
Abstract
P2C-type ATPases are a subfamily of P-type ATPases comprising Na+/K+-ATPase and H+/K+-ATPase. Na+/K+-ATPase is ubiquitously expressed and has been implicated in several neurological diseases, whereas H+/K+-ATPase is found principally in the colon, stomach, and kidney. Both ATPases have two subunits, α and β, but Na+/K+-ATPase also has a regulatory subunit called FXYD, which has an important role in cancer. The most important functions of these ATPases are homeostasis, potassium regulation, and maintaining a gradient in different cell types, like epithelial cells. Na+/K+-ATPase has become a center of attention ever since it was proposed that it might play a crucial role in neurological disorders such as bipolar disorder, mania, depression, familial hemiplegic migraine, rapid-onset dystonia parkinsonism, chronic stress, epileptogenesis, and Alzheimer’s disease. On the other hand, it has been reported that lithium could have a neuroprotective effect against ouabain, which is the best known Na+/K+-ATPase inhibitor, but and high concentrations of lithium could affect negatively H+/K+-ATPase activity, that has a key role in regulating acidosis and potassium deficiencies. Finally, potassium homeostasis regulation is composed of two main mechanisms, extrarenal and renal. Extrarenal mechanism controls plasma levels, shifting potassium from the extracellular to the intracellular, whereas renal mechanism concerns with body balance and is influenced by potassium intake and its urinary excretion. In this article, we discuss the functions, isoforms, and localization of P2C-type ATPases, describe some of their modulators, and discuss their implications in some diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
P2-type constitute a family of ATPases consisting of Ca2+-ATPases (P2A) from sarcoplasmic reticulum (SERCA) and plasma membrane; calmodulin-binding Ca2+-ATPase (P2B); H+/K+-ATPases from stomach, colon, and kidney; and Na+/K+-ATPase (P2C). The family name is related to a transient phosphorylation of an aspartyl residue during cations’ transport [1], such as Cu2+, K+, Mg2+, H+, Ca2+, Cd2+, and Na+, for which these proteins are responsible of. The P-type family members have the following five functional and structural domains: an actuator domain (A), a nucleotide binding domain (N), a phosphorylation domain (P), a transport domain (T), and a class-specific binding domain (S). The first three domains are cytoplasmic, and the other two are membrane-embedded domains. It has been reported that Na+/K+-ATPase and SERCA sequence structures are 30 % identical and 65 % similar [2], this is the reason why SERCA model was used for Na+/K+-ATPase studies until it was crystalized. Also, both proteins have an α subunit, with 10 transmembrane spans, but Na+/K+-ATPase differs in having also β and γ isoforms, with 12 transmembrane spans, so two subunits should affect the α subunit conformation of Na+/K+-ATPase [3, 4]. In this review, we concentrate only in P2C-ATPases: H+/K+-ATPases and Na+/K+-ATPase. The carboxy-terminal domain is essential for the correct assembly and function of both H+/K+-ATPases and Na+/K+-ATPase; if it is not present, the affinity for Na+ and H+ is significantly affected [5, 6]. These enzymes are composed of α and β subunits, but Na+/K+-ATPase has a third subunit called FXYD too [7]. These proteins have different structure since Na+/K+-ATPase is a monomeric heterodimer, which is essential for its catalytic activity and cation translocation, whereas H+/K+-ATPase is a dimeric heterodimer [8].
Na+/K+-ATPase and H+/K+-ATPase are proteins that actively transport either three molecules of Na+ and two molecules of K+ or two molecules of H+ and two molecules of K+, respectively, against a concentration gradient. This transport maintains ionic cellular homeostasis, keeps the extracellular concentrations of Na+ and H+ low and the intracellular concentration of K+ high.
Na+/K+-ATPase uses almost 30 % of the ATP available to the cell and has important roles that include the following: (1) maintaining ionic balance, (2) providing energy through the coupled transport of nutrients, (3) re-establishing the ionic gradient after an action potential in neurons, (4) helping to maintain a gradient in epithelial cells, and (5) activating lymphocytes [9].
The H+/K+-ATPase also uses ATP, but its regulator role is related to gastric processes and maintaining a normal K+ concentration in organs such as the stomach, colon, and kidney. In the stomach, it regulates the HCl concentration, but in altered states, it can function as a Na+/K+-ATPase and as a Ca2+-ATPase in parietal cells [10]; in the kidney, H+/K+-ATPase has a key role in states of altered K+ balance, such as hypokalemia, and in the colon, it is also involved in regulating H+ concentration, and it functions in K+ absorption [11]. Gastric H+/K+-ATPase, it can be stimulated through to the histamine H2 receptor [12].
Na+/K+-ATPase and H+/K+-ATPase bound ATP, but there are alternatives: one is through extracellular signal-regulated kinase (ERK) [13], protein kinase C (PKC), or protein kinase A (PKA) [14–16] and another, which is not well understood, is through the insulin receptor [17]. Through PKC and PKA pathways, the phosphorylation site can stimulate or diminish Na+/K+-ATPase or H+/K+-ATPase activity [14, 16], and ERK activity could be activated by insulin stimulating Na+/K+-ATPase activity [13].
Na+/K+-ATPase can function as a signal transducer, through calcium signaling or Src signaling pathways [18]. The Src pathway is present in the kidney and has an important role in renal function; a decline in the protein expression of Na+/K+-ATPase is sufficient to raise Src activity, and this interaction may depend on the conformational state of the ATPase [19].
Na+/K+-ATPase has endogenous modulators whose expression and activity are regulated by hormones including steroids, thyroid hormones, peptide hormones, and catecholamines. These have different effects; some regulate its gene expression, and others regulate its activity [20]. Exogenous modulators include cardiotonic steroids, such as ouabain and bufadienolides [21], as well as lithium, which has different effects on the protein.
H+/K+-ATPase also has inhibitors, such as benzimidazoles, that are commercially known as esomeprazole and proton pump inhibitors like rabeprazole, omeprazole, lansoprazole, and pantoprazole. Other H+/K+-ATPase inhibitors are K+-competitive reagents, named either acid pump antagonist (APA) or potassium competitive acid blocker (P-CAB) class, which are imidazo-pyridines that are known to react on the outside surface of the pump in competition with potassium ions [22]. Lithium is a cation reported to activate Na+/K+-ATPase [23], that could inhibit H+/K+-ATPase in the cortical collecting tubule in the kidney, which can trigger a severe metabolic acidosis [24].
Isoforms and Localization
P2C-type ATPases are conformed by α and β subunits, and Na+/K+-ATPase has a third subunit called γ or FXYD. α subunit contains the catalytic site, and in Na+/K+-ATPase and one type of H+/K+-ATPase, is the site of ouabain binding. Four isoforms for the α subunit of Na+/K+-ATPase have been reported: α1, α2, α3, and α4. These isoforms occur in different tissues with the fourth found only in sperm.
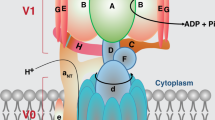
H+/K+-ATPase has only two isoforms, α1 and α2, and one gastric and the other non-gastric. The α subunits of Na+/K+-ATPase and H+/K+-ATPase have close to 60 % similarity [25]. β subunit is a minor subunit that contributes to the K+ affinity in its binding site and may contribute to the trafficking and delivery of Na+/K+-ATPase to the cell membrane [26]. The β subunit also contributes to the function of the α subunit. Na+/K+-ATPase has three β isoforms, β1, β2, and β3, whereas only one β subunit, called βHK, has been reported for H+/K+-ATPase. The third subunit of Na+/K+-ATPase, named FXYD, is also a regulatory unit and contributes to the affinity of Na+ to its binding site; there are seven different isoforms, FXYD1 to FXYD7 [27]. Figure 1 depicts representations of Na+/K+-ATPase and H+/K+-ATPase.

