. The bar diagram depicts the number of TH-positive neurons as % of control. b Effect of BITC on Zn-induced alterations in the protein expression of TH and GST-π. Upper panel shows the representative western blot and lower panel depicts the densitometric analysis of the same. Data are expressed as mean ± SEM (n = 4). The value ***p < 0.001 is expressed as compared with control and ##p < 0.01 and ###p < 0.001 as compared with Zn-treated group
In addition to dopaminergic neuronal loss, a marked attenuation was also observed in the level of TH protein following Zn exposure, which was restored towards normalcy with BITC pre-treatment (Fig. 3b). BITC alone did not alter the expression of TH protein in the exposed groups.
Protein expression of GST-π
Zn-exposed groups exhibited reduction in the protein level of GST-π. However, BITC significantly rescued from Zn-induced change in GST-π content (Fig. 3b). BITC per se also increased the GST- π protein expression (Fig. 3b).
Total GST and GST-π activity
Zn significantly inhibited the activities of total GST and GST-π in the exposed groups. BITC noticeably alleviated Zn-induced reductions of total GST and GST-π activities in exposed groups (Fig. 4a). BITC alone also augmented the activities of total GST and GST-π in the exposed groups as compared with control groups (Fig. 4a).

a Effect of BITC on Zn-induced alterations in the total GST activity and GST-π activity. b Effect of BITC on Zn-induced changes in the oxidative stress indices. Data are expressed as mean ± SEM (n = 4) and the value *p < 0.05, **p < 0.01, and ***p < 0.001 are expressed as compared with control and #p < 0.05, ##p < 0.01, and ###p < 0.001 as compared with Zn-treated group
Oxidative stress indices
Zn exposure resulted in elevated SOD activity and LPO levels while a significant reduction was observed in the catalase activity in the exposed groups. Pre-treatment with BITC significantly ameliorated Zn-induced changes in LPO, SOD, and catalase activities (Fig. 4b). No significant change was observed in the aforementioned oxidative stress indexes in the groups exposed to BITC alone (Fig. 4b).
Activation of JNK and its target protein c-jun
GST-π is known to regulate JNK pathway therefore, JNK activation was assessed in Zn-exposed groups. Zn treatment exhibited drastic reduction in the level of JNK along with simultaneous increase in p-JNK level. BITC treatment markedly prevented the phosphorylation of JNK in Zn-exposed groups (Fig. 5a). BITC per se did not affect JNK activation in the exposed groups.

a Effect of BITC on Zn-induced modulations in the expression of JNK, p-JNK and p–c-jun. b Effect of Zn on the expression of Bcl-2 in the presence and absence of BITC. Upper panels in both a and b illustrate the representative blots and lower panels depict the densitometric analyses of the same. Data are expressed as mean ± SEM (n = 4). The values **p < 0.01 and ***p < 0.001 are expressed as compared with control; ###p < 0.001 as compared with Zn-treated group
Zn exposure increased the level of phosphorylated c-jun (p–c-jun) protein. However, BITC pre-treatment discernibly mitigated Zn-induced increase in the level of p–c-jun protein (Fig. 5a). BITC alone did not alter the expression of p–c-jun protein in the exposed groups.
Protein expression of Bcl-2 and Bax translocation
A noteworthy reduction was observed in the level of Bcl-2 protein in Zn-exposed groups. Pre-treatment with BITC significantly attenuated Zn-induced changes in the level of Bcl-2 (Fig. 5b). The level of Bcl-2 protein was not affected in the groups exposed to BITC alone (Fig. 5b).
Zn exposure resulted in the marked increase in the translocation of Bax from the cytosol to the mitochondria as evident from the reduced level of Bax in the cytosolic fraction with a concomitant increase in the level of Bax associated with the mitochondrial fraction (Fig. 6a). Pre-treatment with BITC noticeably prevented Zn-induced translocation of Bax to the mitochondria (Fig. 6a). BITC alone did not affect the translocation of Bax in the exposed groups.

