
Abstract
Approximately 30 % of epilepsy cases are refractory to current pharmacological treatments. Thus, novel therapeutic approaches that prevent or reverse the molecular and cellular mechanisms of epilepsy are required. 5-HT6 receptor (HTR6) blockade can modulate multiple neurotransmitter systems, and HTR6 may be a potential therapeutic treatment for neurological diseases, including epilepsy. Here, we investigated the role of HTR6 in epilepsy. We detected HTR6 expression both in human epileptic tissues and the pilocarpine rat model by western blotting. We observed behavioral changes after administration of pilocarpine in rats pretreated with a selective HTR6 antagonist, SB-399885, and recorded the electrophysiological index in the pilocarpine rat model pre- or posttreated with SB-399885 by electroencephalogram (EEG) and whole-cell clamp. We measured the activity of mammalian target of rapamycin (mTOR) in the pilocarpine rat model pretreated with the mTOR-specific inhibitor, rapamycin, and SB-399885 using western blotting. We found that HTR6 expression was upregulated in both human tissues and the pilocarpine rat model, and that SB-399885 could suppress epileptic seizures and mTOR activity in epileptic seizures. These results suggest that HTR6 plays an important role in modulating seizure activity and that the blockade of the HTR6/mTOR pathway could be a potential therapeutic target for epilepsy treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epilepsy is a disabling neurological disorder characterized by recurring, unprovoked seizures, which affects approximately 0.5–1 % of the world population. It often requires lifelong medication and places an enormous burden on the individual and society [1]. Approximately 30 % of affected individuals are refractory to current pharmacological treatments [2]. In responsive cases, medication can suppress seizures as symptomatic therapies, but there is minimal evidence that the current anti-epileptic drugs correct the brain abnormalities causing epilepsy or have disease-modifying effects. Therefore, it is now recognized that novel therapeutic approaches that prevent or reverse the molecular and cellular mechanisms of epilepsy are required [3].
Serotonin (5-HT) exerts diverse physiological and pharmacological effects by acting on multiple receptor subtypes. The 5-HT6 receptor (HTR6) is one of the most recent additions to the serotonin receptor family. HTR6 was discovered by using molecular cloning approaches in 1993 [4]. It shares <40 % protein sequence homology with all other known G-protein coupled 5-HT receptors, has no known functional splice variants, and appears to be almost exclusively expressed within the central nervous system (CNS), particularly in the hippocampus, olfactory tubercle, striatum, nucleus accumbens, and cerebral cortex and presents a heterogeneous distribution in different brain areas [5, 6]. HTR6 activation leads to a prolongation of neuronal excitability, an increase in neurotransmitters release to regulate glutamatergic and cholinergic neuronal activity and modulation of synaptic plasticity [7, 8]. Increasing evidence suggests that it may be involved in the regulation of cognition, feeding, and, possibly, affective state and seizures [7]. HTR6 blockade can modulate multiple neurotransmitter systems and therefore enhance cognition in preclinical studies. Thus, HTR6 antagonists may be considered as potential therapeutics for cognitive disorders related to aging and neurodegenerative diseases [8]. In term of seizures, Routledge et al. [9] reported that treatment with the HTR6 antagonists SB-271046, Ro 04–6790, and SB-258510 significantly increased the seizure threshold in the maximal electroconvulsive shock test (MEST) rat model used to study seizure regulation. Subsequently, this finding was also confirmed using the HTR6 antagonist SB-357134, suggesting that HTR6 may regulate seizure threshold [10]. The specific localization of HTR6 in the corticolimbic areas and the anti-seizure effects of HTR6 antagonism suggest that this receptor may be involved in epilepsy.
Meffre et al. [11] reported that HTR6 physically interact with several proteins of the mammalian target of rapamycin (mTOR) pathway, including mTOR, using a proteomic strategy and HTR6 activation increased mTOR signalling. Interestingly, the mTOR pathway represents a potentially novel therapeutic target for epilepsy. In poststatus epilepticus models of temporal lobe epilepsy(TLE), rapamycin, an mTOR inhibitor, may ameliorate the development of epilepsy-related pathology, suppress seizures, and prevent epilepsy in animal models of certain genetic epilepsies, such as tuberous sclerosis complex [12, 13].