Conformation of P2C-type ATPase. Na+/K+-ATPase is composed by three subunits, α, β, and FXYD. Only α subunit is the catalytic one and is the place where ion exchange occurs, in this case is three intracellular Na+ for two extracellular K+. H+/K+-ATPase is a dimer composed of two subunits, α and β, and α is the catalytic subunit too; this enzyme exchanges one intracellular H+ for one extracellular H+, but how it works as a dimer, the exchange is 2H+ by 2K+. β and FXYD subunits are to stabilize the ion binding and the activity of the enzyme
Na+/K+-ATPase
The α subunit for Na+/K+-ATPase, molecular weight of ≈110 kDa, contains the catalytic site and is where Na+ and K+ are exchanged. This exchange occurs against the concentration gradient. The first step occurs when three intracellular Na+ ions bound to the enzyme (E1) and ATP phosphorylates the α subunit to E1P, first releasing one sodium ion into the extracellular space, then releasing the other two (E2P). In this state, the enzyme is phosphorylated and, when no ion is present, can bound ouabain. In the absence of ouabain, two extracellular K+ ions bound to the α subunit (E2P), the phosphorus is released (E2), and finally, potassium enters to the cytoplasm (E1), and the protein can start the cycle again [28].
Only three of the four α isoforms were originally identified [29]. The α4 isoform [30], which is found only in male gametes [31], controls Na+ gradients and plays a role in Ca2+ and H+ homeostasis in cells. Sperm motility is dependent on pH balance and membrane excitement; therefore, regulation by α4 may contribute to the motility of the sperm [32, 33]. The other isoforms are widely found in different tissues; for example, the α1 isoform occurs in every tissue but principally in the kidney, which is considered a model for this isoform. The α2 isoform is found in the brain, heart, and in skeletal muscle, and α3 is also expressed in the heart and brain. An important role for the α subunit in memory and mood is proposed; haploinsufficiency of α2 and α3 generate behavioral alterations [34], and a missense mutation in the α3 isoform was found in a maniac-like mouse model [35].
The different affinities of these isoforms for ouabain, a Na+/K+-ATPase inhibitor, are also interesting. The α1 subunit has a low affinity, whereas α2 and α3 have high affinities: This is an important research subject because the α1 isoform is present in α-motoneurons and α3 is present in γ-motoneurons [36]. The α2 isoform has a more prominent role in regulating Ca2+ release from the sarcoplasmic reticulum of myocytes [37].
The β subunit for Na+/K+-ATPase, molecular weight of ≈55 kDa, is a glycoprotein that lacks a catalytic site and does not participate in ion exchange. Instead, it has regulatory activity that, in MDCK cells, is crucial for the correct maturation of the enzyme [38]. Na+/K+-ATPase is resistant to non-ionic detergents when α and β subunits are joined [39]. Inhibiting the glycosylation of the β subunit reduces the cellular abundance of both subunits, which implies a role in the expression of Na+/K+-ATPase on the cell [40].
There are three β isoforms of β subunit, β1, β2, and β3, where β1 is more expressed in the kidney (where it is part of the model for the α1 β1 isoform) and in organs related to homeostasis [26]. Normal glycosylation of this isoform is needed for the correct stability of adherent and tight junctions in mature epithelia [41]. The reported reduction of the β2 isoform in glioblastomas is interesting because this could be the factor that initiates invasion by these cells [42]. Inactivation of the β3 isoform can inactivate T and B lymphocytes, which is important for immune regulation [9].
Isoforms α1, α2, and α3 are present in the brain, where they have different expression profiles: α1 is ubiquitous, α2 is predominant in astrocytes, and α3 is principally present in neurons. The β1 and β2 isoforms occur in the brain, with astrocytes expressing α1, α2, and β2 and neurons expressing α1, α3 and β1 and β2 [43]
In one study, inhibition of α3 nearly eliminated the ability of hippocampal dendrites to restore the ionic concentration of Na+ [44], and animals with α3 haploinsufficiency had difficulties finding the platform in the Morris Water Maze test, even when it was removed, indicating an impaired short- and long-term memory [34]. In α3 haploinsufficient mice, the NR1 subunit of the NMDA receptor is downregulated [45].
The additional regulatory protein, FXYD, is present in seven isoforms: FXYD1 or phospholemman; FXYD2 or γ subunit; FXYD3 or mammary tumor marker Mat-8; FXYD4 or corticosteroid hormone-induced factor CHIF; FXYD5 or related to ion channel RIC or dysadherin; FXYD6 or phosphohippolin; and FXYD7 [46]. Na+/K+-ATPase might be a target for anticancer treatment because FXYD protein, e.g., FXYD3, is a cancer-related protein [47].
H+/K+-ATPase
The α subunit of H+/K+-ATPase, that has a molecular weight close to 100 kDa, is conformed by 10 transmembrane domains and has two isoforms [25]. It contains the catalytic domain of the protein to which H+ binds, but, interestingly, this ATPase also needs a molecule of Mg2+ for an entire H+–K+ exchange cycle. In the presence of Mg2+, the enzyme binds ATP, then one H+ is added and ADP detaches, leaving a phosphate bound to the enzyme. Next, with the anion still in the ATPase, H+ is exchanged for K+; later, the anion is liberated, and K+ can then be dissociated. Finally, the state in which H+/K+-ATPase is bound to Mg2+ is restored and the cycle can start again [11, 12, 28]. High K+ concentrations are needed for some functions of H+/K+-ATPase; for example, during fetal development, a HKα2-null mice may have problems with placental perfusion or even partial abortion. These may result from failure of compartment volume adjustment, most likely because of a high plasma K+ concentration [48]. Both benign prostate hyperplasia and prostatic tumor tissue had increased non-gastric H+/K+-ATPase compared with normal prostate cells, not only in the basal cells and membrane but also along the epithelium and cytosol [49]. The colonic H+/K+-ATPase acts as an Na+/K+-ATPase in Xenopus leavis oocytes because its function is devoted to K+ transport [50], and it can also transport NH4 + in rat distal colon [51]. Most recently, it was reported to function as a Na+/K+-ATPase and Ca2+-ATPase in parietal cells during altered pH states [10].
Two isoforms of α subunits exist: the gastric or HKα1 H+/K+-ATPase and the non-gastric or colonic H+/K+-ATPase or HKα2 H+/K+-ATPase [52]. The HKα2 gene, however, produces three alternative transcripts. HKα2(a) is found in the colonic H+/K+-ATPase, HKα2b is reported in the rat and has been observed in kidney, and HKα2c has been reported in rabbit and rat but may not be found in humans [53]. Omeprazol is a specific modulator of H+/K+-ATPase [54], and it binds in the α subunit during acid transporter conditions [12]. Ouabain can bind to the HKα2 H+/K+-ATPase, but not the HKα1 H+/K+-ATPase, of guinea pig colon [55]; this is an interesting finding because ouabain is a specific modulator of Na+/K+-ATPase.
