
Abstract
Mitogen-activated protein kinase phosphatase (MKP)-1 provides a negative feedback mechanism for regulating mitogen-activated protein kinase (MAPK) activity and thus a variety of cellular processes such as proliferation, differentiation, growth and apoptosis. MKP-1 is established as a central regulator of a variety of functions in the immune, metabolic and cardiovascular systems, and it is now increasingly acknowledged as having a role to play in the nervous system. It has been implicated in regulating processes of neuronal cell development and death as well as in glial cell function. Reduced MKP-1 levels have been observed in models of neurological conditions including Huntington’s disease, multiple sclerosis, ischemia and cerebral hypoxia. It has also been suggested to have a role to play in psychiatric disorders such as major depressive disorder. Here, we discuss the role of MKP-1 in nervous system development and disease and examine current evidence providing insight into MKP-1 as a potential therapeutic target for various diseases of the central nervous system.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
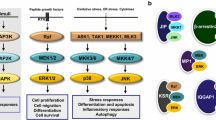
Mitogen-activated protein kinases (MAPKs) are a group of serine/threonine protein kinases that are instrumental in orchestrating a variety of cellular processes such as changes in gene expression, cell proliferation, differentiation and survival [1]. MAPK are present and active in most cell types including neurons and other brain cell types and respond to a range of stimuli including growth factors, cytokines and oxidative stress [2–4]. Four groups of MAPK have been identified to date, namely the extracellular signal-regulated kinases (ERK1 and ERK2), c-JUN N-terminal kinases (JNK1, JNK2 and JNK3), p38 kinases (p38 α, β, γ and δ) and ERK5/Big MAP kinase1 (BMK1) (for review see [5, 6]). The activity of MAPK is controlled by tyrosine-specific phosphatases, threonine-specific phosphatase or dual-specific (tyrosine- and threonine-specific) phosphatases (DUSPs), which are often referred to as MAPK phosphatases (MKPs) [7]. In mammalian cells, MKPs, of which there are 11 members containing a MAPK-binding domain [8], are the principal phosphatases responsible for dephosphorylation and thus deactivation of MAPKs [9]. Indeed, there are many more phosphatases than there are MAPKs, and as the kinetics of MAPK activation are crucial in controlling downstream responses in the cell, phosphatases could be considered as the ultimate controller in MAPK signalling. MKP-1 (also called DUSP1, ERP, CL100, HVH1, PTPN10 and 3CH134 [2, 10]) is the founding member of the MKP family and is a 39-kDa protein which is expressed at basal levels in unstimulated cells. It contains a dual-specificity phosphatase domain (DUSP) on the C-terminus along with a MAPK-binding domain at the N-terminus. MKP-1 was initially believed to specifically dephosphorylate ERK [8] but evidence now suggests that it preferentially inhibits p38 and JNK activity over ERK to provide a negative feedback loop on many cellular processes [11] (Fig. 1).

Schematic representation of MKP-1’s regulatory network. a MKP-1 attenuates the detrimental effects of p38 and JNK. Stress, inflammation or apoptosis leads to the activation of p38 and JNK MAP kinases which contribute to deficits in neural development and an increased stress response, inflammatory response and neuronal cell death in the nervous system (red box). Activated MAP kinases are bound by MKP-1’s MAPK binding domain with decreasing preference (p38 > JNK) and dephosphorylated by the dual specificity phosphatase (DuSP) domain. Inactivation of p38 and JNK through dephosphorylation by MKP-1 attenuates their effects (green box). b ERK MAP kinase contributes to increased MKP-1 response. Growth factor signalling leads to ERK activation which binds and phosphorylates the C-terminus of MKP-1, increasing its cellular half-life. ERK signalling has also been reported to stimulate MKP-1 gene transcription. Increased MKP-1 availability in response to ERK’s activation leads to a decrease in p38 and JNK activity, contributing to improvements in neural development, decreased neuroinflammation and reduced neuronal cell death
An array of recent studies has yielded important insights into the physiological functions of MKP-1, particularly its role in regulating responses in immunological and metabolic diseases, cancer and more recently in the nervous system (reviewed by [6, 12]). For example, it has been shown that MKP-1 is a negative regulator of MAPK-mediated inflammatory responses in macrophages and dendritic cells [13–15] and that it protects mice from endotoxic insult [16]. MKP-1 has a role to play in lipid metabolism in adipose tissue [17], while a pathophysiological involvement of MKP-1 has been suggested in obesity and metabolic syndrome [18, 19]. Indeed, MKP-1 knock-out mice display enhanced levels of inflammation, autoimmunity and metabolic defects [20, 21]. In various cancers such as epithelial carcinogenesis, breast cancer and non-small cell lung cancer, upregulation of MKP-1 has been reported [22–25], which suggests that excess MKP-1 in these cancer types may be responsible for inhibition of apoptotic pathways through its regulation of MAPK and can ultimately lead to excess cell proliferation (for review see [26]). Thus, while researchers are exploring ways of preventing upregulation of MKP-1 in cancer cells as it impedes pro-apoptotic drug therapies, the opposite is required to preserve the integrity of neuronal cells. Indeed, along with the multitude of reports implicating MAPKs in neuronal development, plasticity and survival, as well as learning and memory [27–30], emerging data shows that MKP-1 is involved in regulating many of these processes (Table 1). Thus, current research positions MKP-1 as a potential new drug target not only for the regulation of the immune response, metabolic function and cell proliferation in tumour cells but also for diseases of the nervous system. Accordingly, the evidence to date on the involvement of MKP-1 in preserving the developing and degenerating nervous system as well as its role in glial cell function and the pathophysiology of depression will form the basis of this review.