However, the relationship between HTR6 and mTOR in epilepsy and whether HTR6 regulates mTOR to modulate seizure activity remain unknown. In the present study, we investigated the specific relationship between HTR6 and mTOR and the potential role of these proteins in epilepsy using an HTR6 antagonist to identify whether the HTR6/mTOR pathway is a potential treatment target for epilepsy.
Methods
Human Material
Twenty-five surgical removal temporal neocortex samples obtained from patients with intractable TLE and ten histologically normal anterior temporal neocortex samples obtained from patients who were treated for posttrauma intracranial hypertension were randomly selected from our established brain tissue bank [14]. The criteria, informed consent, and brain tissue processing were referenced in our previous publications [14, 15]. Individual patient details are listed in Supplemental Tables 1 and 2.
Pilocarpine Rat Model and Drug Treatments
Adult male Sprague–Dawley rats were selected and the modeling process was based on our previous experiment [15]. Briefly, 127 mg/kg lithium chloride (Sigma) was intraperitoneally administered 20 h before pilocarpine treatment. pilocarpine (50 mg/kg, i.p.; Sigma) was administered to induce seizures. The behavioral seizures were observed continuously and graded according to the Racine scale [16]. Rats experiencing stage 4 or 5 seizures for 1 h were injected with diazepam (10 mg/kg, i.p.) to terminate status epilepticus and were used for subsequent experiments. To detect the effect between HTR6 and mammalian target of rapamycin (mTOR), the rats were pretreated with HTR6 antagonist SB-399885 (10 mg.kg-1.d-1, i.p.) or mTOR-specific inhibitor rapamycin (6 mg.kg-1.d-1, i.p. LC Labs) for three consecutive days before the induction of seizures through the administration of pilocarpine. The rats were sacrificed at various time points (ranging from 1 to 30 days) after the onset of status epilepticus (each group n = 6). Some of the rats were sacrificed by decapitation after an i.p. administration of chloral hydrate. The hippocampus and neocortex were quickly dissected out on ice, frozen in liquid nitrogen, and stored at −80 °C for western blotting. Some of the rats were anesthetized with chloral hydrate and transcardially perfused with 0.9 % saline followed by 4 % paraformaldehyde (4 g/100 ml) in PBS (pH 7.4). The brains were immediately removed, postfixed in 4 % paraformaldehyde, processed, and embedded in paraffin. Coronal brain sections were then prepared and mounted on polylysine-coated slides for immunohistochemistry and immunofluorescence.
Immunohistochemistry and Double-labeling Immunofluorescence
The processes were based on our previous experiment [15, 14]. For immunohistochemistry, tissue sections were sequentially incubated with the primary anti-HTR6 antibody (1:50; Abcam), a biotinylated secondary antibody, and avidin-biotin complexes. Diaminobenzidine was used for color development, and hematoxylin was used for counterstaining. For immunofluorescence, tissue sections were incubated with an anti-HTR6 antibody (1:400) and anti-MAP2 antibody mixture or an anti-HTR6 antibody and anti-GFAP antibody, and then, incubated with FITC-conjugated antibody (1:100) and TRITC-conjugated antibody (1:100). The sections were observed using laser scanning confocal microscopy (Leica).
Western Blot Analysis
Protein extracts from brain tissues were separated by SDS-PAGE and transferred to nitrocellulose membranes. After blocking, the membranes were incubated at 4 °C overnight with primary antibodies against the following target proteins: HTR6 (1:1,000), phosphorylated S6 (P-S6) (1:1,000; Cell Signaling), S6 (1:1,000; Cell Signaling), and β-actin (1:600; Santa Cruz Biotechnology). The membranes were then washed and incubated with species-specific peroxidase-conjugated secondary antibodies for 2 h at room temperature. Specific bands were detected using the ECL system (Amersham) and a Bio-Rad electrophoresis image analyzer.
Electrophysiology
In chronic period (30 days), after the rats were deeply anaesthetized by intraperitoneal injection of 3.5 % chloral hydrate (1 ml/100 g), one intracranial recording electrode was placed in the CA3 area of the hippocampus. The behavioral changes included seizure activities, incidence and latency of convulsions, and ictal activity over a total period of 2 h. Electroencephalogram (EEG) ictal discharges were defined as respective spiking (amplitude ≥3 times baseline) lasting longer than 15 s.