Four isoforms of β subunit, HKβ, NaKβ1, NaKβ2, and NaKβ3, with molecular weights of ≈35–55 kDa [52], can pair with the HKα subunits. HKα1 is known to pair with HKβ [53], but apparently, the β1 subunit of Na+/K+-ATPase pairs with HKα2 [56]. β subunit lacks of a catalytic site, but it is crucial for H+/K+-ATPase function, localization on the plasma membrane, and for K+ binding to the α subunit, which is similar to Na+/K+-ATPase.
X+/K+-ATPases and Their Modulation
H+/K+-ATPases
H+/K+-ATPases have specific inhibitors that are principally divided into two groups: the cardiac glycosides and the non-cardiac glycosides which are included substituted benzimidazoles, potassium-competitive acid blockers, and vanadate [22] (Table 1). The most common substituted benzimidazole known is omeprazole, and it inhibits proton exchange, thereby reducing stomach acidity and the risk for gastric ulcer. Omeprazole derivatives that share the same action but differ chemically include timoprazole, esomeprazole, rabeprazole, pantoprazole, and lansoprazole [57]. Omeprazole sulfide is a metabolite of omeprazole found in plasma that is eliminated in the urine and feces; it competes with the activated form of omeprazole to reduce its inhibitory effect. This metabolite was used to locate the site of omeprazole binding, and the sulfur moiety has a nucleophilic attraction to Cys813 in the transmembrane (TM) helices TM1 and TM2 [58].
In the potassium-competitive acid blocker class, SCH28080 inhibits ATPase function when potassium is not in the enzyme; other inhibitors in this family are TAK.438, AZD0865, and revaprazan [22]. Vanadate binds to all P-type ATPases in the phosphate binding site and irreversibly inhibits them. The binding affinity of vanadate is higher in the E2 of conformation, that is, after ATP hydrolysis [53]. Finally, of the cardiac glycosides, the best known is ouabain, an inhibitor of Na+/K+-ATPase. Its function in H+/K+-ATPase is controversial because the degree of enzyme inhibition is variable, depending on the cell type and expression system used. For example, HKα2 is insensitive to ouabain in HEK-293 cells [59] but sensitive when it was expressed on Xenopus oocytes [60]. Lithium, a well-characterized cation that is used to treat psychiatric conditions such as bipolar disorder, is known to cause kidney failure at high concentrations. Significantly, lithium can decrease H+/K+-ATPase activity in cortical collecting tubules but not in medullary collecting tubules [24].
Na+/K+-ATPase
Na+/K+-ATPase has several endogenous modulators and non-endogenous modulators. Endogenous modulators include the following: steroidal hormones, endogenous ouabain, non-steroidal peptide hormones, and catecholamines. Non-endogenous modulators include the following: digitalis (ouabain-like cardiotonic steroid modulators), bufadienolides [21], competitors such as chlorpromazine and chloroquine [61], and lithium (Table 1). From all these, we focused on ouabain, which is the most common inhibitor used, and on lithium, which is a very important monovalent cation that is used for treatment of bipolar disorder. In particular, lithium has been observed to have protective effects against ouabain, which is really interesting to understand how lithium could modulate Na+/K+-ATPase (Fig. 2 and 3).

Lithium and its role in P2C-type ATPase. Lithium is a well-known cation that is used for neurological diseases like bipolar disease and depression, and lately, it has been reported a possible role in Alzheimer’s disease, and it has been seen that it stimulates Na+/K+-ATPase activity at the same time that at this concentrations, it affects H+/K+-ATPase activity in kidney and can produce other illness. Now would be really interesting to find the correct lithium concentration that could enhance Na+/K+-ATPase and not affect H+/K+-ATPase to be able to use it to prevent or stabilize aging affections as Alzheimer’s disease

Possible relation between Na+/K+-ATPase and H+/K+-ATPase. Since H+/K+-ATPase has a role in potassium excretion, if this enzyme fails could be possible to have a role in hypertension, an illness that is well-known to be a risk factor in neurological diseases as dementia and Alzheimer’s disease (AD) and is acknowledged that Na+/K+-ATPase activity is reduced in these diseases
Ouabain is an endogenous cardiac glycoside that inhibit Na+/K+-ATPase, is thought that is able to bind the α subunit. It is interesting to note that, in cardiotonic steroids, ouabain’s hydroxyl group position is very important because it has been reported that this position might be involved in the inhibitory activity with the enzyme [62]. As previously mentioned, inhibition could take place when Na+/K+-ATPase is in E2P state, after the first Na+ release, which could allow ouabain binding. In addition, it has been observed that inhibition site ouabain might be in the TM helices TM1, TM2, TM4, and TM6 of Na+/K+-ATPase [63]. However, in another report, results showed that Gln111 and Asn 122 in TM1 and TM2 are key amino acids that may be involved in the cardiac steroid-enzyme interaction [58]. Bonding can occur in two steps: First, the junction bonding occurs, and then, a sugar residue interaction occurs at a slower rate [62].
Ouabain can inhibit different α isoforms depending on its concentration, because it has different affinities as mentioned above, where the α1 subunit has low affinity to ouabain and α2 and α3 have high affinity. Ouabain also has different effects, including promotes apoptosis and necrosis at high concentrations [64], and it is involved in neurological diseases. In fact, it has been reported that ouabain is involved in mood disorders such as depression [65], and, interestingly, ouabain is used to make mania rat models, which is achieved with an intracerebroventricular administration of ouabain, and it was reported that lithium can prevent ouabain’s effect [66].
Notably, because Na+/K+-ATPase could be a potential target for the treatment of neurological diseases, it is important to identify which isoforms are involved in different diseases because it has been observed that different haploinsufficient mice have different deficits [34]. Mice deficient in the α3 isoform have more difficulties in the Morris water maze test, whereas the mice deficient in α2 have less entries in an open field area and do not perform well in the elevated zero maze test. In this context, it is very important to remember that α3 is an isoform found in neurons, α2 in astrocytes, and α1 is ubiquitous. Therefore, α3 issues are related to memory deficits, and α2 issues could be related to the homeostatic stabilization.
It has been reported that insulin promotes translocation of Na+/K+-ATPase [67], which is preventing digoxin toxicity [68], and it has also been observed that insulin reduces Na+/K+-ATPase’s activity in diabetic liver [69]. This traslocation induced by insulin is interesting because in diabetes, it has been observed a cognitive deficiency, as demonstrated by the Morris water maze test [70].
Lithium is a monovalent cation that it is known to replace Na+ in protein, but it protects Na+/K+-ATPase against inhibition by ouabain. However, the mechanisms of action are not understood. Lithium protects human neuroblastoma SH-SY5Y cells from ouabain-induced damage [71], and lithium delays ouabain-stimulated multiple spikes [72]. Lithium also enhances Na+/K+-ATPase’s activity in animal models of bipolar disorder [73] and depression [74].
The main concern with lithium is that its concentration in the human body should be closely monitored because it can cause problems such as hypothyroidism, weight gain, an increased risk of reduced urinary concentrating ability, hyperparathyroidism, and, in some cases, a significant reduction in renal function [75]. Nephrotoxicity can be observed after 2 months of lithium treatment, and lithium can also lead to diabetes insipidus [76]. A recent review reported that a safe long-term treatment dose of lithium is up to 0.8 mmol/L [77], and it has also been reported that lithium treatment of 40 nmol in humans for 15 months did not present any problems in kidney nor thyroid dysfunction [78], which indicates that this dose may be an appropriate limit for human treatment. This is an important point because lithium treatment has been shown to diminish Alzheimer’s disease (AD) risk in patients with bipolar disease [79] and stabilized cognitive impairment in Alzheimer’s disease [78].
