
Abstract
A greater understanding of the mechanisms that promote the survival and growth of dopaminergic neurons is essential for the advancement of cell replacement therapies for Parkinson’s disease (PD). Evidence supports a role for the mitogen-activated protein kinase p38 in the demise of dopaminergic neurons, while mitogen-activated protein kinase phosphatase-1 (MKP-1), which negatively regulates p38 activity, has not yet been investigated in this context. Here, we show that MKP-1 is expressed in dopaminergic neurons cultured from E14 rat ventral mesencephalon (VM). When dopaminergic neurons were transfected to overexpress MKP-1, they displayed a more complex morphology than their control counterparts in vitro. Specifically, MKP-1-transfection induced significant increases in neurite length and branching with a maximum increase observed in primary branches. We demonstrate that inhibition of dopaminergic neurite growth induced by treatment of rat VM neurons with the dopaminergic neurotoxin 6-hydroxydopamine (6-OHDA) in vitro is mediated by p38 and is concomitant with a significant and selective decrease in MKP-1 expression in these neurons. We further show that overexpression of MKP-1 in dopaminergic neurons contributes to neuroprotection against the effects of 6-OHDA. Collectively, we report that MKP-1 can promote the growth and elaboration of dopaminergic neuronal processes and can help protect them from the neurotoxic effects of 6-OHDA. Thus, we propose that strategies aimed at augmenting MKP-1 expression or activity may be beneficial in protecting dopaminergic neurons and may provide potential therapeutic approaches for PD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterised by the selective loss of midbrain dopaminergic neurons from the substantia nigra (SN). This leads to gradual degeneration of the nigrostriatal pathway resulting in primary motor and eventually secondary cognitive and psychiatric symptoms. However, despite intensive research, the aetiology of the disease still remains unclear and a cure remains elusive. Because of the specific loss of dopaminergic neurons in a discreet region of the brain, PD is considered a suitable disease candidate for cell replacement therapy, and in the past number of years, much research has gone into optimising the generation of dopaminergic neurons as a potential cell replacement therapy for PD (Toulouse and Sullivan 2008). To date, however, this therapy is limited by the poor survival and minimal reinnervation of the transplanted dopaminergic neurons (Kim 2011). Therefore, a deeper understanding of the mechanisms that promote the survival and growth of dopaminergic neurons is essential for the advancement of cell replacement therapies.
One such class of regulatory proteins which are instrumental in orchestrating a variety of cellular processes such as proliferation, differentiation, growth, cell cycle arrest and apoptosis are the mitogen-activated protein kinases (MAPKs) (Raman et al. 2007). These signal-transducing enzymes, including p38, ERK and JNK, are activated by a wide range of stimuli such as hormones, growth factors, cytokines and environmental stresses. Mitogen-activated protein kinase phosphatases (MKPs or DUSP) provide a negative feedback mechanism for regulating MAPK activity by de-phosphorylating these kinases at threonine and tyrosine residues (Farooq and Zhou 2004). Evidence now exists to support a role for MKP-1 as a regulator of a variety of physiological functions in the immune, metabolic, cardiovascular and musculoskeletal systems but little is yet known of its role in the nervous system [for reviews see (Lawan et al. 2012; Wancket et al. 2012)]. In the brain, MKP-1 expression has been reported in the rodent hippocampus (Gass et al. 1996), cortex (Choi et al. 2006), striatum, thalamus (Takaki et al. 2001) and ventral tegmental area (Rajadhyaksha et al. 2004), as well as in embryonic sympathetic neurons (Peinado-Ramon et al. 1998). However, while the involvement of MAPK in the brain is well established (Nolan et al. 2003; Valjent et al. 2001), there have been very few reports on the role of MKP-1 in the brain. Specifically, MKP-1 mRNA is induced by long-term potentiation in the dentate gyrus (Davis et al. 2000), and the endocannabinoid anandamide has been shown to protect neurons from inflammatory damage by a rapid induction of MKP-1 in microglia (Eljaschewitsch et al. 2006). More recently, MKP-1 has been shown to be essential for BDNF-induced axon branching and may therefore be an essential regulator of synaptogenesis (Jeanneteau et al. 2010). Thus, strategies targeting MKP-1 have been proposed as potential neuroprotective approaches (Doddareddy et al. 2012).