a Effect of BITC on Zn-induced translocation of Bax. Upper panel shows representative western blot while lower panel depicts the densitometric analysis of the same. b It depicts the level of cyt c in the cytosolic and mitochondrial fractions of the nigrostriatal tissue in control and Zn-treated rats in the presence or absence of BITC. Upper panel and lower panel show representative western blots and densitometric analysis, respectively. c Effect of BITC on Zn-induced changes in pro-caspase-3/9 level. Upper panel shows representative western blot and lower panel illustrates the densitometric analysis of the same. Data are expressed as mean ± SEM (n = 4). The values **p < 0.01 and ***p < 0.001 are expressed as compared with control; ###p < 0.001 as compared with Zn-treated group

Schematic representation of role of GST-pi in Zn-induced dopaminergic neurodegeneration. [BITC benzyl isothiocyanate, GST-pi glutathione S-transferase-pi, JNK c-Jun N-terminal Kinases, cyt c cytochrome c. Positive sign (+) denotes induction and negative sign (−) denotes inhibition]
Cyt c release and caspase activation
Zn exposure exhibited decrease in the level of mitochondrial cyt c with a corresponding increase in the cytosolic cyt c level indicating increased mitochondrial cyt c release into cytosol (Fig. 6b). Pre-treatment with BITC significantly ameliorated Zn-induced mitochondrial cyt c release. BITC per se did not affect the cyt c levels in the exposed groups (Fig. 6b).
Zn exposure caused decrease in the level of pro-caspase 3 and pro-caspase 9 showing their activation into respective caspases (Fig. 6c). Pre-treatment with BITC significantly mitigated Zn-induced alterations in the level of pro-caspase 3 and pro-caspase 9 (Fig. 6c). BITC alone did not affect the activation of pro-caspase 3/9 in the exposed groups.
Discussion
Oxidative stress is one of the pioneer perpetrators in the onset and progression of PD while GSTs provide defence against oxidative stress-mediated damage [15,16,17,18, 28, 29]. Moreover, free radicals are established as the key players in Zn-induced neuronal cell death leading to PD phenotype in rodents so the current study was performed to delineate the role of GST-π in Zn-induced Parkinsonism [7,8,9]. Time-dependent reduction in CDNB-related total GST activity and GST-π activity without any change in GST-α or GST-µ activity with concurrent attenuation in GST-π protein level in the nigrostriatal tissue of Zn-exposed groups was found in this study. The results were in concurrence with many previous reports [15, 30] suggesting that GST-π might be involved in Zn-induced Parkinsonism. Reduced GST-π activity and protein expression are also reported in the brain of PD patients and toxin-based models [14, 15, 31] implicating its role in PD pathogenesis.
Neurobehavioral deficits in Zn-exposed groups were in agreement with earlier studies documenting that Zn induces PD-like features in rodents [6, 32]. The role of GST-π was explored by evaluating the effect of GST-π inducer on Zn-induced dopaminergic neurodegeneration. Marked rescue from the motor impairments in groups pre-treated with BITC, a GST-π inducer, indicated the protective effect of GST-π in Zn-induced Parkinsonism. Depletion of the striatal dopamine, DOPAC, HVA, TH protein expression and number of TH-positive neurons and unaltered serotonin level in exposed groups reaffirmed that Zn leads to PD phenotype [7,8,9]. Restoration of neurotransmitter levels, the number of TH-positive neurons, TH-protein expression by BITC suggested the protective role of GST-π in Zn-induced PD. These results are supported by the previous reports where over-expression of GST-π and its homologue are found to protect against dopaminergic neuronal death [15, 19, 22, 30].
In this study, BITC alleviated Zn-induced changes in GST activity/expression as also documented by other investigators [23]. Zn-induced inhibition of the total GST and GST-π activities along with diminished protein level of GST-π in the brain of exposed groups was in concurrence with the previous reports that have shown the reduced GST-π activity and protein expression in the brain of PD patients and toxin models [15, 30]. Increased LPO levels and SOD activity along with reduced catalase activity found in Zn-exposed brain tissue suggest the involvement of the oxidative stress in Zn-induced dopaminergic neurodegeneration [7,8,9]. Pre-treatment with BITC significantly prevented Zn-induced oxidative stress indexes indicating that antioxidant property of GST-π could be responsible for reduced oxidative stress [18, 33, 34]. BITC augmented the GST-π activity and expression in the Zn-exposed group with concomitant restoration in Zn-induced neurobehavioral deficits, neurodegenerative indices and oxidative stress implying that GST-π provided protection against Zn-induced neuronal death [19, 22, 30].