Na+/K+ ATPase and Neurological Diseases
Several studies have related Na+/K+-ATPase to neurological disorders. In this paper, we discuss several examples of this relationship, and then, we focus on mood disorders and Alzheimer’s disease. A study has suggested that Na+/K+-ATPase and endogenous digitalis-like compounds are involved in mood disorders, similar to that reported in an animal model of depression [65]. For bipolar disorder, a genetic variation related to the α isoform has been observed [80], and a mania-like behavior has been reported in Myshkin mice that are heterozygous for the α3 isoform [35]. Familial hemiplegic migraine has been related to deficiencies in and mutations of the α2 isoform [81, 82], and mice with the same isoform deficiency have shown similar behavior [82]. In rapid-onset dystonic parkinsonism (RPD), six missense mutations have been found in the α3 isoform of Na+/K+-ATPase (ATP1A3) [81, 83] in seven unrelated families [84]. The same mutation in the α3 subunit has also been reported in two cases of RPD [85], and it has also been proposed that a possible mutation in D923N could be involved in this illness [86]. In other illnesses, such as alternating hemiplegia of childhood, patients have shown de novo mutations in ATP1A3, which do not affect the protein expression but do cause a reduction in the ATPase activity [87]. In Alzheimer’s disease that is principally related with aging, Na+/K+-ATPase activity is lower [88]. It is thought that lithium, a treatment for bipolar disorders, could be acting through Na+/K+-ATPase to repair neurological damage because an improvement in the activity of Na+/K+-ATPase has been observed in patients with Alzheimer’s disease [78].
By contrast, studies have shown a relationship between neurological diseases and a reduction in intracellular Na+ that could be associated with a decreased affinity of Na+ to this ATPase. This is particularly important because [Na+]i is reduced when Na+ affinity increases, and [Na+]i rises when Na+ affinity is reduced, suggesting that affinity itself could be regulating [Na+]I [89].
Na+/K+-ATPase and Mood States
Na+/K+ ATPase is altered in mood-related illnesses such as depression and bipolar disease. It has also been shown that early life experiences, e.g., neonatal handling in rats, have significant effects on Na+/K+-ATPase activity, which in rats results in a model of depression during adulthood [90]. In bipolar disease, there is evidence that Na+/K+-ATPase activity is significantly reduced together with an increase in lipid peroxidation [73], and two single nucleotide polymorphisms (SNPs) are significantly associated with this illness. Both SNPs are related to the α3 isoform, and one SNP is related to the α1 isoform [80].
Bipolar disease is known for episodes of mania and episodes of depression. It has been observed that intra-cerebroventricular administration of ouabain, which decreases Na+/K+-ATPase activity, can produce mania episodes, detectable by motoric hyperactivity [66]. A mania-like behavior can also be produced in mice heterozygous for the α3 isoform of Na+/K+-ATPase, in Myshkin mice (Atp1a3 Myk/+; Myk/+) [35]. In double transgenic mice that are heterozygous for the α3 subunit of Na+/K+-ATPase (MyK/+) and agrin (Agrn/+), a reduced mania-like behavior has been observed, which could indicate an interesting relation between agrin and Na+/K+-ATPase that could function as a target for mood stabilizers [91].
Does Na+/K+ ATPase Have a Role in Alzheimer’s Disease?
In different models of AD, the activity of Na+/K+-ATPase is affected by different inhibitors. First, a 40 % decrease in ouabain binding in the brains of patients with AD was described [92]; then, it was found that amyloid β-peptide impairs Na+/K+-ATPase [93] and that the activity of Na+/K+-ATPase was reduced in the brains of AD patients [88, 94]. A review also reported that Na+/K+-ATPase has a function in aging and energy balance [95]. In oocytes of X. leavis, an inhibition of Na+/K+-ATPase activity by β-amyloid was observed [96], and in the APP + PS1 double transgenic mice model of AD, a reduced activity of Na+/K+-ATPase was observed, particularly in the areas bordering the β-amyloid plaques [97]. Moreover, Zhang et al. proposed that Na+/K+-ATPase is an important factor to account for in the investigation of neuroprotection in AD [98], and, more recently, it was shown that microinjections of β-amyloid into the rat brain produces an impairment in Na+/K+-ATPase activity [99].
H+/K+-ATPases, Kidney Function, and Regulation
In the kidney, there are three isoforms of H+/K+-ATPases: HKα1, HKα2, and HKα2b. These three enzymes are expressed in the collecting duct, which deals with the renal regulation of pH and potassium secretion; HKα1 has also been reported on the ascending limb of Henle. It is likely that HKα1 and HKα2 may have different functions; thus, HKα1 and HKα2 are both upregulated during acidosis and cellular metabolism, whereas during cellular respiration, only the HKα1 is upregulated. HKα2 has increased expression during chronic hypokalemia in the renal medulla [100] [101]. During metabolic acidosis, it has been described an increase in enzyme activity but without change in gene expression. During alkalosis, there is no agreement whether it is increased or decreased since there are studies reporting both. Interestingly, vasopressin and aldosterone can modulate proton secretion of H+/K+-ATPase and H+-ATPase through the mineralocorticoid receptor and V1aR, and both aldosterone and vasopressin can also stimulate proton secretion [102].
By contrast, it is well known that urinary K+ excretion has a circadian profile, although the pathway involved is not elucidated. HKα2 also has a circadian profile regulated by Nrf2, so this could be the protein responsible for the excretion of K+ in a circadian rhythm [103]. Furthermore, in one study, serum hypokalemia was reported in two women taking omeprazole, and the hypokalemia was reversed by administration of potassium supplements and discontinuation of the H+/K+-ATPase inhibitor [104].
Importance of Dietary Potassium Intake in Health and Disease
The regulation of potassium homeostasis is composed of two main mechanisms: extrarenal and renal. The extrarenal mechanism controls very tightly plasma levels shifting potassium from the extracellular to the intracellular, whereas the renal mechanism deals with the body balance and is largely influenced by the intake and urinary excretion.
In the kidney, the distal nephron is a main site for the regulation of potassium handling; in particular, the connecting tubule cells (CNTc) highly specialized on potassium secretion [105]. The CNTc contains Na,K-ATPase in the basolateral side and a potassium channel (ROMK) in the apical side, whereas the neighboring intercalated cells (Ic) display H,K-ATPase in the apical side (Figs. 4 and 5).