In animal models of PD, a terminal injection of 6-hydroxydopamine (6-OHDA), the dopaminergic neurotoxin which is commonly used to model PD, initially targets the dopaminergic nerve terminals (Walsh et al. 2011) and leads to the activation of phospho-p38 in dopaminergic neurons (Crotty et al. 2008). As MKP-1 preferentially de-phosphorylates p38 (Hutter et al. 2000) and has been implicated in regulating axonal branching, we decided to explore a role for MKP-1 in regulating the growth of dopaminergic neurons during development and to examine whether its overexpression could protect the cytoarchitecture of these vulnerable cells from 6-OHDA-induced degeneration. Based on these findings, we propose that therapeutic strategies aimed at augmenting MKP-1 expression may be a useful approach to protect dopaminergic neurons terminals or to stimulate innervation of the host parenchyma by transplanted cells in PD.
Materials and Methods
Preparation of E14 Rat Ventral Mesencephalic Cultures
Ventral mesencephalon (VM) cultures were prepared at embryonic (E) day 14 and E18 from the embryos of time-mated Sprague–Dawley rats and from postnatal (P) day 1, 11, 31, 60, 90 Sprague–Dawley rat pups (Biological Services Unit, University College Cork) as previously described (Long-Smith et al. 2010). The cells were allowed to differentiate for 3 days in vitro (DIV) in a humidified atmosphere containing 5 % CO2 at 37 °C. Where indicated, the p38 inhibitor, SB203580 (10 μM; Calbiochem), was added to the culture medium at 3 DIV for 60 min prior to 6-OHDA (40 μM; pre-stabilised in 0.01 % ascorbic acid, Sigma) treatment. 6-OHDA was added for 30 min immediately before preparation of the cells for immunocytochemical or western blotting analysis. Previous studies have shown that concentrations of 6-OHDA >40 μM act non-selectively (Long-Smith et al. 2010; Michel and Hefti 1990).
qPCR
5 μl of total RNA, isolated from VM and striatum using the RNeasy mini lipid kit (Qiagen), was reverse-transcribed using Affinity Script reverse-transcriptase (Agilent Technologies, UK), for 45 min at 45 °C, in a 20 μl reaction containing 5 mM dNTPs, 10 mM DTT and 10 μM random hexamer primers. 2 μl of cDNA template was used to amplify each of the normalising reference genes, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), succinate dehydrogenase complex, subunit A (SDHA) and ubiquitin C (UBQC), in 20 μl reactions using Brilliant III Ultra-Fast SYBR® Green QPCR Master Mix reagents (Agilent Technologies) containing 150 nM each of forward and reverse primers. The PCR primers were as follows: GAPDH forward, 5′-GCC TTC CGT GTT CCT ACC-3′ and reverse, 5′-TAG CCA TAT TCA TTG TCA TAC CA-3′; SDHA forward, 5′-GCT CTT TCC TAC CCG CTC AC-3′ and reverse, 5′-GTG TCA TAG AAA TGC CAT CTC CAG-3′; UBQC forward, 5′-CTT TGT GAA GAC CCT GAC-3′ and reverse, 5′-CCT TCT GGA TGT TGT AGT C-3′. Cycling parameters for GAPDH, SHDA and UBQC cDNA amplification on a Stratagene MX3000P thermal cycler were 3 min at 95 °C followed by 45 cycles of: 95 °C for 12 s; 60 °C for 35 s. GAPDH, SDHA and UBQC amplification products were verified as accurate via melting-curve analysis of the completed PCR (melting temperatures of 83.5, 80 and 85 °C, respectively). For MKP-1, 2 μl of cDNA was amplified in a 20 μl PCR using Brilliant III Ultra-Fast QPCR Master Mix reagents (Agilent Technologies) containing 150 nM each or forward and reverse primers and 300 nM of a FAM/BHQ1 dual-labelled hybridisation probe (Eurofins). The MKP-1-specific primers were as follows: forward, 5′-CTA CTA CAA CGG TCT TCA A-3′ and reverse, 5′-CTC TCC CAG AGT TAT TGC-3′, and the MKP-1-specific probe was 5′-FAM-TTC CCT GTC TCC ATC CCT GT-BHQ1-3′. Cycling parameters for MKP-1 were 3 min at 95 °C followed by 45 cycles of: 95 °C for 12 s; 60 °C for 35 s. The initial quantities of each cDNA in each PCR were determined by comparison with standard curves that were generated by serial dilutions of reverse-transcribed RNA purified from either the striatum or VM. Each PCR run for every cDNA included triplicate standard curve samples for each cDNA dilution. Values for each gene of interest were normalised to a geometrical mean of the three reference genes. Appropriate no template and no RT controls were performed for each cDNA. Each assay was repeated three times and in technical triplicate.