MKP-1 and CNS Development
Accumulating evidence now points to MKP-1 as an essential regulator of neural cell development. It is expressed in embryonic chicken dorsal root and sympathetic ganglia [31], in neuronal cells differentiated from the mouse embryonic carcinoma cell line P19 [32], in inhibitory and excitatory neurons during embryogenesis in the rat prefrontal cortex, hippocampus and striatum [33], and in dopaminergic neurons in the embryonic rat ventral mesencephalon (VM) [34]. A study carried out to show that MKP-1 mRNA was induced in embryonic chicken sympathetic neurons after nerve growth factor stimulation was the first indication of a capacity for MKP-1 to be modulated in the developing nervous system [31]. This was followed by a report demonstrating that P19 cells induced by retinoic acid to differentiate along a neuronal pathway expressed elevated levels of MKP-1, which was coincident with inactivation of its substrate ERK [32]. This demonstrated a regulatory role for MKP-1 on stem cells under neuronal differentiation conditions. More recently, MKP-1 has been specifically implicated in neuronal axonal development in the rat cortex [33]. The study reported by Jeanneteau et al. (2010) demonstrated that in utero electroporation to deliver MKP-1 overexpression or knockout constructs to cortical neurons at embryonic day (E)15 in rat resulted in increased morphological complexity of dendritic axonal arbors when examined at postnatal (P) day 7. Meanwhile, downregulation of MKP-1 resulted in less dendritic arborisation compared to GFP-transfected control cells. Axons from MKP-1 overexpressing cells entered the white matter with aberrant branching compared to the GFP- and small hairpin RNA (shRNA)-expressing cells, and the number of total axon branches within the contralateral layers I–V was increased or decreased, respectively, by upregulation or downregulation of MKP-1. This report also showed that an MKP-1 mutant specifically targeting the dephosphorylation of JNK attenuated stathmin-induced phosphorylation and restricted axon outgrowth, indicating that MKP-1 activity was sufficient to destabilise cortical neurons in culture. In addition, expression of a JNK mutant with decreased sensitivity to MKP-1 was sufficient to rescue the observed axonal defects. Furthermore, neurons from mkp1 −/− mice were unable to form axon branches in response to brain-derived neurotrophic factor (BDNF) [33]. As the morphological development of neural cells is critical for successful generation and delivery of cells aimed at cell replacement therapy for neurodegenerative diseases such as Huntington’s disease (HD) and Parkinson’s disease (PD), the results from this study suggest that MKP-1 may play a role in the generation of cells for replacement therapy. In relation to this, recent work from our group has shown that dopaminergic neurons (the loss of which is responsible for the pathological and motor symptoms of PD) cultured from embryonic rat VM and transfected to overexpress MKP-1 displayed a more complex morphology than their control counterparts [34]. Inhibition of p38 (but not JNK) prevented the MKP-1-induced increase in neurite branching and length suggesting that p38 signalling is required for MKP-1-mediated function in dopaminergic neurons. When dopaminergic neurite growth was inhibited in these cells by treatment with the dopaminergic neurotoxin 6-hydroxydopamine (6-OHDA), we observed a selective decrease in MKP-1 expression. Furthermore, overexpression of MKP-1 was neuroprotective against 6-OHDA-induced decrease in neurite length of dopaminergic neurons. While the studies to date identify MKP-1 as a promising growth-promoting mediator of neuronal processes and a potential neuroprotective protein, further work needs to be done to determine its potential beneficial effect in disease models in vivo and particularly in facilitating newly transplanted neurons to establish appropriate connections in the host brain.
MKP-1 and Neuronal Cell Survival and Death
While much evidence exists in support of a role for MAPKs in neuronal cell survival and/or death following injury or degenerative stimuli, investigation into the mechanism of involvement of their upstream phosphatase MKP-1 has not been as prolific. It has been shown however that knockdown of endogenous MKP-1 resulted in an enhancement of glutamate-induced oxidative death of HT22 hippocampal neuronal cells and primary mouse cortical neurons [35]. Upregulation of hippocampal MKP-1 along with transcription factors involved in cell survival was shown to be associated with ischemic tolerance in a rat global ischemia model [36], while a more recent study implicated increased MKP-1 preceding induction of apoptotic factors such as BCL2 and cleaved poly-ADP ribose polymerase (PARP) in response to hypoxic exposure of cortical neurons [37]. These findings suggest that MKP-1 promotes death in these cells as well as in the hippocampus of mice subjected to transient forebrain ischemia by priming apoptotic signalling cascades to become activated after prolonged ischemic stress. On the other hand, conditional overexpression of MKP-1 suppressed neuronal death along with phosphorylated JNK and the expression of pro-apoptotic genes including Bim, DP5 and FasL downstream of JNK activation in neuroblastoma N1E115 cells exposed to hypoxia-re-oxygenation in vitro [38]. This is also in agreement with a report which demonstrates that a nitric oxide-mediated decrease in MKP-1 activity contributes to neuronal cell death in newborn piglets exposed to cerebral hypoxia [39] and that MKP-1 overexpression suppresses apoptotic genes in the nerve growth factor-withdrawal-induced apoptosis of sympathetic neurons [40]. Agents showing neuroprotective properties, namely erythropoietin protection against spinal cord trauma injury in rats [41] and ginsenoside Rg1 protection against amyloid-β-induced apoptosis in rat cerebrocortical neurons [42], have also demonstrated that their beneficial effects are concomitant with an induction of MKP-1 protein and mRNA expression, respectively. Thus, for the most part, it appears that MKP-1 plays a protective role in neuronal viability, although variations in tissue type, the nature, timing and duration of insult and injury, as well as the transient nature of MAPK signalling may account for the discrepancies reported.