Localization of Na,K-ATPase, H,K-ATPase, and kallikrein in the kidney. In the panel is shown the localization of Na,K-ATPase in the basolateral side of distal tubule cell (a), the localization of H,K-ATPase in the apical or luminal side of Intercalated cells (b), and the localization of kallikrein in the connecting tubule cells (c). The insets in each photography display at higher magnification their subcellular distribution. A cartoon showing the distribution polarized distribution of Na,K-ATPase and ROMK in the CNTc is a key regulator of potassium and the site of origin of kallikrein

Cell types of the distal nephron, a main site for regulation of potassium by the kidney. The distal nephron is composed of four different cell types: the distal convoluted tubule cell (DCTc), the connecting tubule cell (CNTc), the intercalated cell (Ic), and the collecting duct cell (CDc). Na,K-ATPase is located in the basolateral side of DCTc, CNTc, and CDc, whereas H,K-ATPase is located in the apical or luminal side of Ic. The CNTc contains Na,K-ATPase and ROMK and is a key regulator of potassium by the kidney and the site of origin of kallikrein
The CNTc is the site of origin of renal kallikrein [106]; this enzyme is the main component of the renal kallikrein-kinin system, a vasoactive system with vasodilatory and sodium excretory function, which is stimulated by increased potassium intake [105]. In response to a diet high in potassium, the CNTc cell hypertrophy increases the basolateral membrane and the Na,K-ATPase as a compensatory mechanism to secrete increased potassium intake. This mechanism is known as potassium adaptation and participates in the potassium balance. Along with cell hypertrophy, CNTc increased renal kallikrein synthesis and kallikrein system stimulation by high potassium diet increases the excretion of sodium and water with a net effect on decreasing high blood pressure [105]. This points to the kallikein system as a protective vasoactive system against hypertension, renal diseases, and stroke. Moreover, previous studies from our group have demonstrated in hypertensive human potassium supplementation in the diet significantly increased kallikrein, urinary sodium excretion and lower blood pressure [107].
On the contrary, we have demonstrated that during hypokalemia, there is a reduction of renal kallikrein, and despite of the mechanism underlying the decreased kallirein, an impaired kallikrein system contributed to salt sensitivity, hypertension, and renal damage [108].
The importance placed on the relevance of dietary potassium on health and disease [109, 110] is underscored compared with that placed on dietary sodium. Much effort has been placed on dietary sodium reduction on the control of several diseases, but much less have been placed on the adequate dietary potassium intake. Despite that natural food contains 10–15 times more potassium than sodium, however, our intake has ≈3 times more sodium (186 mmol/day) than potassium (65 mmol/day) due to the main component of processed food in our diet. Moreover, although it is known for decades that adequate potassium diet is beneficial to health, particularly for the control of blood pressure, stroke, and kidney disease [109] which has recently been confirmed, there are few studies on biomedical bases explaining this beneficial effect, and the cellular and molecular mechanisms responsible for the beneficial effects of increased dietary potassium remain unknown [111], and they are subject on active research by our group.
Concluding Remarks
P2C-type ATPases have different but important functions and expression. Na+/K+-ATPase has an important function in potassium regulation in neurons and is able to re-establish the membrane potential of neurons to produce regenerative action potentials and restore neuronal communication. H+/K+-ATPase is focused on the kidney, stomach, and colon; it is crucial for potassium serum concentrations in the kidney and for re-establishing acid–base homeostasis in the stomach and colon. It would be very interesting to evaluate the impact that H+/K+-ATPase could have in hypertension because Na+ and K+ homeostasis are key factors in this disease. Moreover, Na+ also influences urinary K+ secretion [112]. Because hypertension is a very common illness induced by an unhealthy diet, altogether with aging are risk factors for Alzheimer’s disease, another disease that might be influenced by a Na+/K+-ATPase defficient activity (Fig. 3). Finally, these two enzymes are more closely related than previously supposed, not only because of homology. It is possible that H+/K+-ATPase dysfunction in the maintenance of potassium homeostasis could affect neuronal activity and survival, as well as affect Na+/K+-ATPase activity.
References
Köksoy AA (2002) Na, K-ATPase: a review. J ANKARA Med Sch 24:73–82
Sweadner KJ, Donnet C (2001) Structural similarities of Na, K-ATPase and SERCA, the Ca(2+)-ATPase of the sarcoplasmic reticulum. Biochem J 356:685–704
Abe K, Tani K, Friedrich T, Fujiyoshi Y (2012) Cryo-EM structure of gastric H+, K+-ATPase with a single occupied cation-binding site. Proc Natl Acad Sci U S A 109:18401–18406. doi:10.1073/pnas.1212294109
Nyblom M, Poulsen H, Gourdon P et al (2013) Crystal structure of Na+, K(+)-ATPase in the Na(+)-bound state. Science 342:123–127. doi:10.1126/science.1243352
Li J, Codina J, Petroske E et al (2004) The carboxy terminus of the colonic H(+), K(+)-ATPase alpha-subunit is required for stable beta subunit assembly and function. Kidney Int 65:1301–1310. doi:10.1111/j.1523-1755.2004.00507.x
Toustrup-Jensen MS, Holm R, Einholm AP et al (2009) The C terminus of Na+, K+-ATPase controls Na+ affinity on both sides of the membrane through Arg935. J Biol Chem 284:18715–18725. doi:10.1074/jbc.M109.015099
Mishra NK, Peleg Y, Cirri E et al (2011) FXYD proteins stabilize Na, K-ATPase: amplification of specific phosphatidylserine-protein interactions. J Biol Chem 286:9699–9712. doi:10.1074/jbc.M110.184234
Hall K, Perez G, Sachs G, Rabon E (1991) Identification of H+/K(+)-ATPase alpha, beta-heterodimers. Biochim Biophys Acta 1077:173–179
Chiampanichayakul S, Szekeres A, Khunkaewla P et al (2002) Engagement of Na, K-ATPase beta3 subunit by a specific mAb suppresses T and B lymphocyte activation. Int Immunol 14:1407–1414
Ray T (2013) The parietal cell gastric H, K-ATPase also functions as the Na, K-ATPase and Ca-ATPase in altered states. F1000 Res 2:165–175. doi:10.12688/f1000research.2-165.v2