GST-π negatively regulates JNK activation via protein–protein interaction thereby controls the phosphorylation of the downstream target protein, c-jun [19, 21, 35, 36]. The JNK pathway represents a central oxidative stress-activated response, which contributes to apoptotic cell death. The increased level of p-JNK accompanied by increased phosphorylation of its target protein c-jun in Zn-exposed groups clearly pointed towards the activation of the JNK pathway following Zn exposure. Alleviation of Zn-induced increase in the levels of p-JNK and p–c-jun in BITC-treated groups suggested that reduced GST-π could be the plausible reason for increased oxidative stress and subsequently activation of JNK and phosphorylation of c-jun in Zn-exposed animals. Increased levels of p-JNK and p–c-jun are already reported in the brain of PD patients and toxin-based models [12, 19, 37, 38]. Increased resistance in JNK null mice and protection afforded by selective JNK inhibitors in MPTP-induced neurodegeneration further substantiated its role in dopaminergic neuronal death [39, 40].
JNK signaling results in modulation of anti-apoptotic B-cell lymphoma-2 (Bcl-2) protein and mitochondrial translocation of Bcl-2 associated X protein (Bax) that cause cytochrome c (cyt c) release and caspase cascade activation leading to apoptotic cell death [39, 41]. The reduced levels of Bcl-2 with concurrent increase in translocation of the Bax, mitochondrial cyt c release and activation of pro-caspase 3/9 in the Zn-exposed groups implied that Zn induces dopaminergic neuronal death via apoptotic pathway [9, 32] and suggested the involvement of JNK-mediated apoptosis. Furthermore, a marked amelioration in Zn-induced decrease in Bcl-2 levels, increased Bax translocation to mitochondria along with mitochondrial cyt c release and pro-caspase 3/9 activation with concomittant inhibition of JNK activation by BITC signifies the role of GST-π in JNK-mediated apoptotic dopaminergic neuronal cell death in Zn-induced Parkinsonism in concurrence with other toxin-based PD models [19, 42] as illustrated in the schematic diagram (Fig. 7).
Conclusion
The results of the study demonstrate that Zn inhibits GST-π, increases oxidative stress, and activates JNK pathway that eventually lead to Bax-dependent cell death. On the other hand, BITC restores GST-π activity/expression and protects from Zn-induced oxidative stress and JNK-mediated intrinsic apoptotic cell death. Taken as a whole, the study indicates that GST-π protects from Zn-induced dopaminergic neurodegeneration leading to Parkinsonism.
References
Dardiotis E, Xiromerisiou G, Hadjichristodoulou C, Tsatsakis AM, Wilks MF, Hadjigeorgiou GM (2013) The interplay between environmental and genetic factors in Parkinson’s disease susceptibility: the evidence for pesticides. Toxicology 307:17–23
Goldman SM (2014) Environmental toxins and Parkinson’s disease. Annu Rev Pharmacol Toxicol 54:141–164
Singh C, Ahmad I, Kumar A (2007) Pesticides and metals induced Parkinson’s disease: involvement of free radicals and oxidative stress. Cell Mol Biol (Noisy-le-grand) 53(5):19–28
Fleming SM (2017) Mechanisms of gene-environment interactions in Parkinson’s Disease. Curr Environ Health Rep 4:192–199
Dexter DT, Carayon A, Javoy-Agid F, Agid Y, Wells FR, Daniel SE, Lees AJ, Jenner P, Marsden CD (1991) Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 114(Pt 4):1953–1975
Singh BK, Kumar A, Ahmad I, Kumar V, Patel DK, Jain SK, Singh C (2011) Oxidative stress in zinc-induced dopaminergic neurodegeneration: implications of superoxide dismutase and heme oxygenase-1. Free Radic Res 45(10):1207–1222
Kumar A, Singh BK, Ahmad I, Shukla S, Patel DK, Srivastava G, Kumar V, Pandey HP, Singh C (2012) Involvement of NADPH oxidase and glutathione in zinc-induced dopaminergic neurodegeneration in rats: similarity with paraquat neurotoxicity. Brain Res 1438:48–64