Recent evidence has demonstrated a downregulation of mkp1 mRNA in human post-mortem HD caudate samples, as well as in the striatum and cortex of R6/2 mice (a transgenic mouse model of HD) and in E16 rat primary cortical and striatal neurons expressing polyglutamine expanded proteolytic fragments of huntingtin (Htt 171-82Q), compared to neurons expressing wild-type Htt fragment (Htt 171-18Q) [43]. Co-expression of lentiviral Htt 171-82Q and MKP-1 resulted in the neuroprotection of primary striatal and cortical neurons as assessed by the preservation of neuronal nuclear antigen (NeuN: a marker of neuronal maturation)-positive cells compared to cultures expressing Htt 171-18Q. In addition, MKP-1 silencing with shRNAs resulted in the enhancement of Htt 171-82Q-induced neurodegeneration demonstrating that MKP-1 downregulation may contribute to HD pathogenesis. These findings were corroborated in an in vivo model of HD where it was observed that the polyglutamine-expanded lesion size was significantly reduced by co-expression of MKP-1. Thus, while a substantial number of reports point to an upregulation of MKP-1 as a potential therapeutic strategy to ameliorate neuronal loss as a result of injury, hypoxia, ischemia or degeneration, much work has yet to be done to establish distinct mechanisms of endogenous MKP-1 activity in the various cell death paradigms.
MKP-1 and Glial Cell Function
A substantial body of evidence indicates that MKP-1 is expressed in glial cells and that MKP-1 regulation in glia may impact neuroinflammatory processes. Indeed, Ndong et al. (2012) demonstrate that MKP-1 overexpression in BV-2 microglial cells prevented lipopolysaccharide (LPS)-induced p38 and JNK phosphorylation, in addition to release of pro-inflammatory cytokine and nitric oxide [44]. Furthermore, the synthetic glucocorticoid dexamethasone has been shown to blunt nitric oxide and reactive oxygen species (ROS) release from LPS-stimulated BV-2 cells, and this effect was mediated by MKP-1 upregulation in microglial cells [45]. Similarly, the plant-derived sesquiterpene lactone costunolide [46], and liquorice-derived dehydroglyasperin C [47], enhances MKP-1 expression in BV-2 microglia and prevents LPS-induced inflammatory signalling, thus supporting its role as an anti-inflammatory agent. Crittenden et al. (2011) also demonstrate that exposure of the N9 microglial cell line to the metal manganese potentiates LPS-induced MAPK activation and pro-inflammatory cytokine production and that this effect is mediated by manganese-induced downregulation of MKP-1 protein and mRNA [48].
Elsewhere, it has been reported that MKP-1 is expressed in microglia during CNS inflammation and that MKP-1, but not MKP-2, co-localises with microglia in inflammatory lesions in multiple sclerosis (MS) post-mortem tissue [49]. Furthermore, Eljaschewitsch et al. (2006) demonstrates that the endocannabinoid anandamide (AEA) can rapidly induce MKP-1 in microglia and that this represents a mechanism of neuro-immune communication to limit immune-mediated damage following CNS injury. Similarly, recent findings indicate that AEA targets MKP-1 in human retinal Muller glia, which limits inflammation induced by the HIV-1 Tat protein, suggesting a role for endocannabinoid-induced regulation of glial MKP-1 in inflammatory eye disease [50]. Exposure of primary microglia to the cannabinoid receptor type 2 (CB2) agonist, JWH015, inhibits LPS-induced microglial migration, by JWH015-mediated induction of both MKP-1 and MKP-3 [51]. In support of these findings, recent microarray analysis of genome-wide mRNA levels in LPS-treated BV-2 cells identified MKP-1 as a target of plant-derived cannabinoids, in addition to endogenous ligands [52], confirming that CB1/2 receptors couple to MKP-1 regulation in microglia, and thus broadening the therapeutic window for cannabinoid-based compounds. Some limited evidence directly links MKP-1 regulation in astrocytes with oxidative stress and ischemia. For example, MKP-1 associates with the gap junction protein connexin43 in astrocytes [53]. An increased association of connexin43 with MKP-1 occurs following chemical ischemia and may impact homeostasis and glucose metabolism in the CNS, in addition to influencing neuronal viability during ischemia [53]. Exposure of primary astrocytes to ROS has also been shown to induce the expression of MKP-1, with an associated modulation of JNK/ERK and oxidative stress responses [54], and hence further implicates the role of astrocytic MKP-1 in ischemic events in the CNS.
Transforming growth factor β1 (TGFβ1) plays a pivotal role in neuroinflammation, and evidence indicates that TGFβ1 can control the production of MKP-1 in glia. Indeed, Herrera Molina et al. (2012) have recently demonstrated a role for MKP-1 in glial activation in mixed glial cultures. Their findings indicate that TGFβ1 deactivates MAPKs by increasing the expression of MKP-1, with resulting modulation of interferon-γ (IFN-γ)-induced glial activation [55]. TGFβ1 has also been shown to inhibit inflammatory signalling in primary glial cells exposed to amyloid-β, via an MKP-1 mechanism [56]. Furthermore, IFN-γ-mediated induction of the inflammatory chemokine monocyte chemoattractant protein-1 (MCP-1) in primary microglia and astrocytes is prevented by the peroxisome proliferator-activated receptor (PPAR)-α agonist 5,8,11,14-eicosatetraynoic acid (ETYA) [57]. Interestingly, ETYA-induced MCP-1 suppression is reliant on the inhibition of JNK signalling via the promotion of glial MKP-1. Using a similar approach, Lee et al. (2008) demonstrate that the anti-inflammatory mechanisms of the PPAR ligand 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) in microglia and astrocytes are reliant on MKP-1. Specifically, the inhibitory action of 15d-PGJ2 on IFN-γ induction of MCP-1 is mediated by MKP-1 activity in both glial cell types [58, 59].