Plasmids and Electroporation
For each group, 2 × 106 freshly dissociated E14 VM cells (0 DIV) were transfected in suspension using the Neontm transfection system (Invitrogen). Cells were centrifuged, washed with PBS, and resuspended in 10 μl Resuspension Buffer (Invitrogen) containing 1 μg plasmid DNA. Cells were transfected with the MKP-1 overexpression plasmid pWAY21-MKP-1-FL (Wu et al. 2005) or an eGFP expression control plasmid pCX-eGFP (Gutierrez et al. 2008). Two transient electrical pulses (30 ms, 1,100 V) were applied to the cell–DNA mixture in an electrode tip; the cells were transferred to poly-d-lysine-coated 24-well plates containing culture medium and were allowed to differentiate for 3 DIV. Where indicated, the p38 inhibitor, SB203580 (10 μM; Calbiochem), and JNK inhibitor, SP600125 (10 μM; Sigma), were added to the cells 24 h before preparation of the cells for immunocytochemical analysis at 3 DIV (Bennett et al. 2001; Crampton et al. 2012).
Immunocytochemistry
The cultures were fixed in ice-cold methanol for 10 min, and the cells were immunocytochemically stained as described previously (Crampton et al. 2012; Long-Smith et al. 2010) using antibodies that target tyrosine hydroxylase (TH) (1:200; goat polyclonal; Millipore), βIII-tubulin (1:300; mouse monoclonal; Chemicon), doublecortin (DCX) (1:300; goat polyclonal; Santa Cruz Biotechnology), dopamine transporter (DAT) (1:200; goat polyclonal; Santa Cruz Biotechnology), MKP-1 (1:200; rabbit; Santa Cruz Biotechnology) and phospho-p38 (1:200; rabbit; Cell Signalling). The cells were washed and incubated with the appropriate secondary antibodies (Alexa Fluor 594-conjugated donkey anti-mouse IgG, anti-goat IgG or anti-rabbit IgG, all 1:2,000; Invitrogen). All cells were counterstained with DAPI (1:3,000; Sigma) and imaged under an Olympus IX70 Provis Inverted microscope or an Olympus AX70 upright fluorescence microscope. Individual cell fluorescence intensity was measured using the ImageJ software (version 1.38X, NIH, USA). For each treatment, cells from four individual wells were stained and analysed, and each experiment was repeated three times.
Immunohistochemistry
Rats were anaesthetised with pentobarbital and transcardially perfused with 100 ml of ice-cold heparinised saline followed by 150 ml of 4 % paraformaldehyde (pH 7.4). Serial sections (30 μm) from the adult SN or E14 VM were prepared from the fixed and cryoprotected tissue. Following quenching of endogenous peroxidase activity (0.3 % H2O2–10 % methanol in dH2O) and blocking (3 % normal horse or goat serum in 0.2 % Triton-X-TBS), the sections were incubated overnight with primary antibodies against MKP-1 (1:200; rabbit polyclonal; Santa Cruz Biotechnology) diluted in 1 % normal horse or goat serum. The sections were incubated with biotinylated secondary antibodies (Vectastain® ABC Kit peroxidase mouse or rabbit IgG), followed by a streptavidin–biotin–horseradish peroxidase solution (Vector Laboratories). Immunolabelling was revealed by incubating the sections in a 0.05 % solution of diaminobenzidine tetrahydrochloride—0.015 % H2O2 in TBS.
Analysis of Neuronal Complexity
The total neurite length and the number of branch points of neurons were measured at 1 and 3 DIV using Sholl analysis of embryonic neurons as previously described (Gutierrez and Davies 2007; Nolan et al. 2011). Traces of GFP+/TH+ neurons were carried out using the CorelDRAW x4 software and analysed as previously described (O’Keeffe et al. 2004). Briefly, neurite length was calculated using the formula; n × T × π/2, where n = the number of times the neurites intersect the grid lines and T = the distance between the gridlines (taking the magnification into account). The number of neurons with intact processes was calculated in six random fields per coverslip and on 3–4 coverslips per condition where any neuron with a process which was at least one and half times the length of the soma was determined as an intact process. This is based on the information reported by Rayport et al. and O’Keeffe et al. demonstrating that the average size of dopaminergic neuron soma is 10–20 μm, while the average length of primary neurites is ~ 85 ± 10 μm (O’Keeffe et al. 2004; Rayport et al. 1992). By considering only neurons with a neuritic length >1.5 times the size of the soma in our cell count, we account for diversity in somatic shape and neuritic length but most importantly, we exclude cells that are likely committed to the apoptotic pathway. These cells while still present within the culture dish have nonetheless initiated the process of axonal retraction (as illustrated by their shortened processes).