Coronal brain slices (350 μm) were obtained in ice-cold sterile slice solution (containing in mM: KCl, 2.5; NaH2PO4.2H2O, 1.25; MgCl2.H2O, 6; CaCl2, 1; NaHCO3, 26; sucrose, 220; glucose, 10). Slices was perfused with artificial cerebral spinal fluid (ACSF) at 35 °C for 1 h and then at room temperature. The whole-cell current-clamp technique [17, 18] was used to record action potentials. The internal solution contained (in mM): 60 K2SO4, 60 NMG, 40 HEPES, 4 MgCl2, 0.5 BAPTA, 12 phosphocreatine, 2 Na2ATP and 0.2 Na3GTP, pH 7.2-7.3, and 265–270 mOsm. Neurons in CA1 were selected and perfused with ACSF to record AP, and then perfused with ACSF + SB-399885, the data were then recorded. Once the recording data were stable, the slice was bathed with ACSF for the remainder of the recording. Multiclamp 700B amplifier (Axon, USA) and Digidata 1322A were used to collect and analyze data, which were filtered at 10 KHz and low-pass filtered at 2 KHz, then recorded with pClamp 9.2 software (Molecular Devices, Sunnyvale, CA, USA).
Statistical Analysis
The data are reported as the means ± SD. We analyzed the data using one-way ANOVA and Student’s t-test using SPSS 13.0 software. We defined statistical significance as P < 0.05.
Results
HTR6 Expression is Upregulated in Human and Pilocarpine Rat Model of TLE
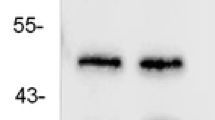
To demonstrate the relationship between HTR6 and epilepsy, we first assessed HTR6 expression in the temporal neocortices from TLE subjects and control individuals by western blot analysis. Notably, HTR6 protein level in the TLE samples was significantly higher than HTR6 protein level in the control group (Fig. 1a). To clearly determine the HTR6 alterations that occur during epilepsy, we turned to the pilocarpine epilepsy model. HTR6 expression was assayed by western blotting at different time points (1, 3, 7, 14, and 30 days) after the onset of status epilepticus. Notably, 3 d after pilocarpine-induced status epilepticus, HTR6 expression increased and remained elevated until day 30 (Fig. 1b). This increase was observed both in the hippocampus and neocortex. The HTR6 immunoreactivity was faint in the hippocampus and neocortex neurons of the control group (Fig. 2a and c) but relatively strong in the hippocampus and neocortex neurons of the model group (Fig. 2b and d). The localization of HTR6 in the rat brain was estimated by double-labeling the sections with the neuronal marker microtubule-associated protein 2 (MAP2) and the astrocyte marker glial fibrillary acidic protein (GFAP) antibodies. HTR6 (green) was co-expressed with MAP2 (red) in neurons but not with GFAP (red) in astrocytes both in the hippocampus and neocortex (Fig. 3).

HTR6 expression is upregulated in human and pilocarpine rat model of TLE. a The representative western blot shows HTR6 expression in the temporal neocortex of control individuals (lanes 1, 3, 5, and 7) or individuals with TLE (lanes 2, 4, 6, and 8). *P < 0.05, compared to the control group. b Three days after status epilepticus, HTR6 expression increased and remained elevated until 30 days both in the hippocampus and the neocortex of rats. *P < 0.05, compared to nontreated rats (0 day/control)

HTR6 immunohistochemical staining in rat brain sections. a, c HTR6 immunoreactivity was faintly visible in the hippocampus (a) and neocortex (c) of the control group. b, d HTR6 immunoreactivity was relatively strong in the hippocampus (b) and neocortex (d) of the pilocarpine group. As indicated by the arrows, HTR6 was expressed in the cytoplasm and cell membrane of neurons in hippocampus and neocortex. Scale bar is 40 μm

HTR6 double-labeling immunofluorescence in the brain sections of pilocarpine-treated rats. a, ) In the hippocampus, HTR6 (green) was co-expressed with MAP2 (red) in neurons (a) but not with GFAP (red) in astrocytes (b). c, d In the neocortex, HTR6 (green) was co-expressed with MAP2 (red) in neurons (c) but not with GFAP (red) in astrocytes (d). Scale bar is 75 μm