Several studies have identified a role for MKP-1 in disorders of the temporomandibular joint (TMJ), focusing on the role of trigeminal ganglia relaying nociceptive signals to the CNS. For example, investigation of NO-induced inflammatory signalling in rat trigeminal neurons identified MKP-1 expression in satellite glial cells from ganglia, which may be linked with pain and inflammation in the TMJ [59]. Recently, similar studies by Cady et al. (2010, 2013) investigated the role of dietary cocoa [60] and grape seed polyphenols [61] as therapeutic strategies for orofacial pain conditions using bilateral injection of complete Freund’s adjuvant in the TMJ. Interestingly, basal expression of MKP-1 was increased in glia in the spinal trigeminal nucleus of rats on cocoa and grape seed supplemented diets, which may be linked with anti-inflammatory properties in this model [61, 60]. MAPKs have a defined role in spinal cord plasticity, and existing evidence indicates that peripheral nerve injury downregulates MKP-1 expression in the L5–L6 spinal cord [44]. In addition, using an in vivo nanoparticle approach, Ndong et al. (2012) demonstrated that overexpression of MKP-1 in the spinal cord (in microglia, astrocytes and neurons) had anti-inflammatory propensity in a model of nerve-injury-induced hypersensitivity. Studies linking MKP-1 with oligodendrocytes and Schwann cell progenitors are lacking, even though MKP-1 is involved in activating the myelin basic protein promoter [62], indicating a potential role for this phosphatase in myelination in both the CNS and PNS.
MKP-1, Learning and Memory
Given the evidence of a role for MKP-1 in neuronal development, survival and prevention of neuronal cell death mechanisms, especially in hippocampal and cortical tissue, it is not surprising that there is also physiological and behavioural evidence for its involvement in learning and memory. MKP-1 has been shown to be upregulated at the same time as MAPK/ERK phosphorylation has returned to basal levels after induction of long-term potentiation in the dentate gyrus of the hippocampus in vivo [63]. It has thus been proposed that MKP-1 is involved in a negative feedback loop to regulate deactivation of MAPK/ERK, and thus, their results highlight the complexity of this signalling system as well as intimating a role for MKP-1 in hippocampal synaptic plasticity and long-term memory formation. In support of this finding, streptozotocin-induced diabetes in rats gave rise to deficits in the radial arm maze test indicative of impairment of hippocampal-dependent learning and memory [64], and the behavioural result was concomitant with decreased MKP-1 expression in the dentate gyrus of these rats. Recent evidence also points to a role for MKP-1 in hippocampal-dependent memory; a closed head injury (CHI) paradigm in mice resulted in a loss of object recognition memory, hippocampal neuronal loss and decreased expression of BDNF and MKP-1 in the hippocampus [65]. Interestingly, when mice were exposed to a post-injury exercise schedule, neurite regeneration, BDNF and MKP-1 levels were restored in the hippocampus. When the MKP-1 synthesis inhibitor triptolide was administered before each bout of exercise, the exercise-induced restoration of MKP-1 expression as well as the object recognition memory in CHI mice was blocked. This study further supports the evidence by Jeanneteau et al. (2010) showing that BDNF facilitates axonal branching and remodelling via upregulation of MKP-1 [33] and by Collins et al. (2013) who found that MKP-1 can promote the growth and elaboration of dopaminergic neuronal processes [34].
MKP-1 and the Pathophysiology of Stress-Related Depression
While deficits in MKP-1 in neurons and glia have for the most part been associated with developmental problems and neurodegenerative disorders, it has also been reported that there is an upregulation of MKP-1 in post-mortem hippocampal samples from patients with major depressive disorder (MDD) [66]. In this context, mkp1 mRNA has been shown to be elevated in the dentate gyrus and pyramidal CA3 cell layer of rats subjected to chronic unpredictable stress (CUS), whereby rats display depressive-like behaviours such as helplessness and anhedonia (inability to experience pleasure). Administration of the anti-depressant fluoxetine blocked the CUS-induced anhedonia and helpless behaviour and reversed mkp1 mRNA upregulation in the dentate gyrus and partially normalised this increase in the CA1 cell layer. It should be noted however that these effects were correlative rather than causative. In addition, enhanced MKP-1 expression was not a global effect and remained specific to the hippocampus as neither stress nor fluoxetine altered mkp1 mRNA expression in the cortex. Unstressed rats administered with a hippocampal-targeted adenovirus vector expressing MKP-1 displayed anhedonia (as assessed by decreased sucrose preference) and an increase in escape failures in active avoidance test (similar to that shown by CUS-exposed rats), increased latency to feed in the novelty suppressed feeding test and a significant