Western Blotting
Western blotting was carried out as previously described (Crampton et al. 2012). The cells were lysed in RIPA buffer, and insoluble debris was removed by centrifugation. Samples were run on a gel and transferred to nitrocellulose membranes using a Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad, CA, USA). The membranes were incubated with primary antibodies against p38 (1:1,000; Cell Signalling), phospho-p38 (1:1,000; Cell Signalling), MKP-1 (1:100; Santa Cruz Biotechnology) or βIII-tubulin (1:10,000; Promega) overnight at 4 °C, washed, incubated with horseradish peroxidase-labelled anti-rabbit IgG (1:2,000; Promega), washed and developed with ECL-Plus (Amersham).
Statistical Analyses
Statistical analyses were performed using unpaired Student’s t test or ANOVA with a post hoc Newman–Keuls’ test as appropriate. Results are expressed as mean ± s.e.m and deemed significant when p < 0.05.
Results and Discussion
MKP-1 Overexpression Regulates the Growth of Dopaminergic Neurons in the Developing VM
We examined MKP-1 gene expression during the development of the nigrostriatal pathway and show that MKP-1 expression is highest in the VM and striatum during E14 to E18, the peak period of striatal axonogenesis (Fig. 1a), supporting a potential role for MKP-1 in the growth of dopaminergic neurons. We carried out MKP-1 immunohistochemistry in the VM tissue from E14 rat (which is the age of highest MKP-1 mRNA expression—Fig. 1a) and in the adult substantia nigra at postnatal day 90 (age of lowest level of mRNA expression—Fig. 1a) and found clear differences in the expression levels of MKP-1 between embryonic and adult tissue that agree with the qPCR data of MKP-1 mRNA levels (Fig. 1b). We next prepared cultures of E14 rat VM and found that MKP-1 is expressed in βIII-tubulin+ VM neurons (Fig. 1c). Cells in a negative control, in which the primary antibody was omitted from the staining protocol, displayed no immunoreactivity for MKP-1, indicating the specificity of the staining (Fig. 1c). To determine the functional significance of MKP-1 expression in these neurons, E14 rat VM cultures were electroporated with a MKP-1 overexpression plasmid or a control plasmid together with a pCX-GFP plasmid to visualise the transfected cells (Fig. 1d, e). Our results showed that MKP-1 overexpression increased neuronal complexity when compared to controls. Specifically, a significant increase in neurite length and in the degree of neuronal branching was observed after 3 DIV compared to controls at the same time point (p < 0.01), while no difference was observed after 1 DIV (Fig. 1e, f, g). Sholl analysis, which provides a graphic illustration of neurite length and branching with respect to distance from the cell body (Gutierrez and Davies 2007), was also performed on MKP-1-transfected cultures, and it revealed that MKP-1 overexpression significantly increased the degree of neuronal complexity at 3 DIV (p < 0.05) (Fig. 1h) and thus suggests that MKP-1 promotes neurite growth. Interestingly, it has recently been suggested that while transient induction of MKP-1 in the developing cortex promotes axonal branching, prolonged expression of MKP-1 may impede axonal growth (Jeanneteau and Deinhardt 2011), and so the temporal regulatory nature of MKP-1 must be considered in future research in this area.

MKP-1 overexpression increases the neurite growth of VM neurons. a mRNA expression profile of MKP-1 in vivo in the embryonic and postnatal rat VM and striatum. b Immunohistochemical staining showing MKP-1 expression in E14 rat VM tissue and in adult rat substantia nigra (SN) at postnatal day 90. c Representative photomicrographs showing MKP-1 expression (green) in βIII-tubulin+ neurons (red) in cultures of E14 rat VM. Cells were counterstained with DAPI (blue). A negative control for MKP-1 is also shown. d, e Representative images of GFP-transfected neurons at 3 DIV in primary cultures of E14 rat VM that were co-transfected with either a control plasmid or a MKP-1 expression plasmid. f The total length of the neurite arbours of MKP-1 and control-transfected neurons at 1 and 3 DIV. g The number of neurite branches in MKP-1 and control-transfected neurons at 1 and 3 DIV. h Sholl analyses of MKP-1 and control-transfected neurons at 3 DIV. Results are presented as the mean ± SEM of data from 21 to 34 neurons in each condition from 3 independent experiments (statistical comparison with control at the same time point, *p < 0.05, **p < 0.01) (Color figure online)
In contrast to a previous report demonstrating that MKP-1 could not be detected in neurons in the SN (Winter et al. 1998), our immunocytochemical staining revealed that MKP-1 is expressed in TH+ dopaminergic neurons in E14 rat VM cultures under basal conditions in vitro (Fig. 2a). MKP-1 staining was prominent in the soma and around the nucleus rather than in the neurites. It has previously been shown that MKP-1 was present in the nuclear fraction of brain extracts of animals subjected to kainate-induced limbic seizures (Gass et al. 1996) and that it localises to the nucleus via its N-terminus (Wu et al. 2005). However, it has also been described as being able to trap p38 in the cytosol (Pratt et al. 2003). The appearance of aggregated MKP-1 staining in the present study suggests that MKP-1 is either sequestered into vesicles within the cell or that it associates with organelles, which may be indicative of a processing function of the enzyme. While MKP-1 staining is mostly absent from neurites, this does not mean that MKP-1 cannot affect neuronal complexity. Indeed, there are a variety of molecules specifically expressed on the neuronal soma, and not the neurite that promote neurite growth, for example, transmembrane receptors such as the glucocorticoid-induced tumour necrosis factor receptor (McKelvey et al. 2012; O’Keeffe et al. 2008).