Effect of HTR6 Antagonist on Epileptic Seizure Activity
To test whether HTR6 interferes with the seizure phenotype, we measured the effect of the HTR6 antagonist, SB-399885, on seizure activities. SB-399885 (10 mg.kg-1.d-1, i.p.) was intraperitoneally administered for three consecutive days before the induction of seizures through the administration of pilocarpine. SB-399885 significantly increased seizure latency in pilocarpine-treated rats. The differences in latency between the SB-399885 group and control group were statistically significant (39.8 ± 7.5 min in SB-399885, n = 18; 26.4 ± 6.5 min in control, n = 18.) (Fig. 4a). In terms of seizure severity, the Racine score was estimated once, 10 min from after the rats were treated with pilocarpine only or pilocarpine and SB-399885 for 10 to 60 min. SB-399885 reduced the Racine score (seizure severity) in pilocarpine-treated rats, compared to the control group, *P < 0.05 (Fig. 4b). To test whether the effect of SB-399885 on behavioral activities was due to the inhibition of hyperexcitability, we measured spontaneous action potentials in the hippocampal CA1 neurons. As shown in Fig. 4d, slices from the pilocarpine-treated rats presented outbursts of action potentials, and slices from treated with SB-399885 showed significantly reduced spontaneous action potentials frequencies. Consistently, EEG results showed similar changes in the SB-399885 pretreated group (Fig. 4c).

The effect of HTR6 antagonist SB-399885 on seizure activities. a After pretreatment with SB-399885, seizure latency was significantly increased in pilocarpine-treated rats. *P < 0.05, compared to the control group. (each group n = 12); b After pretreatment with SB-399885, the Racine score (seizure severity) was reduced in pilocarpine-treated rats. *P < 0.05, compared to the control group. (each group n = 12). c In chronic period (30 days), intensive spontaneous action potentials in CA1 neurons on coronal brain slices (350 μm) of pilocarpine-treated rats were detected by whole-cell patch clamp. After posttreatment with SB-399885, spontaneous action potentials in CA1 neurons were significantly decreased. After being washed with ACSF, spontaneous action potentials were increased to some extent. (n = 6). d In chronic period (30 days), EEG ictal discharges were reduced in rats pretreated with SB-399885 when compared with pilocarpine-treated rats only (each group n = 6)
mTOR is Activated and HTR6 Mediates the Activation of mTOR in Pilocarpine-treated Rats
To determine whether mTOR activity was changed in the pilocarpine-treated rats, we used western blot analysis to assay the ratio of P-S6 protein to total S6 protein at different time intervals (1, 3, 7 days) after status epilepticus. This ratio is a marker of mTOR activity [19]. Notably, by 3 days after status epilepticus, the mTOR activity in the hippocampus increased significantly and remained elevated 7 days after status epilepticus. This finding was reflected by an increase in the P-S6/total S6 ratio (Fig. 5a). This increase indicates mTOR activation in the pilocarpine epilepsy model. When the rats were pretreated with the mTOR specific inhibitor rapamycin, the activation of mTOR was inhibited (Fig. 5b). The HTR6 antagonist SB-399885 was administered intraperitoneally for three consecutive days prior to pilocarpine injection. We found that pretreatment with SB-399885 effectively prevented the increase of HTR6 expression that occurred in the hippocampus after pilocarpine-induced seizures (Fig. 6a).We also examined the ability of SB-399885 pretreatment to block mTOR activation in the pilocarpine-treated rats by assaying the P-S6 to total S6 ratio. Interestingly, SB-399885 pretreatment significantly inhibited the increase of mTOR activity that occurred in the hippocampus after epileptic seizures (Fig. 6b).