increase in immobility in the forced swim test (FST), which is used to model depressive-like behaviours. Interestingly, CUS-exposed mkp1 −/−mice did not develop anxiety and depressive-like behaviour and maintained normal sucrose consumption, while CUS-exposed wild-type mice developed a progressive and significant reduction in sucrose consumption. MKP-1 has also been implicated in the depressive-like changes in rats subjected to a chronic social defeat stress as decreased levels of phosphorylated MEK1/2 and ERK1/2, and increased MKP-1 expression was evident in the hippocampus of these rats [67]. Changes in MAPK and MAPK phosphatase expression were concomitant with reduced body weight gain, enlarged adrenal glands and an increase in the immobility time in the FST. Interestingly, another study has shown that MKP-1 levels are unaltered in the hippocampus or the cortex of pre-natally stressed rats suggesting that the changes in MKP-1 expression in response to stress may be specific to the type and timing of the stress [68]. Collectively, these data indicate that induction of MKP-1 is not only a direct consequence of certain types of stressors but may contribute to the expression of depressive symptoms. A number of other studies have supported this notion and have thus endorsed the disruption of MKP-1 expression as a therapeutic strategy for mood disorders. Specifically, it has been reported that an increase in MKP-1 protein expression has been observed in the cortex of reserpine-induced MDD mice [69]. An intraventrolateral orbital cortex (VLO) infusion of sanguinarine (a selective MKP-1 inhibitor [70]) to rats significantly reduced immobility time in the FST in a dose-dependent manner compared to vehicle-treated controls [71]. The sanguinarine-induced decrease in MKP-1 expression was concomitant with an increase in ERK activation in the VLO. Moreover, the behavioural effect was similar to that observed after systemic administration of fluoxetine indicating that at least within the VLO, inhibition of MKP-1 may have anti-depressant effects. In a more recent study, hippocampal MKP-1 has been shown to be increased in morphine withdrawal-induced depressive-like behaviours in mice including reduced body weight gain, decreased social interaction time and increased immobility in the tail suspension test [72]. Intrahippocampal infusion of sanguinarine prevented these behavioural effects as well as attenuating the withdrawal-induced decrease in hippocampal levels of phospho-ERK and suggests that MKP-1 is a positive regulator of depressive behaviour through its regulation of ERK. Sasaki et al. (2013) have also demonstrated a positive correlation between a decrease in immobility time in the tail suspension test and a decrease in cerebral levels of MKP-1 in mice administered with the antidepressant-like polyphenol Rosmarinus officinalis [73], further implicating MKP-1 as having a role to play in the pathophysiology of depression.
Concluding Remarks
A substantial body of published evidence suggests that MKP-1 plays a diversity of roles in a number of CNS diseases. The complexity of the cellular mechanisms underlying its action in the brain is also starting to be realised, although much has yet to be done to understand the specific role that MKP-1 plays in disease states, especially in the clinical setting. One conundrum that has emerged is the apparent discrepancy in the contribution of MKP-1 to stress-induced depressive disorder compared to its role in neuronal development and survival. Reconciling these opposing roles will thus pose a challenge to the research community when targeting MKP-1 for therapeutic development. Nevertheless, it is clear that MKP-1 is a key player in the regulation of glial cell-mediated neuroinflammatory processes, and its manipulation represents a promising target to limit neuronal damage which occurs as a result of neuroinflammation. Considering the lack of current efficacious interventions for the neurological and psychiatric disorders, and based on the central regulatory role of MKP-1 in the pathophysiology of these disorders, development of therapeutic strategies targeting MKP-1 may prove to be worthy ventures.
References
Nishida E, Gotoh Y (1993) The MAP kinase cascade is essential for diverse signal transduction pathways. Trends Biochem Sci 18(4):128–131
Boutros T, Chevet E, Metrakos P (2008) Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev 60(3):261–310
Duan W, Wong WSF (2006) Targeting mitogen-activated protein kinases for asthma. Curr Drug Targets 7(6):691–698
Jeffrey KL, Camps M, Rommel C, Mackay CR (2007) Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat Rev Drug Discov 6(5):391–403
Theodosiou A, Ashworth A (2002) MAP kinase phosphatases. Genome Biol 3(7):1–10
Wancket LM, Frazier WJ, Liu Y (2012) Mitogen-activated protein kinase phosphatase (MKP)-1 in immunology, physiology, and disease. Life Sci 90(7–8):237–248
Farooq A, Zhou MM (2004) Structure and regulation of MAPK phosphatases. Cell Signal 16(7):769–779