MKP-1 overexpression increases the neurite length and branching of VM dopaminergic neurons. a Representative photomicrographs showing MKP-1 expression (green) in TH+ dopaminergic neurons (red) in E14 rat VM cultures. Cells were counterstained with DAPI (blue). b Representative images at 3 DIV of TH+ cells (red) from E14 rat VM primary cultures electroporated with a control plasmid or a MKP-1 expression plasmid together with GFP. c Traces of the GFP+/TH+ cells transfected with either a control or MKP-1 expression after 3 DIV. d The total length of the neurite arbours of MKP-1 and control-transfected neurons at 3 DIV. e The number of neurite branches in MKP-1 and control-transfected neurons at 3 DIV. f The number of branches at the primary, secondary and tertiary levels of the neurite field of MKP-1 and control-transfected dopaminergic neurons at 3 DIV. Results are presented as the mean ± SEM of data from each condition from 3 independent experiments (**p < 0.01, ***p < 0.001) (Color figure online)
The expression of MKP-1 in VM dopaminergic neurons under basal conditions suggests that MKP-1 may play a regulatory role in these neurons. Thus, we transfected cultures of embryonic rat VM with MKP-1 and immunocytochemically stained them for TH. Using this approach, we were able to identify GFP+/TH+ dopaminergic neurons after 3 DIV, indicating they had been transfected (Fig. 2b). In control experiments using GFP and RFP plasmids, we achieved a >98 % co-transfection efficiency. We also prepared traces of the neurons for analysis of neuronal complexity which revealed that the dopaminergic neurons overexpressing MKP-1 were more complex than their control counterparts in vitro (Fig. 2c). Specifically, a significant increase in the total neurite length was observed in MKP-1-transfected cultures, when compared to controls (p < 0.01, Fig. 2d). MKP-1 overexpression also significantly increased the degree of neuronal branching (p < 0.001, Fig. 2e), with a maximum increase observed at the level of primary branches (p < 0.001, Fig. 2f). Similarly, a recent study has demonstrated increased morphological complexity of E18 rat cortical neurons following MKP-1 overexpression (Jeanneteau et al. 2010). This group also report that a BDNF-induced increase in the intricacy of dendritic and axonal arbours in cortical neurons is dependent on MKP-1 activity; cortical neurons prepared from MKP-1−/− mice were unable to produce axon branches in response to BDNF, and MKP-1 down-regulation resulted in reduced dendritic arbourisation compared to GFP-transfected cells. Interestingly, analysis of postnatal rat brains that had been electroporated with MKP-1 in utero showed that axons from MKP-1-overexpressing cells entered the white matter with aberrant branching and innervations into the dorso-lateral striatum and contralateral hemisphere (Jeanneteau et al. 2010). Together with the evidence we present here, this data demonstrate the potential involvement of MKP-1 expression for host reinnervation by transplanted dopaminergic neurons.