The effect of rapamycin pretreatment on mTOR activation in the pilocarpine rat model. a Three days after status epilepticus, mTOR activity was significantly increased and remained elevated 7 days after status epilepticus. *P < 0.05, compared to nontreated rats (0 day). b Representative western blots show the hippocampus of nontreated rats (pilo−, rapamycin−), pilocarpine-treated rats (pilo+, rapamycin−), and rapamycin-pretreated rats prior to pilocarpine (pilo+, rapamycin+). Pretreatment with rapamycin inhibited mTOR activation, as reflected by the P-S6 to total S6 ratio that was assayed 3 and 7 days after pilocarpine-induced seizures, *P < 0.05 . (each group n = 6)

HTR6 mediates the activation of mTOR in the pilocarpine rat model. Representative western blots show the hippocampus of nontreated rats (pilo−, SB-399885−), pilocarpine-treated rats (pilo+, SB-399885−), and SB-399885-pretreated rats prior to pilocarpine (pilo+, SB-399885+). a Pretreatment with SB-399885 inhibited the increase in HTR6 expression, as shown 3 and 7 days after pilocarpine-induced seizures, *P < 0.05. b SB-399885 pretreatment also inhibited mTOR activation, as reflected by the P-S6 to total S6 ratios that were determined 3 and 7 days after pilocarpine-induced seizures. *P < 0.05. (each group n = 6)
Discussion
In the present study, we detected an increase in HTR6 expression in human brain tissues and in the TLE rat model and verified that the HTR6 antagonist SB-399885 could suppress epileptic seizures in pilocarpine rat model. We also demonstrated that HTR6 might effect on mTOR in epilepsy. To our knowledge, this is the first demonstration of a relationship between HTR6 and mTOR in epilepsy.
HTR6 is one of the most recently cloned receptors among the known serotonin receptors. It is almost exclusively localized in the CNS and is abundantly distributed in the limbic and cortical regions. Thus, new therapeutic agents targeting HTR6 may have relatively few peripheral side effects. Consequently, HTR6 has generated much interest for potential drug discovery and as a biological research target involved in neurological disorders. Using immunohistochemistry, Gerard et al. [20] found that HTR6 was highly expressed in the olfactory tubercles, nucleus accumbens, striatum and hippocampus, and moderately expressed in the cerebellum, cerebral cortex, and substantia nigra in the rat brain, while, Marazziti et al. [6] showed the presence of HTR6 in both pyramidal and glial cells in the prefrontal cortex, using immunohistochemistry and double-label immunofluorescence microscopy experiments, positive cells were mainly identified as pyramidal neurons in the hippocampus. In our study, we detected HTR6 in the cytoplasm and cell membrane of neurons in the hippocampus and neocortex of rats, but HTR6 was not expressed in astrocytes. Interestingly, epileptic seizures could induce HTR6 upregulation. In the TLE rat model, HTR6 expression was highest during spontaneous recurrent seizures period, which was consistent with that of human TLE. The temporal and spatial variation of HTR6 provides a preliminary explanation that it may play a role in epilepsy.
HTR6 appears to be a promising target for treating some neurological disorders, and HTR6 ligands, particularly selective antagonists, have shown beneficial effects in multiple studies, and some of them are currently used in clinical trials [21, 7]. They have a beneficial effect on neuronal plasticity and modulated the function of neurotransmitters, such as cholinergic and glutamatergic systems. For instance, HTR6 blockade can improve memory processes and reverse age-related spatial episodic like memory deficits [8]. Treatment with an HTR6 antagonist, such as SB-271046, Ro 04–6790, SB-258510, and SB 357134, increase seizure threshold in the MEST rat model [7, 9, 10]. Various selected HTR6 antagonists have different affinities for HTR6. SB-399885 is a potent, selective, brain penetrant HTR6 antagonist, which displays over 200-fold selectivity for HTR6 over all other receptors and has a high affinity for human recombinant and native HTR6 [22]. In this study, we selected SB-399885 to antagonize HTR6 activity. Pretreatment with SB-399885, significantly increased seizure latency and relieved seizure severity in rats after pilocarpine intraperitoneal injection. During chronic period, pretreatment of the pilocarpine rat models with SB-399885 alleviated spontaneous recurrent seizures as observed by EEG recording and posttreatment with SB-399885 could also mitigate hippocampal neurons discharge. These results suggest that HTR6 blockade might have anti-seizures effect and SB-399885 might be a good choice to achieve this effect.