Lang R, Hammer M, Mages J (2006) DUSP meet immunology: dual specificity MAPK phosphatases in control of the inflammatory response. J Immunol 177(11):7497–7504
Keyse SM (2000) Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr Opin Cell Biol 12(2):186–192
Sun H, Charles CH, Lau LF, Tonks NK (1993) MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell 75(3):487–493
Franklin CC, Kraft AS (1997) Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J Biol Chem 272(27):16917–16923
Lawan A, Shi H, Gatzke F, Bennett AM (2013) Diversity and specificity of the mitogen-activated protein kinase phosphatase-1 functions. Cell Mol Life Sci 70(2):223–237. doi:10.1007/s00018-012-1041-2
Chen P, Li J, Barnes J, Kokkonen GC, Lee JC, Liu Y (2002) Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. J Immunol 169(11):6408–6416
Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA, Bennett AM, Flavell RA (2006) Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci U S A 103(7):2274–2279. doi:10.1073/pnas.0510965103
Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T (2006) Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J Immunol 176(3):1899–1907
Hammer M, Mages J, Dietrich H, Servatius A, Howells N, Cato AC, Lang R (2006) Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J Exp Med 203(1):15–20. doi:10.1084/jem.20051753
Sakaue H, Ogawa W, Nakamura T, Mori T, Nakamura K, Kasuga M (2004) Role of MAPK phosphatase-1 (MKP-1) in adipocyte differentiation. J Biol Chem 279(38):39951–39957. doi:10.1074/jbc.M407353200
Roth RJ, Le AM, Zhang L, Kahn M, Samuel VT, Shulman GI, Bennett AM (2009) MAPK phosphatase-1 facilitates the loss of oxidative myofibers associated with obesity in mice. J Clin Invest 119(12):3817–3829. doi:10.1172/JCI39054
Wu JJ, Roth RJ, Anderson EJ, Hong EG, Lee MK, Choi CS, Neufer PD, Shulman GI, Kim JK, Bennett AM (2006) Mice lacking MAP kinase phosphatase-1 have enhanced MAP kinase activity and resistance to diet-induced obesity. Cell Metab 4(1):61–73. doi:10.1016/j.cmet.2006.05.010
Bennett AM, Tonks NK (1997) Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science 278(5341):1288–1291
Liu Y, Shepherd EG, Nelin LD (2007) MAPK phosphatases—regulating the immune response. Nat Rev Immunol 7(3):202–212. doi:10.1038/nri2035
Loda M, Capodieci P, Mishra R, Yao H, Corless C, Grigioni W, Wang Y, Magi-Galluzzi C, Stork PJ (1996) Expression of mitogen-activated protein kinase phosphatase-1 in the early phases of human epithelial carcinogenesis. Am J Pathol 149(5):1553–1564
Rojo F, Gonzalez-Navarrete I, Bragado R, Dalmases A, Menendez S, Cortes-Sempere M, Suarez C, Oliva C, Servitja S, Rodriguez-Fanjul V, Sanchez-Perez I, Campas C, Corominas JM, Tusquets I, Bellosillo B, Serrano S, Perona R, Rovira A, Albanell J (2009) Mitogen-activated protein kinase phosphatase-1 in human breast cancer independently predicts prognosis and is repressed by doxorubicin. Clin Cancer Res 15(10):3530–3539. doi:10.1158/1078-0432.CCR-08-2070
Wang HY, Cheng Z, Malbon CC (2003) Overexpression of mitogen-activated protein kinase phosphatases MKP1, MKP2 in human breast cancer. Cancer Lett 191(2):229–237
Vicent S, Garayoa M, Lopez-Picazo JM, Lozano MD, Toledo G, Thunnissen FB, Manzano RG, Montuenga LM (2004) Mitogen-activated protein kinase phosphatase-1 is overexpressed in non-small cell lung cancer and is an independent predictor of outcome in patients. Clin Cancer Res 10(11):3639–3649. doi:10.1158/1078-0432.CCR-03-0771
Wu GS (2007) Role of mitogen-activated protein kinase phosphatases (MKPs) in cancer. Cancer Metastasis Rev 26(3–4):579–585. doi:10.1007/s10555-007-9079-6
Valjent E, Caboche J, Vanhoutte P (2001) Mitogen-activated protein kinase/extracellular signal-regulated kinase induced gene regulation in brain: a molecular substrate for learning and memory? Mol Neurobiol 23(2–3):83–99. doi:10.1385/MN:23:2-3:083
Nolan Y, Vereker E, Lynch AM, Lynch MA (2003) Evidence that lipopolysaccharide-induced cell death is mediated by accumulation of reactive oxygen species and activation of p38 in rat cortex and hippocampus. Exp Neurol 184(2):794–804. doi:10.1016/S0014-4886(03)00301-7
Roth TL, Sweatt JD (2008) Rhythms of memory. Nat Neurosci 11(9):993–994. doi:10.1038/nn0908-993
Kim EK, Choi EJ (2010) Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta 1802(4):396–405. doi:10.1016/j.bbadis.2009.12.009
Peinado-Ramon P, Wallen A, Hallbook F (1998) MAP kinase phosphatase-1 mRNA is expressed in embryonic sympathetic neurons and is upregulated after NGF stimulation. Brain Res Mol Brain Res 56(1–2):256–267
Reffas S, Schlegel W (2000) Compartment-specific regulation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) mitogen-activated protein kinases (MAPKs) by ERK-dependent and non-ERK-dependent inductions of MAPK phosphatase (MKP)-3 and MKP-1 in differentiating P19 cells. Biochem J 352(Pt 3):701–708
Jeanneteau F, Deinhardt K, Miyoshi G, Bennett AM, Chao MV (2010) The MAP kinase phosphatase MKP-1 regulates BDNF-induced axon branching. Nat Neurosci 13(11):1373–1379
Collins LM, O’Keeffe GW, Long-Smith CM, Wyatt SL, Sullivan AM, Toulouse A, Nolan YM (2013) Mitogen-activated protein kinase phosphatase (MKP)-1 as a neuroprotective agent: promotion of the morphological development of midbrain dopaminergic neurons. Neuromol Med 15(2):435–446. doi:10.1007/s12017-013-8230-5