Inhibition of p38 but not JNK Prevents the MKP-1-Induced Increase in Neurite Branching and Length in the Developing VM
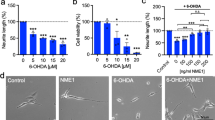
We examined neurite branching and neurite length in dopaminergic neurons that had been transfected to overexpress MKP-1 in the presence or absence of pharmacological inhibitors of p38 (SB203580) or JNK (SP600125) (Fig. 3). The data show that p38 inhibition but not JNK inhibition prevents the MKP-1-induced increase in neurite branching (Fig. 3a) and length (Fig. 3b) of dopaminergic neurons. Specifically, MKP-1 overexpression in TH-positive neurons cultured from E14 rat VM induced a significant increase in branching (p < 0.01) and length (p < 0.05) of neurites. Treatment with SP600125 did not induce any significant change in either morphological parameter measured in the presence or absence of MKP-1-transfection. However, in cells treated with SB203580, there was no significant effect of MKP-1 on neurite branching or neurite length. These data indicate that there is an inductive effect of p38 on MKP-1-induced morphological development of dopaminergic neurons. While these data may appear counter-intuitive, it is well established that the activity of MKP-1 is regulated through its direct interaction with MAP kinases (Hutter et al. 2000; Slack et al. 2001) and that the expression of MKP-1 is induced by stimuli that activate MAPK pathways (Camps et al. 2000; Owens and Keyse 2007). A number of studies have now demonstrated a role for phospho-p38, but not phospho-JNK or phospho-Erk in the upregulation of MKP-1 mRNA (Che et al. 2012; Manetsch et al. 2012; Staples et al. 2010). In agreement with these findings, we found that the p38 (SB203580) but not the JNK (SP600125) inhibitor prevented the effects of MKP-1 on neuronal growth, suggesting that a basal level of phospho-p38 signalling but not phospho-JNK signalling is required for MKP-1 expression and thus function, including in cells transfected with MKP-1. These data suggest a functionally reciprocal link between MKP-1 mRNA expression and p38 phosphorylation, which is in agreement with the data showing that p38 is the preferred substrate of MKP-1 (Franklin and Kraft 1997).

Inhibition of p38 but not JNK prevents the MKP-1-induced increase in neurite branching and length of VM dopaminergic neurons. The total number of neurite branches (a) and the total length of the neurite arbours (b) in MKP-1 and control-transfected VM dopaminergic neurons at 3 DIV treated for 24 h in the presence or absence of pharmacological inhibitors of p38 (SB203580) or JNK (SP600125). Results are presented as the mean ± SEM of data from each condition from 3 independent experiments (*p < 0.05, **p < 0.01)
6-OHDA Selectively Inhibits Neurite Growth of Dopaminergic Neurons by Activating p38
We and others have previously shown that 6-OHDA-induced activation of p38 (the upstream dephosphorylation target of MKP-1) is detrimental to dopaminergic neurons (Choi et al. 2004; Crotty et al. 2008). However, there is no evidence to date showing an effect of 6-OHDA on MKP-1 expression. Here, we show by immunoblotting that treatment of E14 VM cultures with 6-OHDA for 0, 5, 15 or 30 min co-inducted both MKP-1 and phospho-p38 with increasing time (Fig. 4a). We probed for βIII-tubulin to ensure equal concentration of protein between samples. In order to examine the specific, direct effects of 6-OHDA on dopaminergic neurons, a 30-min incubation period was chosen to account for the non-specific extracellular effects of reactive oxygen species formed in the culture medium at 15 min (Ding et al. 2004). Moreover, a 30-min incubation with 6-OHDA has been shown to decrease the percentage composition of TH-positive cells without affecting the total number of cells (Long-Smith et al. 2010). Immunocytochemical staining of cultures after treatment with 6-OHDA for 30 min revealed a significant increase in the intensity of phospho-p38 within dopaminergic neurons (p < 0.001; Fig. 4b, c). 6-OHDA treatment resulted in a decrease in the percentage of dopaminergic neurons with intact processes (p < 0.001), which was prevented by pre-treating the cultures with the p38 inhibitor, SB203580 (Fig. 4d, e). It has been shown also that 6-OHDA increases the phosphorylation of JNK (the other main substrate of MKP-1) in SH-SY5Y cells (Chambers et al. 2013) but not in rat nigral tissue after intrastriatal 6-OHDA lesion (Chu et al. 2012). These mixed results are compounded by a report showing that JNK inhibition protected dopaminergic neurons after 6-OHDA lesion to rats (Crocker et al. 2011). Interestingly, it has also been shown that homozygous null mutations of jnk2 and jnk3 isoforms abrogated the apoptotic cell death induced in dopamine neurons by intrastriatal 6-OHDA to adult rats, but did not protect against axonal degeneration (Ries et al. 2008). This evidence is in line with the thesis that molecular mechanisms of axon degeneration are distinct from those underlying the destruction of the cell soma (Coleman 2005; Raff et al. 2002) and supports our hypothesis (and data presented in Fig. 3) that JNK is not that principal MAPK involved in MKP-1-induced protective effects on the neurite outgrowth of dopaminergic neurons in the presence of 6-OHDA.