Some recent experiments have indicated that HTR6 may participate in the modulation of mTOR, which is a potential downstream component of the HTR6 signaling pathway [11]. mTOR is an evolutionarily highly conserved serine/threonine kinase belonging to the phosphoinositide 3-kinase-related kinase family and represents one of the most recently studied pathways in relation to seizures and epileptogenesis and a potential novel therapeutic target for epilepsy, due to its suggested crucial role in many aspects of cellular proliferation and growth also including neurodegeneration, neurogenesis, and synaptic plasticity [23]. In the present study, we found that mTOR was activated in the pilocarpine epilepsy model and that this activation could be abrogated through HTR6 antagonism. As reflected by the P-S6 to total S6 ratio, mTOR was markedly activated after pilocarpine-induced status epilepticus. Pretreatment with rapamycin, an mTOR specific inhibitor, significantly inhibited the activation of mTOR after seizure activity. More importantly, mTOR activation was effectively blocked by pretreatment with a selective HTR6 antagonist SB-399885, indicating that HTR6 might be upstream of mTOR in pilocarpine-induced seizures.
In conclusion, we found that HTR6 expression was upregulated in both the epileptic foci of individuals with TLE and the pilocarpine rat model. HTR6 antagonist SB-399885 can suppress seizure activities, and HTR6 may mediate epileptic seizures by activating mTOR. The HTR6/mTOR signaling pathway could be a potential therapeutic target for epilepsy. Further studies are needed to thoroughly elucidate the role of the HTR6/mTOR signaling pathway in epilepsy.
References
Ngugi AK, Kariuki SM, Bottomley C, Kleinschmidt I, Sander JW, Newton CR (2011) Incidence of epilepsy: a systematic review and meta-analysis. Neurology 77(10):1005–1012. doi:10.1212/WNL.0b013e31822cfc90
Perucca E, French J, Bialer M (2007) Development of new antiepileptic drugs: challenges, incentives, and recent advances. Lancet Neurol 6(9):793–804. doi:10.1016/S1474-4422(07)70215-6
Stefan H, da Silva FH L, Loscher W, Schmidt D, Perucca E, Brodie MJ, Boon PA, Theodore WH, Moshe SL (2006) Epileptogenesis and rational therapeutic strategies. Acta Neurol Scand 113(3):139–155. doi:10.1111/j.1600-0404.2005.00561.x
Ruat M, Traiffort E, Arrang JM, Tardivel-Lacombe J, Diaz J, Leurs R, Schwartz JC (1993) A novel rat serotonin (5-HT6) receptor: molecular cloning, localization and stimulation of cAMP accumulation. Biochem Biophys Res Commun 193(1):268–276
Yoshioka M, Matsumoto M, Togashi H, Mori K, Saito H (1998) Central distribution and function of 5-HT6 receptor subtype in the rat brain. Life Sci 62(17–18):1473–1477
Marazziti D, Baroni S, Pirone A, Giannaccini G, Betti L, Testa G, Schmid L, Palego L, Borsini F, Bordi F, Piano I, Gargini C, Castagna M, Catena-Dell'osso M, Lucacchini A (2013) Serotonin receptor of type 6 (5-HT6) in human prefrontal cortex and hippocampus post-mortem: an immunohistochemical and immunofluorescence study. Neurochem Int 62(2):182–188. doi:10.1016/j.neuint.2012.11.013
Woolley ML, Marsden CA, Fone KC (2004) 5-ht6 receptors. Curr Drug Targets CNS Neurol Disord 3(1):59–79
Quiedeville A, Boulouard M, Da Silva Costa-Aze V, Dauphin F, Bouet V, Freret T (2014) 5-HT6 receptor antagonists as treatment for age-related cognitive decline. Rev Neurosci. doi:10.1515/revneuro-2014-0013
Routledge C, Bromidge SM, Moss SF, Price GW, Hirst W, Newman H, Riley G, Gager T, Stean T, Upton N, Clarke SE, Brown AM, Middlemiss DN (2000) Characterization of SB-271046: a potent, selective and orally active 5-HT(6) receptor antagonist. Br J Pharmacol 130(7):1606–1612. doi:10.1038/sj.bjp.0703457
Stean TO, Hirst WD, Thomas DR, Price GW, Rogers D, Riley G, Bromidge SM, Serafinowska HT, Smith DR, Bartlett S, Deeks N, Duxon M, Upton N (2002) Pharmacological profile of SB-357134: a potent, selective, brain penetrant, and orally active 5-HT(6) receptor antagonist. Pharmacol Biochem Behav 71(4):645–654