Shin JM, Munson K, Sachs G (2011) Gastric H+, K+-ATPase. Compr Physiol 1:2141–2153. doi:10.1002/cphy.c110010
Shin JM, Munson K, Vagin O, Sachs G (2009) The gastric HK-ATPase: structure, function, and inhibition. Pflugers Arch 457:609–622. doi:10.1007/s00424-008-0495-4
Al-Khalili L, Kotova O, Tsuchida H et al (2004) ERK1/2 mediates insulin stimulation of Na(+), K(+)-ATPase by phosphorylation of the alpha-subunit in human skeletal muscle cells. J Biol Chem 279:25211–25218. doi:10.1074/jbc.M402152200
Cornelius F, Mahmmoud YA (2003) Direct activation of gastric H, K-ATPase by N-terminal protein kinase C phosphorylation. Comparison of the acute regulation mechanisms of H, K-ATPase and Na, K-ATPase. Biophys J 84:1690–1700. doi:10.1016/S0006-3495(03)74977-7
Poulsen H, Morth P, Egebjerg J, Nissen P (2010) Phosphorylation of the Na+, K+−ATPase and the H+, K+−ATPase. FEBS Lett 584:2589–2595. doi:10.1016/j.febslet.2010.04.035
Mahmmoud YA, Cornelius F (2003) PKA and PKC phosphorylation of gastric H, K-ATPase. Ann N Y Acad Sci 986:548–549
Frindt G, Palmer LG (2012) Effects of insulin on Na and K transporters in the rat CCD. Am J Physiol Renal Physiol 302:F1227–F1233. doi:10.1152/ajprenal.00675.2011
Aperia A (2007) New roles for an old enzyme: Na, K-ATPase emerges as an interesting drug target. J Intern Med 261:44–52. doi:10.1111/j.1365-2796.2006.01745.x
Xie JX, Li X, Xie Z (2013) Regulation of renal function and structure by the signaling Na/K-ATPase. IUBMB Life 65:991–998. doi:10.1002/iub.1229
Lingrel JB, Orlowski J, Shull MM, Price EM (1990) Molecular genetics of Na, K-ATPase. Prog Nucleic Acid Res Mol Biol 38:37–89
Wang H-YL, O’Doherty GA (2012) Modulators of Na/K-ATPase: a patent review. Expert Opin Ther Pat 22:587–605. doi:10.1517/13543776.2012.690033
Li H, Meng L, Liu F et al (2013) H+/K+−ATPase inhibitors: a patent review. Expert Opin Ther Pat 23:99–111. doi:10.1517/13543776.2013.741121
Beaugé L (1978) Activation by lithium ions of the inside sodium sites in Na, K-ATPase. Niochimica Biophys Acta 527:472–484
Dafnis E, Kurtzman NA, Sabatini S (1993) H-K-ATPase in distal renal tubular acidosis: urinary tract obstruction, lithium, and amiloride. Am J Physiol 265:F875–F880
Crowson MS, Shull GE (1992) Isolation and characterization of a cDNA encoding the putative distal colon H+, K(+)-ATPase. Similarity of deduced amino acid sequence to gastric H+, K(+)-ATPase and Na+, K(+)-ATPase and mRNA expression in distal colon, kidney, and uterus. J Biol Chem 267:13740–13748
Horisberger JD, Lemas V, Kraehenbühl JP, Rossier BC (1991) Structure-function relationship of Na, K-ATPase. Annu Rev Physiol 53:565–584. doi:10.1146/annurev.ph.53.030191.003025
Kaplan JH (2002) Biochemistry of Na, K-ATPase. Annu Rev Biochem 71:511–535. doi:10.1146/annurev.biochem.71.102201.141218
Apell H-J (2004) How do P-type ATPases transport ions? Bioelectrochemistry 63:149–156. doi:10.1016/j.bioelechem.2003.09.021
Herrera VL, Emanuel JR, Ruiz-Opazo N et al (1987) Three differentially expressed Na, K-ATPase alpha subunit isoforms: structural and functional implications. J Cell Biol 105:1855–1865
Woo AL, James PF, Lingrel JB (1999) Characterization of the Fourth alpha Isoform of the Na, K-ATPase. J Membr Biol 169:39–44
McDermott JP, Sánchez G, Chennathukuzhi V, Blanco G (2012) Green fluorescence protein driven by the Na, K-ATPase α4 isoform promoter is expressed only in male germ cells of mouse testis. J Assist Reprod Genet 29:1313–1325. doi:10.1007/s10815-012-9876-x
Jimenez T, Sánchez G, Wertheimer E, Blanco G (2010) Activity of the Na, K-ATPase alpha4 isoform is important for membrane potential, intracellular Ca2+, and pH to maintain motility in rat spermatozoa. Reproduction 139:835–845. doi:10.1530/REP-09-0495
Jimenez T, Mcdermott JP, Sánchez G, Blanco G (2010) Na, K-ATPase α4 isoform is essential for sperm fertility. Proc Natl Acad Sci U S A 108:644–649. doi:10.1073/pnas.1016902108/-/DCSupplemental.www.pnas.org/cgi/doi/10.1073/pnas.1016902108
Lingrel JB, Williams MT, Vorhees CV, Moseley AE (2007) Na, K-ATPase and the role of alpha isoforms in behavior. J Bioenerg Biomembr 39:385–389. doi:10.1007/s10863-007-9107-9
Kirshenbaum GS, Clapcote SJ, Duffy S et al (2011) Mania-like behavior induced by genetic dysfunction of the neuron-specific Na, K-ATPase α3 sodium pump. Proc Natl Acad Sci U S A 108:18144–188149. doi:10.1073/pnas.1108416108/-/DCSupplemental.www.pnas.org/cgi/doi/10.1073/pnas.1108416108
Edwards IJ, Bruce G, Lawrenson C et al (2013) Na+/K+ ATPase α1 and α3 isoforms are differentially expressed in α- and γ-motoneurons. J Neurosci 33:9913–9919. doi:10.1523/JNEUROSCI. 5584-12.2013
Despa S, Lingrel JB, Bers DM (2012) Na(+)/K)+)-ATPase α2-isoform preferentially modulates Ca2(+) transients and sarcoplasmic reticulum Ca2(+) release in cardiac myocytes. Cardiovasc Res 95:480–486. doi:10.1093/cvr/cvs213
Clifford RJ, Kaplan JH (2008) B-Subunit overexpression alters the stoicheometry of assembled Na-K-ATPase subunits in MDCK cells. Am J Physiol Ren Physiol 295:1314–1323. doi:10.1152/ajprenal.90406.2008
Chow DARC, Forte JG (1995) Functional significance of the beta subunit for heterodimeric P-type ATPases. J Exp Biol 198:1–17
McDonough AA, Geering K, Farley RA (1990) The sodium pump needs its beta subunit. FASEB J 4:1598–1605
Vagin O, Sachs G, Tokhtaeva E (2007) The roles of the Na, K-ATPase beta 1 subunit in pump sorting and epithelial integrity. J Bioenerg Biomembr 39:367–372. doi:10.1007/s10863-007-9103-0
Sun MZ, Kim JM, Oh MC et al (2013) Na+/K+-ATPase β2-subunit (AMOG) expression abrogates invasion of glioblastoma-derived brain tumor-initiating cells. Neuro Oncol 15:1518–1531. doi:10.1093/neuonc/not099
Peng L, Martin-Vasallo P, Sweadner KJ (1997) Isoforms of Na, K-ATPase alpha and beta subunits in the rat cerebellum and in granule cell cultures. J Neurosci 17:3488–3502
Azarias G, Kruusmägi M, Connor S et al (2013) A specific and essential role for Na, K-ATPase α3 in neurons co-expressing α1 and α3. J Biol Chem 288:2734–2743. doi:10.1074/jbc.M112.425785
Moseley AE, Williams MT, Schaefer TL et al (2007) Deficiency in Na, K-ATPase alpha isoform genes alters spatial learning, motor activity, and anxiety in mice. J Neurosci 27:616–626. doi:10.1523/JNEUROSCI. 4464-06.2007
Geering K (2005) Function of FXYD proteins, regulators of Na, K-ATPase. J Bioenerg Biomembr 37:387–392. doi:10.1007/s10863-005-9476-x
Mijatovic T, Ingrassia L, Facchini V, Kiss R (2008) Na+/K+−ATPase alpha subunits as new targets in anticancer therapy. Expert Opin Ther Targets 12:1403–1417. doi:10.1517/14728222.12.11.1403