6-OHDA induces phosphorylation of p38 MAPK kinase in VM dopaminergic neurons. a Representative immunoblots of MKP-1, phospho-p38 and βIII-tubulin expression in protein extracts from E14 rat VM cultures treated with 6-OHDA (40 μM) for 0, 5, 15 and 30 min at 3 DIV. b Densitometric analysis of phospho-p38 expression in control and 6-OHDA-treated (40 μM, 30 min) dopaminergic neurons at 3 DIV. c Representative images of phospho-p38 and TH in control and 6-OHDA-treated (40 μM, 30 min) E14 rat VM cultures at 3 DIV. Cultures were counterstained with DAPI (blue). d Representative images of TH+ cells in control, 6-OHDA, SB203580 and 6-OHDA/SB203580 treatment groups. e The percentage of TH+ cells with intact processes in control E14 rat VM cultures treated with SB203580 (30 μM, 60 min) or pre-treated with SB203580 (30 μM, 60 min) prior to the addition of 6-OHDA (40 μM, 30 min). f Densitometric analysis of phospho-p38 levels in control and MKP-1-transfected cells treated with or without 6-OHDA (40 μM, 30 min) relative to their non-transfected counterparts (the red line represents the normalised levels of phospho-p38 in non-transfected cells). Results are presented as the mean ± SEM of data from each condition from 3 independent experiments (*p < 0.05, ***p < 0.001, +++p < 0.001) (Color figure online)
To demonstrate that overexpression of MKP-1 can reduce the phosphorylation of p38 induced by 6-OHDA, we assessed the intensity levels of phospho-p38 by immunocytochemistry in non-transfected, control-transfected and MKP-1-transfected dopaminergic neurons. Figure 4F shows that there is a significant reduction in the percentage of phospho-p38 levels in MKP-1-transfected cells relative to their non-transfected counterparts (the red line is the normalised levels of phospho-p38 in non-transfected cells) (p < 0.001), that there is no reduction in the percentage of phospho-p38 levels relative to non-transfected controls in control-transfected cells treated with 6-OHDA, and that there is a significant reduction in the percentage of phospho-p38 levels compared to MKP-1-transfected cells treated with 6-OHDA (p < 0.05). Thus, it is feasible to propose that 6-OHDA-induced p38 activation mediates the reduction in the complexity of dopaminergic neurite architecture, which may contribute to the onset of neuronal degeneration. Indeed, we have previously shown that 6-OHDA treatment caused a significant decrease in the percentage of dopaminergic neurons in these cultures (Long-Smith et al. 2010). Our data showing that MKP-1 can enhance the reduction in phosphorylation levels of p38 in the presence of 6-OHDA contribute to the hypothesis that MKP-1 may protect against 6-OHDA-induced inhibition of dopaminergic neurite growth by inhibiting p38 activation. Thus, we next carried out experiments to assess whether MKP-1 overexpression is neuroprotective against 6-OHDA in dopaminergic neurons.
6-OHDA Selectively Decreases MKP-1 Expression in Dopaminergic Neurons
To determine whether 6-OHDA reduces the expression of MKP-1 in cultured dopaminergic neurons, control and 6-OHDA-treated E14 VM cultures were fixed and labelled with antibodies against MKP-1 in combination with antibodies against DCX, TH or DAT. Densitometric analyses of double-stained cultures revealed that 6-OHDA induced a decrease in MKP-1 expression in TH+ (p < 0.05, Fig. 5c) and DAT+ (p < 0.001, Fig. 5e) dopaminergic neurons but not in newly formed (DCX+) neurons (Fig. 5a) when compared to untreated control cultures. These data suggest that within the defined anatomical area of the VM, a selective decrease in MKP-1 expression in the dopaminergic subpopulation may be involved in the specific vulnerability of these cells to 6-OHDA. MKP-1 expression has previously been shown to be decreased in response to other neurotoxic insults such as hypoxia in newborn piglet cortical neurons (Mishra and Delivoria-Papadopoulos 2004) and glutamate in HT22 hippocampal cells and primary mouse cortical neurons (Choi et al. 2006). Both of these studies also reported that a decrease in MKP-1 activity contributed to the death of the respective neurons. Furthermore, Choi et al. reported that siRNA knockdown of endogenous MKP-1 resulted in an increase in glutamate-induced cytotoxicity (Choi et al. 2006). Interestingly, glutamate-induced oxidative stress and excitotoxicity have long been thought to be key mechanisms responsible for the degeneration of dopaminergic cells [reviewed by (Blandini et al. 1996)]. This opens up another potential mechanistic avenue for a role of MKP-1 in the neuroprotection of dopaminergic neurons.