Kumar V, Singh BK, Chauhan AK, Singh D, Patel DK, Singh C (2016) Minocycline rescues from zinc-induced nigrostriatal dopaminergic neurodegeneration: biochemical and molecular interventions. Mol Neurobiol 53(5):2761–2777
Chauhan AK, Mittra N, Kumar V, Patel DK, Singh C (2016) Inflammation and B-cell lymphoma-2 associated X protein regulate zinc-induced apoptotic degeneration of rat nigrostriatal dopaminergic neurons. Mol Neurobiol 53(8):5782–5795
Dias V, Junn E, Mouradian MM (2013) The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis 3:461–491
Kwakye GF, McMinimy RA, Aschner M (2017) Disease-toxicant interactions in Parkinson’s neuropathology. Neurochem Res 42:1772–1786
Peng J, Andersen JK (2003) The role of c-Jun N-terminal kinase (JNK) in Parkinson’s disease. IUBMB Life 55(4–5):267–271
Puspita L, Chung SY, Shim JW (2017) Oxidative stress and cellular pathologies in Parkinson’s disease. Mol Brain 10:53. https://doi.org/10.1186/s13041-017-0340-9
Smeyne M, Smeyne RJ (2013) Glutathione metabolism and Parkinson’s Disease. Free Radic Biol Med 62:13–25
Smeyne M, Boyd J, Raviie Shepherd K, Jiao Y, Pond BB, Hatler M, Wolf R, Henderson C, Smeyne RJ (2007) GSTpi expression mediates dopaminergic neuron sensitivity in experimental parkinsonism. Proc Natl Acad Sci USA 104(6):1977–1982
Kelada SN, Stapleton PL, Farin FM, Bammler TK, Eaton DL, Smith-Weller T, Franklin GM, Swanson PD, Longstreth WT Jr, Checkoway H (2003) Glutathione S-transferase M1, T1, and P1 polymorphisms and Parkinson’s disease. Neurosci Lett 337(1):5–8
Vilar R, Coelho H, Rodrigues E, Gama MJ, Rivera I, Taioli E, Lechner MC (2007) Association of A313 G polymorphism (GSTP1*B) in the glutathione-S-transferase P1 gene with sporadic Parkinson’s disease. Eur J Neurol 14(2):156–161
Shi M, Bradner J, Bammler TK, Eaton DL, Zhang J, Ye Z, Wilson AM, Montine TJ, Pan C (2009) Identification of glutathione S-transferase pi as a protein involved in Parkinson disease progression. Am J Pathol 175(1):54–65
Castro-Caldas M, Carvalho AN, Rodrigues E, Henderson C, Wolf CR, Gama MJ (2012) Glutathione S-transferase pi mediates MPTP-induced c-Jun N-terminal kinase activation in the nigrostriatal pathway. Mol Neurobiol 45:466–477
Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ, Wolf CR, Davis RJ, Ronai Z (1999) Regulation of JNK signaling by GSTp. EMBO J 18(5):1321–1334
Wang T, Arifoglu P, Ronai Z, Tew KD (2001) Glutathione S-transferase P1-1 (GSTP1-1) inhibits c-Jun N-terminal kinase (JNK1) signaling through interaction with the C terminus. J Biol Chem 276(24):20999–21003
Lin CY, Fu RH, Chou RH, Chen JH, Wu CR, Chang SW, Tsai CW (2017) Inhibition of JNK by pi class of glutathione S-transferase through PKA/CREB pathway is associated with carnosic acid protection against 6-hydroxydopamine-induced apoptosis. Food Chem Toxicol 103:194–202
Tan XL, Shi M, Tang H, Han W, Spivack SD (2010) Candidate dietary phytochemicals modulate expression of phase II enzymes GSTP1 and NQO1 in human lung cells. J Nutr 140(8):1404–1410
Kumar A, Ahmad I, Shukla S, Singh BK, Patel DK, Pandey HP, Singh C (2010) Effect of zinc and paraquat co-exposure on neurodegeneration: modulation of oxidative stress and expression of metallothioneins, toxicant responsive and transporter genes in rats. Free Radic Res 44(8):950–965
Habig WH, Pabst MJ, Jacoby WB (1974) Glutathione S-transferases: the first enzymatic step in mercapturic acid formation. J Biol Chem 249:7130–7139
Ahmad I, Shukla S, Singh D, Chauhan AK, Kumar V, Singh BK, Patel DK, Pandey HP, Singh C (2014) CYP2E1-mediated oxidative stress regulates HO-1 and GST expression in maneb-and paraquat-treated rat polymorphonuclear leukocytes. Mol Cell Biochem 393:209–222
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193(1):265–275
Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT (2003) Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci 23(34):10756–10764
Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, Marras C, Bhudhikanok GS, Kasten M, Chade AR, Comyns K, Richards MB, Meng C, Priestley B, Fernandez HH, Cambi F, Umbach DM, Blair A, Sandler DP, Langston JW (2011) Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect 119(6):866–872
Settivari R, VanDuyn N, LeVora J, Nass R (2013) The Nrf2/SKN-1-dependent glutathione S-transferase pi homologue GST-1 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of manganism. Neurotoxicology 38:51–60
Chauhan AK, Mittra N, Patel DK, Singh C (2017) Cyclooxygenase-2 directs microglial activation-mediated inflammation and oxidative stress leading to intrinsic apoptosis in Zn-induced Parkinsonism. Mol Neurobiol 55(3):2162–2173
Lin CY, Chen JH, Fu RH, Tsai CW (2014) Induction of Pi form of glutathione S-transferase by carnosic acid is mediated through PI3K/Akt/NF-kB pathway and protects against neurotoxicity. Chem Res Toxicol 27:1958–1966
Salinas AE, Wong MG (1999) Glutathione S-transferases—a review. Curr Med Chem 6(4):279–309
Yin Z, Ivanov VN, Tew HK, Ronai Z (2000) Glutathione S-transferase p elicits protection against H2O2-induced cell death via coordinated regulation of stress kinases. Cancer Res 60:4053–4057
Adler V, Pincus MR (2004) Effector peptides from glutathione-S-transferase-pi affect the activation of jun by jun-N-terminal kinase. Ann Clin Lab Sci 34(1):35–46
Castro-Caldas M, Neves Carvalho A, Peixeiro I, Rodrigues E, Lechner MC, Gama MJ (2009) GST-pi expression in MPTP-induced dopaminergic neurodegeneration of C57BL/6 mouse midbrain and striatum. J Mol Neurosci 38(2):114–127
Nishi K (1997) Expression of c-Jun in dopaminergic neurons of the substantia nigra in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mice. Brain Res 771(1):133–141
Saporito MS, Thomas BA, Scott RW (2000) MPTP activates c-Jun NH(2)-terminal kinase (JNK) and its upstream regulatory kinase MKK4 in nigrostriatal neurons in vivo. J Neurochem 75(3):1200–1208
Wang W, Shi L, Xie Y, Ma C, Li W, Su X, Huang S, Chen R, Zhu Z, Mao Z, Han Y, Li M (2004) SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Neurosci Res 48(2):195–202
Pan J, Qian J, Zhang Y, Ma J, Wang G, Xiao Q, Chen S, Ding J (2009) Small peptide inhibitor of JNKs protects against MPTP-induced nigral dopaminergic injury via inhibiting the JNK-signaling pathway. Lab Investig 90(2):156–167
Wang Y, Zhang Y, Wei Z, Li H, Zhou H, Zhang Z, Zhang Z (2009) JNK inhibitor protects dopaminergic neurons by reducing COX-2 expression in the MPTP mouse model of subacute Parkinson’s disease. J Neurol Sci 285:172–177
Elsby R, Kitteringham NR, Goldring CE, Lovatt CA, Chamberlain M, Henderson CJ, Wolf CR, Park BK (2003) Increased constitutive c-Jun N-terminal kinase signalling in mice lacking glutathione S-transferase Pi. J Biol Chem 278:22243–22249
Acknowledgements
The research scholarship provided by the Council of Scientific and Industrial Research (CSIR), New Delhi, Department of Science and Technology (DST), New Delhi and University Grants Commission (UGC), New Delhi to Amit Kumar Chauhan, Namrata Mittra and Brajesh Kumar Singh respectively, are sincerely acknowledged. The financial aid rendered by the CSIR to Chetna Singh through CSIR-network programme ‘Integrated NextGen Approaches in Health, Disease and Environmental Toxicity’ [INDEPTH (BSC-0111)] is highly appreciated. The CSIR-IITR communication number of this article is 3546.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interests.
Ethical approval
The institutional animal ethics committee approved the present study. All the experiments were performed as per the guidelines of the committee for the purpose of control and supervision of experiments on animals (CPCSEA).
Rights and permissions
About this article

Cite this article
Chauhan, A.K., Mittra, N., Singh, B.K. et al. Inhibition of glutathione S-transferase-pi triggers c-jun N-terminal kinase-dependent neuronal death in Zn-induced Parkinsonism. Mol Cell Biochem 452, 95–104 (2019). https://doi.org/10.1007/s11010-018-3415-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-018-3415-8