Choi BH, Hur EM, Lee JH, Jun DJ, Kim KT (2006) Protein kinase Cdelta-mediated proteasomal degradation of MAP kinase phosphatase-1 contributes to glutamate-induced neuronal cell death. J Cell Sci 119(Pt 7):1329–1340. doi:10.1242/jcs.02837
Kawahara N, Wang Y, Mukasa A, Furuya K, Shimizu T, Hamakubo T, Aburatani H, Kodama T, Kirino T (2004) Genome-wide gene expression analysis for induced ischemic tolerance and delayed neuronal death following transient global ischemia in rats. J Cereb Blood Flow Metab 24(2):212–223. doi:10.1097/01.WCB.0000106012.33322.A2
Rininger A, Dejesus C, Totten A, Wayland A, Halterman MW (2012) MKP-1 antagonizes C/EBPbeta activity and lowers the apoptotic threshold after ischemic injury. Cell Death Differ 19(10):1634–1643. doi:10.1038/cdd.2012.41
Koga S, Kojima S, Kishimoto T, Kuwabara S, Yamaguchi A (2012) Over-expression of map kinase phosphatase-1 (MKP-1) suppresses neuronal death through regulating JNK signaling in hypoxia/re-oxygenation. Brain Res 1436:137–146. doi:10.1016/j.brainres.2011.12.004
Mishra OP, Delivoria-Papadopoulos M (2004) Effect of hypoxia on the expression and activity of mitogen-activated protein (MAP) kinase-phosphatase-1 (MKP-1) and MKP-3 in neuronal nuclei of newborn piglets: the role of nitric oxide. Neuroscience 129(3):665–673
Kristiansen M, Hughes R, Patel P, Jacques TS, Clark AR, Ham J (2010) Mkp1 is a c-Jun target gene that antagonizes JNK-dependent apoptosis in sympathetic neurons. J Neurosci 30(32):10820–10832. doi:10.1523/jneurosci.2824-10.2010
Huang H, Fan S, Ji X, Zhang Y, Bao F, Zhang G (2009) Recombinant human erythropoietin protects against experimental spinal cord trauma injury by regulating expression of the proteins MKP-1 and p-ERK. J Int Med Res 37(2):511–519
Wu J, Pan Z, Wang Z, Zhu W, Shen Y, Cui R, Lin J, Yu H, Wang Q, Qian J, Yu Y, Zhu D, Lou Y (2012) Ginsenoside Rg1 protection against beta-amyloid peptide-induced neuronal apoptosis via estrogen receptor alpha and glucocorticoid receptor-dependent anti-protein nitration pathway. Neuropharmacology 63(3):349–361. doi:10.1016/j.neuropharm.2012.04.005
Taylor DM, Moser R, Régulier E, Breuillaud L, Dixon M, Beesen AA, Elliston L, Silva Santos MF, Kim J, Jones L, Goldstein DR, Ferrante RJ, Luthi-Carter R (2013) MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in Huntington’s disease via additive effects of JNK and p38 inhibition. J Neurosci 33(6):2313–2325. doi:10.1523/jneurosci.4965-11.2013
Ndong C, Landry RP, DeLeo JA, Romero-Sandoval EA (2012) Mitogen activated protein kinase phosphatase-1 prevents the development of tactile sensitivity in a rodent model of neuropathic pain. Mol Pain 8:34. doi:10.1186/1744-8069-8-34
Huo Y, Rangarajan P, Ling EA, Dheen ST (2011) Dexamethasone inhibits the Nox-dependent ROS production via suppression of MKP-1-dependent MAPK pathways in activated microglia. BMC Neurosci 12:49. doi:10.1186/1471-2202-12-49
Rayan NA, Baby N, Pitchai D, Indraswari F, Ling EA, Lu J, Dheen T (2011) Costunolide inhibits proinflammatory cytokines and iNOS in activated murine BV2 microglia. Front Biosci 3:1079–1091, Elite Ed
Kim J, Shim J, Lee S, Lim SS, Lee KW, Lee HJ (2013) Licorice-derived dehydroglyasperin C increases MKP-1 expression and suppresses inflammation-mediated neurodegeneration. Neurochem Int 63(8):732–740. doi:10.1016/j.neuint.2013.09.013
Crittenden PL, Filipov NM (2011) Manganese modulation of MAPK pathways: effects on upstream mitogen activated protein kinase kinases and mitogen activated kinase phosphatase-1 in microglial cells. J Appl Toxicol 31(1):1–10. doi:10.1002/jat.1552
Eljaschewitsch E, Witting A, Mawrin C, Lee T, Schmidt PM, Wolf S, Hoertnagl H, Raine CS, Schneider-Stock R, Nitsch R, Ullrich O (2006) The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron 49(1):67–79
Krishnan G, Chatterjee N (2014) Endocannabinoids affect innate immunity of Muller glia during HIV-1 Tat cytotoxicity. Mol Cell Neurosci 59C:10–23. doi:10.1016/j.mcn.2014.01.001
Romero-Sandoval EA, Horvath R, Landry RP, DeLeo JA (2009) Cannabinoid receptor type 2 activation induces a microglial anti-inflammatory phenotype and reduces migration via MKP induction and ERK dephosphorylation. Mol Pain 5:25. doi:10.1186/1744-8069-5-25
Juknat A, Pietr M, Kozela E, Rimmerman N, Levy R, Gao F, Coppola G, Geschwind D, Vogel Z (2013) Microarray and pathway analysis reveal distinct mechanisms underlying cannabinoid-mediated modulation of LPS-induced activation of BV-2 microglial cells. PLoS ONE 8(4):e61462. doi:10.1371/journal.pone.0061462
Li W, Hertzberg EL, Spray DC (2005) Regulation of connexin43-protein binding in astrocytes in response to chemical ischemia/hypoxia. J Biol Chem 280(9):7941–7948. doi:10.1074/jbc.M410548200
Tournier C, Thomas G, Pierre J, Jacquemin C, Pierre M, Saunier B (1997) Mediation by arachidonic acid metabolites of the H2O2-induced stimulation of mitogen-activated protein kinases (extracellular-signal-regulated kinase and c-Jun NH2-terminal kinase). Eur J Biochem 244(2):587–595