Salhi A, Lamouroux C, Pestov NB et al (2013) A link between fertility and K+ homeostasis: role of the renal H, K-ATPase type 2. Pflugers Arch 465:1149–1158. doi:10.1007/s00424-013-1252-x
Streif D, Iglseder E, Hauser-kronberger C et al (2011) Expression of the non-gastric H+/K+−ATPase ATP12A in normal and pathological human prostate tissue. Cell Physiol Biochem 28:1287–1294
Cougnon M, Bouyer P, Planelles G, Jaisser F (1998) Does the colonic H, K-ATPase also act as an Na, K-ATPase? Proc Natl Acad Sci U S A 95:6516–6520
Codina J (1999) The colonic H+, K+-ATPase functions as a Na+-dependent K+(NH4+)-ATPase in apical membranes from rat distal colon. J Biol Chem 274:19693–19698. doi:10.1074/jbc.274.28.19693
Gumz ML, Lynch IJ, Greenlee MM et al (2010) The renal H+-K+-ATPases: physiology, regulation, and structure. Am J Physiol Ren Physiol 298:12–21. doi:10.1152/ajprenal.90723.2008
Zies DL, Gumz ML, Wingo CS, Cain BD (2007) The renal H+, K+-ATPases as therapeutic targets. Expert Opin Ther Targets 11:881–890. doi:10.1517/14728222.11.7.881
Fryklund J, Wallmark B, Larsson H, Helander HF (1984) Effect of omeprazole on gastric secretion in H+, K+-ATPase and in pepsinogen-rich cell fractions from rabbit gastric mucosa. Biochem Pharmacol 33:273–280
Belisario DC, Rocafull MA, del Castillo JR (2010) Purification and characterization of the ouabain-sensitive H+/K+-ATPase from guinea-pig distal colon. Arch Biochem Biophys 496:21–32. doi:10.1016/j.abb.2010.01.014
Codina J (1998) The alpha subunit of the colonic H+, K+-ATPase assembles with beta 1-Na+, K+-ATPase in kidney and distal colon. J Biol Chem 273:7894–7899. doi:10.1074/jbc.273.14.7894
Bamford M (2009) 3H+/K+ ATPase inhibitors in the treatment of acid-related disorders. Prog Med Chem 47:75–162. doi:10.1016/S0079-6468(08)00203-8
Yatime L, Buch-Pedersen MJ, Musgaard M et al (2009) P-type ATPases as drug targets: tools for medicine and science. Biochim Biophys Acta Bioenerg 1787:207–220. doi:10.1016/j.bbabio.2008.12.019
Rajendran VM (2000) Ouabain-sensitive H, K-ATPase functions as Na, K-ATPase in apical membranes of rat distal colon. J Biol Chem 275:13035–13040. doi:10.1074/jbc.275.17.13035
Sangan P, Thevananther S, Sangan S et al (2000) Colonic H-K-ATPase alpha- and beta-subunits express ouabain-insensitive H-K-ATPase. Am J Physiol Cell Physiol 278:C182–C189
Bhattacharyya D, Sen PC (1999) The effect of binding of chlorpromazine and chloroquine to ion transporting ATPases. Mol Cell Biochem 198:179–185
Gupta SP (2012) Quantitative structure-activity relationship studies on Na+, K(+)-ATPase inhibitors. Chem Rev 112:3171–3192. doi:10.1021/cr200097p
Sandtner W, Egwolf B, Khalili-Araghi F et al (2011) Ouabain binding site in a functioning Na+/K+ ATPase. J Biol Chem 286:38177–38183. doi:10.1074/jbc.M111.267682
Yu SP (2003) Na+, K+-ATPase: the new face of an old player in pathogenesis and apoptotic/hybrid cell death. Biochem Pharmacol 66:1601–1609. doi:10.1016/S0006-2952(03)00531-8
Goldstein I, Levy T, Galili D et al (2006) Involvement of Na(+), K(+)-ATPase and endogenous digitalis-like compounds in depressive disorders. Biol Psychiatr 60:491–499. doi:10.1016/j.biopsych.2005.12.021
Herman L, Hougland T, El-Mallakh RS (2007) Mimicking human bipolar ion dysregulation models mania in rats. Neurosci Biobehav Rev 31:874–881. doi:10.1016/j.neubiorev.2007.04.001
Rosta K, Tulassay E, Enzsoly A et al (2009) Insulin induced translocation of Na+/K+-ATPase is decreased in the heart of streptozotocin diabetic rats. Acta Pharmacol Sin 30:1616–1624. doi:10.1038/aps.2009.162
Oubaassine R, Weckering M, Kessler L et al (2012) Insulin interacts directly with Na+/K+ATPase and protects from digoxin toxicity. Toxicology 299:1–9. doi:10.1016/j.tox.2012.04.013
Haritha C, Reddy AG, Reddy YR et al (2013) Evaluation of protective action of fenugreek, insulin and glimepiride and their combination in diabetic Sprague Dawley rats. J Nat Sci Biol Med 4:207–212. doi:10.4103/0976-9668.107292
Liu J, Feng L, Zhang M et al (2013) Neuroprotective effect of Liuwei Dihuang decoction on cognition deficits of diabetic encephalopathy in streptozotocin-induced diabetic rat. J Ethnopharmacol 150:371–381. doi:10.1016/j.jep.2013.09.003
Hennion JP, el-Masri MA, Huff MO, el-Mailakh RS (2002) Evaluation of neuroprotection by lithium and valproic acid against ouabain-induced cell damage. Bipolar Disord 4:201–206
El-Mallakh RS, Schurr A, Payne RS, Li R (2000) Ouabain induction of cycling of multiple spike responses in hippocampal slices is delayed by lithium. J Psychiatr Res 34:115–120
Banerjee U, Dasgupta A, Rout JK, Singh OP (2012) Effects of lithium therapy on Na+−K+-ATPase activity and lipid peroxidation in bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatr 37:56–61. doi:10.1016/j.pnpbp.2011.12.006
De Vasconcellos APS, Zugno AI, Dos Santos AHDP et al (2005) Na+, K(+)-ATPase activity is reduced in hippocampus of rats submitted to an experimental model of depression: effect of chronic lithium treatment and possible involvement in learning deficits. Neurobiol Learn Mem 84:102–110. doi:10.1016/j.nlm.2005.05.002
McKnight RF, Adida M, Budge K et al (2012) Lithium toxicity profile: a systematic review and meta-analysis. Lancet 379:721–728. doi:10.1016/S0140-6736(11)61516-X
Wu JY, Wadhwa N (2013) Case 192: lithium-induced nephropathy. Radiology 267:308–312. doi:10.1148/radiol.13111801
Rej S, Looper K, Segal M (2013) The effect of serum lithium levels on renal function in geriatric outpatients: a retrospective longitudinal study. Drugs Aging 30:409–415. doi:10.1007/s40266-013-0068-x
Andrade Nunes M, Araujo Viel T, Sousa Buck H (2013) Microdose lithium treatment stabilized cognitive impairment in patients with Alzheimer’s disease. Curr Alzheimer Res 10:104–107. doi:10.2174/156720513804871354
Nunes PV, Forlenza OV, Gattaz WF (2007) Lithium and risk for Alzheimer’s disease in elderly patients with bipolar disorder. Br J Psychiatry 190:359–360. doi:10.1192/bjp.bp.106.029868
Goldstein I, Lerer E, Laiba E et al (2009) Association between sodium- and potassium-activated adenosine triphosphatase alpha isoforms and bipolar disorders. Biol Psychiatry 65:985–991. doi:10.1016/j.biopsych.2008.10.033
Bøttger P, Doğanlı C, Lykke-Hartmann K (2012) Migraine- and dystonia-related disease-mutations of Na+/K+-ATPases: relevance of behavioral studies in mice to disease symptoms and neurological manifestations in humans. Neurosci Biobehav Rev 36:855–871. doi:10.1016/j.neubiorev.2011.10.005