Meffre J, Chaumont-Dubel S, Mannoury La Cour C, Loiseau F, Watson DJ, Dekeyne A, Seveno M, Rivet JM, Gaven F, Deleris P, Herve D, Fone KC, Bockaert J, Millan MJ, Marin P (2012) 5-HT(6) receptor recruitment of mTOR as a mechanism for perturbed cognition in schizophrenia. EMBO Mol Med 4(10):1043–1056. doi:10.1002/emmm.201201410
Russo E, Citraro R, Donato G, Camastra C, Iuliano R, Cuzzocrea S, Constanti A, De Sarro G, Russo E, Citraro R, Donato G, Camastra C, Iuliano R, Cuzzocrea S, Constanti A, De Sarro G (2013) mTOR inhibition modulates epileptogenesis, seizures and depressive behavior in a genetic rat model of absence epilepsy. Neuropharmacology 69:25–36. doi:10.1016/j.neuropharm.2012.09.019
Wong M (2012) mTOR as a potential treatment target for epilepsy. Future Neurol 7(5):537–545. doi:10.2217/fnl.12.45
Yin H, Wang L, Xiao F, Huang Z, Huang Y, Zhou C, Han Y, Tao S, Yang H, Wang X (2011) Upregulation of liprin-alpha1 protein in the temporal neocortex of intractable epileptic patients and experimental rats. Synapse 65(8):742–750. doi:10.1002/syn.20894
Wang L, Zhou C, Zhu Q, Luo J, Xu Y, Huang Y, Zhang X, Wang X (2010) Up-regulation of serum- and glucocorticoid-induced protein kinase 1 in the brain tissue of human and experimental epilepsy. Neurochem Int 57(8):899–905. doi:10.1016/j.neuint.2010.09.009
Racine RJ (1972) Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32(3):281–294
Chen G, Kittler JT, Moss SJ, Yan Z (2006) Dopamine D3 receptors regulate GABAA receptor function through a phospho-dependent endocytosis mechanism in nucleus accumbens. J Neurosci 26(9):2513–2521
Zhong P, Gu Z, Wang X, Jiang H, Feng J, Yan Z (2003) Impaired modulation of GABAergic transmission by muscarinic receptors in a mouse transgenic model of Alzheimer's disease. J Biol Chem 278(29):26888–26896
Talos DM, Sun H, Zhou X, Fitzgerald EC, Jackson MC, Klein PM, Lan VJ, Joseph A, Jensen FE (2012) The interaction between early life epilepsy and autistic-like behavioral consequences: a role for the mammalian target of rapamycin (mTOR) pathway. PLoS One 7(5):e35885. doi:10.1371/journal.pone.0035885 PONE-D-12-02255
Gerard C, Martres MP, Lefevre K, Miquel MC, Verge D, Lanfumey L, Doucet E, Hamon M, el Mestikawy S (1997) Immuno-localization of serotonin 5-HT6 receptor-like material in the rat central nervous system. Brain Res 746(1–2):207–219
Riccioni T (2010) 5-HT6 receptor characterization. Int Rev Neurobiol 94:67–88. doi:10.1016/B978-0-12-384976-2.00003-4
Hirst WD, Stean TO, Rogers DC, Sunter D, Pugh P, Moss SF, Bromidge SM, Riley G, Smith DR, Bartlett S, Heidbreder CA, Atkins AR, Lacroix LP, Dawson LA, Foley AG, Regan CM, Upton N (2006) SB-399885 is a potent, selective 5-HT6 receptor antagonist with cognitive enhancing properties in aged rat water maze and novel object recognition models. Eur J Pharmacol 553(1–3):109–119. doi:10.1016/j.ejphar.2006.09.049
Russo E, Citraro R, Constanti A, De Sarro G (2012) The mTOR signaling pathway in the brain: focus on epilepsy and epileptogenesis. Mol Neurobiol 46(3):662–681. doi:10.1007/s12035-012-8314-5
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant numbers are 81201003, 81271445, and 81071040) and funding from the Chongqing Municipal Education Commission (No. KJ 080307). We thank the patients and their families for their participation in this study.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors Liang Wang and Yaodong Lv contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 16 kb)
Rights and permissions
About this article

Cite this article
Wang, L., Lv, Y., Deng, W. et al. 5-HT6 Receptor Recruitment of mTOR Modulates Seizure Activity in Epilepsy. Mol Neurobiol 51, 1292–1299 (2015). https://doi.org/10.1007/s12035-014-8806-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-8806-6