Herrera-Molina R, Flores B, Orellana JA, von Bernhardi R (2012) Modulation of interferon-gamma-induced glial cell activation by transforming growth factor beta1: a role for STAT1 and MAPK pathways. J Neurochem 123(1):113–123. doi:10.1111/j.1471-4159.2012.07887.x
Flores B, von Bernhardi R (2012) Transforming growth factor beta1 modulates amyloid beta-induced glial activation through the Smad3-dependent induction of MAPK phosphatase-1. J Alzheimers Dis 32(2):417–429. doi:10.3233/JAD-2012-120721
Lee JH, Kim H, Woo JH, Joe EH, Jou I (2012) 5, 8, 11, 14-eicosatetraynoic acid suppresses CCL2/MCP-1 expression in IFN-gamma-stimulated astrocytes by increasing MAPK phosphatase-1 mRNA stability. J Neuroinflammation 9:34. doi:10.1186/1742-2094-9-34
Lee JH, Woo JH, Woo SU, Kim KS, Park SM, Joe EH, Jou I (2008) The 15-deoxy-delta 12,14-prostaglandin J2 suppresses monocyte chemoattractant protein-1 expression in IFN-gamma-stimulated astrocytes through induction of MAPK phosphatase-1. J Immunol 181(12):8642–8649
Freeman SE, Patil VV, Durham PL (2008) Nitric oxide-proton stimulation of trigeminal ganglion neurons increases mitogen-activated protein kinase and phosphatase expression in neurons and satellite glial cells. Neuroscience 157(3):542–555. doi:10.1016/j.neuroscience.2008.09.035
Cady RJ, Denson JE, Durham PL (2013) Inclusion of cocoa as a dietary supplement represses expression of inflammatory proteins in spinal trigeminal nucleus in response to chronic trigeminal nerve stimulation. Mol Nutr Food Res 57(6):996–1006. doi:10.1002/mnfr.201200630
Cady RJ, Hirst JJ, Durham PL (2010) Dietary grape seed polyphenols repress neuron and glia activation in trigeminal ganglion and trigeminal nucleus caudalis. Mol Pain 6:91. doi:10.1186/1744-8069-6-91
Clark R, Stewart M, Miskimins WK, Miskimins R (1998) Involvement of MAP kinase in the cyclic AMP induction of myelin basic protein gene expression. Int J Dev Neurosci 16(5):323–331
Davis S, Vanhoutte P, Pages C, Caboche J, Laroche S (2000) The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J Neurosci 20(12):4563–4572
Zhou J, Wang L, Ling S, Zhang X (2007) Expression changes of growth-associated protein-43 (GAP-43) and mitogen-activated protein kinase phosphatase-1 (MKP-1) and in hippocampus of streptozotocin-induced diabetic cognitive impairment rats. Exp Neurol 206(2):201–208. doi:10.1016/j.expneurol.2007.04.013
Chen MF, Huang TY, Kuo YM, Yu L, Chen HI, Jen CJ (2013) Early postinjury exercise reverses memory deficits and retards the progression of closed-head injury in mice. J Physiol 591(Pt 4):985–1000. doi:10.1113/jphysiol.2012.241125
Duric V, Banasr M, Licznerski P, Schmidt HD, Stockmeier CA, Simen AA, Newton SS, Duman RS (2010) A negative regulator of MAP kinase causes depressive behavior. Nat Med 16(11):1328–1332
Iio W, Matsukawa N, Tsukahara T, Kohari D, Toyoda A (2011) Effects of chronic social defeat stress on MAP kinase cascade. Neurosci Lett 504(3):281–284
Budziszewska B, Szymanska M, Leskiewicz M, Basta-Kaim A, Jaworska-Feil L, Kubera M, Jantas D, Lason W (2010) The decrease in JNK-and p38-MAP kinase activity is accompanied by the enhancement of PP2A phosphatase level in the brain of prenatally stressed rats. J Physiol Pharmacol 61(2):207
Lee H-R, Hwang I-S, Kim J-E, Choi S-I, Lee Y-J, Goo J-S, Lee E-P, Choi H-W, Kim H-S, Lee J-H (2012) Altered expression of γ-secretase components in animal model of major depressive disorder induced by reserpine administration. Lab Anim Res 28(2):109
Vogt A, Tamewitz A, Skoko J, Sikorski RP, Giuliano KA, Lazo JS (2005) The benzo[c]phenanthridine alkaloid, sanguinarine, is a selective, cell-active inhibitor of mitogen-activated protein kinase phosphatase-1. J Biol Chem 280(19):19078–19086. doi:10.1074/jbc.M501467200
Chen Y, Wang H, Zhang R, Wang H, Peng Z, Sun R, Tan Q (2012) Microinjection of sanguinarine into the ventrolateral orbital cortex inhibits Mkp-1 and exerts an antidepressant-like effect in rats. Neurosci Lett 506(2):327–331. doi:10.1016/j.neulet.2011.11.038
Jia W, Liu R, Shi J, Wu B, Dang W, Du Y, Zhou Q, Wang J, Zhang R (2013) Differential regulation of MAPK phosphorylation in the dorsal hippocampus in response to prolonged morphine withdrawal-induced depressive-like symptoms in mice. PLoS ONE 8(6):e66111. doi:10.1371/journal.pone.0066111
Sasaki K, El Omri A, Kondo S, Han J, Isoda H (2013) Rosmarinus officinalis polyphenols produce anti-depressant like effect through monoaminergic and cholinergic functions modulation. Behav Brain Res 238:86–94. doi:10.1016/j.bbr.2012.10.010
Acknowledgments
Work in the authors’ laboratories is supported by grants from Science Foundation Ireland (12/IA/1537 and RFP/NSC1298; YN) and the College of Medicine and Health, UCC (LC/AT/ED/YN).
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Collins, L.M., Downer, E.J., Toulouse, A. et al. Mitogen-Activated Protein Kinase Phosphatase (MKP)-1 in Nervous System Development and Disease. Mol Neurobiol 51, 1158–1167 (2015). https://doi.org/10.1007/s12035-014-8786-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-8786-6