Gritz SM, Radcliffe RA (2013) Genetic effects of ATP1A2 in familial hemiplegic migraine type II and animal models. Hum Genom 7:8. doi:10.1186/1479-7364-7-8
Heinzen EL, Arzimanoglou A, Brashear A et al (2014) Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol 13:503–514. doi:10.1016/S1474-4422(14)70011-0
De Carvalho AP, Sweadner KJ, Penniston JT et al (2004) Mutations in the Na+/K+-ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron 43:169–175. doi:10.1016/j.neuron.2004.06.028
Brashear A, Mink JW, Hill DF et al (2012) ATP1A3 mutations in infants: a new rapid-onset dystonia-Parkinsonism phenotype characterized by motor delay and ataxia. Dev Med Child Neurol 54:1065–1067. doi:10.1111/j.1469-8749.2012.04421.x
Einholm AP, Toustrup-Jensen MS, Holm R et al (2010) The rapid-onset dystonia parkinsonism mutation D923N of the Na+, K+-ATPase alpha3 isoform disrupts Na+ interaction at the third Na+ site. J Biol Chem 285:26245–26254. doi:10.1074/jbc.M110.123976
Heinzen EL, Swoboda KJ, Hitomi Y et al (2012) De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat Genet 44:1030–1034. doi:10.1038/ng.2358
Hattori N, Kitagawa K, Higashida T et al (1998) Cl-ATPase and Na, K-ATPase activities in Alzheimer’s disease brains. Neurosci Lett 254:141–144
Toustrup-Jensen MS, Einholm AP, Schack VR et al (2013) Relationship between intracellular Na+ concentration and reduced Na+ affinity in Na+, K+-ATPase mutants causing neurological disease. J Biol Chem. doi:10.1074/jbc.M113.543272
Silveira PP, Portella AK, Benetti CDS et al (2011) Association between Na+, K+-ATPase activity and the vulnerability/resilience to mood disorders induced by early life experience. Neurochem Res 36:2075–2082. doi:10.1007/s11064-011-0531-1
Kirshenbaum GS, Clapcote SJ, Petersen J et al (2012) Genetic suppression of agrin reduces mania-like behavior in Na+, K+−ATPase α3 mutant mice. Genes Brain Behav 11:436–443. doi:10.1111/j.1601-183X.2012.00800.x
Harik SI, Mitchell MJ, Kalaria RN (1989) Ouabain binding in the human brain. Effects of Alzheimer’s disease and aging. Arch Neurol 46:951–954
Mark J, Allan D, Mattson P (1995) Amyloid P-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci 15:6239–6249
Kairane C, Mahlapuu R, Ehrlich K et al (2014) The effects of different antioxidants on the activity of cerebrocortical MnSOD and Na, K-ATPase from post mortem Alzheimer’s disease and age-matched normal brains. Curr Alzheimer Res 11:79–85. doi:10.2174/15672050113106660179
Wilson M-MG, Morley JE (2003) Invited review: aging and energy balance. J Appl Physiol 95:1728–1736. doi:10.1152/japplphysiol.00313.2003
Gu QB, Zhao JX, Fei J, Schwarz W (2004) Modulation of Na(+), K(+) pumping and neurotransmitter uptake by beta-amyloid. Neuroscience 126:61–67. doi:10.1016/j.neuroscience.2004.03.022
Dickey CA, Gordon MN, Wilcock DM et al (2005) Dysregulation of Na+/K+ ATPase by amyloid in APP+PS1 transgenic mice. BMC Neurosci 6:7. doi:10.1186/1471-2202-6-7
Zhang L-N, Sun Y-J, Pan S et al (2013) Na+-K+-ATPase, a potent neuroprotective modulator against Alzheimer disease. Fundam Clin Pharmacol 27:96–103. doi:10.1111/fcp.12000
Kreutz F, Scherer EB, Ferreira AGK et al (2013) Alterations on Na+, K+-ATPase and acetylcholinesterase activities induced by amyloid-β peptide in rat brain and GM1 ganglioside neuroprotective action. Neurochem Res 38:2342–2350. doi:10.1007/s11064-013-1145-6
Eiam-ong S, Jerawatana R, Kittikowit W, Mannontarat R (2009) Effects of vanadate and potassium depletion on renal H. K-ATPase Protein Expr 3:517–523
Codina J, Delmas-Mata JT, DuBose TD (1998) Expression of HKalpha2 protein is increased selectively in renal medulla by chronic hypokalemia. Am J Physiol 275:F433–F440
Izumi Y, Hori K, Nakayama Y et al (2011) Aldosterone requires vasopressin V1a receptors on intercalated cells to mediate acid–base homeostasis. J Am Soc Nephrol 22:673–680. doi:10.1681/ASN.2010050468
Salhi A, Centeno G, Firsov D, Crambert G (2012) Circadian expression of H, K-ATPase type 2 contributes to the stability of plasma K+ levels. FASEB J 26:2859–2867. doi:10.1096/fj.11-199711
Maeda Y, Kojima N, Araki Y et al (2011) Does a proton pump inhibitor cause hypokalemia? Intern Med 50:1045–1050. doi:10.2169/internalmedicine.50.4877
Vío CP, Figueroa CD (1987) Evidence for a stimulatory effect of high potassium diet on renal kallikrein. Kidney Int 31:1327–1334
Vío CP, Figueroa CD (1985) Subcellular localization of renal kallikrein by ultrastructural immunocytochemistry. Kidney Int 28:36–42
Valdés G, Vio CP, Montero J, Avendaño R (1991) Potassium supplementation lowers blood pressure and increases urinary kallikrein in essential hypertensives. J Hum Hypertens 5:91–96
Suga SI, Phillips MI, Ray PE et al (2001) Hypokalemia induces renal injury and alterations in vasoactive mediators that favor salt sensitivity. Am J Physiol Renal Physiol 281:F620–F629
He FJ, MacGregor GA (2001) Fortnightly review: beneficial effects of potassium. BMJ 323:497–501
Aburto NJ, Hanson S, Gutierrez H et al (2013) Effect of increased potassium intake on cardiovascular risk factors and disease: systematic review and meta-analyses. BMJ 346:f1378. doi:10.1136/bmj.f1378
McDonough AA, Nguyen MTX (2012) How does potassium supplementation lower blood pressure? Am J Physiol Renal Physiol 302:F1224–F1225. doi:10.1152/ajprenal.00429.2011
Ogiyama Y, Miura T, Watanabe S, et al (2013) Circadian rhythm of urinary potassium excretion during treatment with an angiotensin receptor blocker. J Renin Angiotensin Aldosterone Syst:1–6. doi: 10.1177/1470320313475909
Acknowledgments
This work was supported by grants from the Basal Center of Excellence in Aging and Regeneration (CONICYT-PFB 12/2007) to NCI and CV, FONDECYT No. 1120156 to NCI and FONDECYT No. 1130741. Rocío Retamales-Ortega was a SQM associate researcher.
We thank the Sociedad Química y Minera de Chile (SQM) for grants on the “Role of Potassium in Hypertension and Cognition” and on “The Effect of Lithium on Human Health” to the CARE Biomedical Center.
Graphic work was carried out using Graphique-Science (http://graphiquescience.blogspot.com). We thank Felipe Serrano for his help with the drawing of our figures.
Conflict of interest
The authors report no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Retamales-Ortega, R., Vio, C.P. & Inestrosa, N.C. P2C-Type ATPases and Their Regulation. Mol Neurobiol 53, 1343–1354 (2016). https://doi.org/10.1007/s12035-014-9076-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-9076-z