6-OHDA selectively decreases MKP-1 expression in VM dopaminergic neurons. MKP-1 expression in DCX+ (a, b) TH+ (c, d) and DAT+ (e, f) cells in cultures of E14 rat VM after treatment with 6-OHDA for 30 min. Mean ± SEM of data from 3 independent experiments are shown (*p < 0.05, ***p < 0.01)
MKP-1 Overexpression Attenuates the 6-OHDA-Induced Decrease in Neurite Length in Dopaminergic Neurons
As MKP-1 was selectively decreased in dopaminergic neurons following 6-OHDA treatment, we hypothesised that overexpression of MKP-1 in these neurons could potentially protect them from the neurotoxic effects of 6-OHDA. We transfected E14 rat VM cultures with a MKP-1 expression plasmid (Fig. 6a) and traces of these GFP+/TH+-transfected dopaminergic neurons were prepared (Fig. 6b). MKP-1 overexpression in dopaminergic neurons significantly increased neurite length (p < 0.001) (Fig. 6c) and branching (p < 0.01) (Fig. 6d) compared to controls. While 6-OHDA treatment (30 min) significantly decreased the total neurite length of dopaminergic neurons in control-transfected cultures compared to non-transfected controls (p < 0.01), neurite length remained unaffected in MKP-1-transfected cultures treated with 6-OHDA compared to controls (Fig. 6c). The number of neuronal branches was also unaffected by 6-OHDA in the absence or presence of MKP-1 (Fig. 6d). Thus, these data reveal that overexpression of MKP-1 promotes outgrowth and branching of dopaminergic neurons but only partially overrides the degenerating effect of 6-OHDA treatment. While we did find that 6-OHDA was not able to increase phospho-p38 levels in MKP-1 expressing cells (presented in Fig. 4f as a significant reduction in the percentage of phospho-p38 levels in MKP-1-transfected cells treated with 6-OHDA compared to control-transfected cells), it is likely that other cell death processes such as oxidative stress, inflammatory processes, glutamatergic excitotoxicity and apoptosis (Blum et al. 2001; Collins et al. 2012; Mandel et al. 2003) also contribute to the neurotoxic effect of 6-OHDA on dopaminergic neurite degeneration and thus warrant further investigation.

MKP-1 overexpression is neuroprotective against 6-OHDA in VM dopaminergic neurons. a Representative photomicrographs of control and 6-OHDA-treated E14 rat VM cultures which were electroporated at 0 DIV with a MKP-1 expression or control plasmid and imaged for TH (red) at 3 DIV. b Traces of GFP+/TH+-transfected dopaminergic neurons from each group of transfected cells. c The total length of the neurite arbours of MKP-1 and control-transfected neurons in each group at 3 DIV. d The number of neurite branches in MKP-1 and control-transfected neurons in each group at 3 DIV. e The number of branches at the primary, secondary and tertiary levels of the neurite field of MKP-1 and control-transfected dopaminergic neurons in the presence of 6-OHDA at 3 DIV. Results are presented as the mean ± SEM of data from each condition from 3 independent experiments (**p < 0.01, ***p < 0.001) (Color figure online)
On analysis of the degree of branching at the primary, secondary and tertiary levels, we observed a significant increase in the number of primary neurites in MKP-1-transfected cultures compared to control-transfected cultures that had been treated with 6-OHDA (p < 0.01, Fig. 6e), which is consistent with MKP-1-induced effects on neuronal branching in the absence of 6-OHDA observed in Fig. 2f. It is established that the mechanisms of neurite extension and branching are not identical [reviewed by (Gallo 2011)], and thus, it is plausible to suggest that MKP-1 specifically targets primary neurites under neurotoxic insult, although further research is required to decipher the signalling mechanisms involved in its contribution to dopaminergic neuronal development.
In summary, our data show that MKP-1 promotes the growth and elaboration of dopaminergic neuronal processes and can protect against the neurotoxic effects of 6-OHDA. Thus, strategies aimed at augmenting MKP-1 expression or activity may be beneficial in protecting dopaminergic neurons as well as helping newly transplanted neurons to establish appropriate connections in the striatum.
References
Bennett, B. L., Sasaki, D. T., Murray, B. W., O’Leary, E. C., Sakata, S. T., Xu, W., et al. (2001). SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proceedings of the National Academy of Sciences of the United States of America, 98(24), 13681–13686.
Blandini, F., Porter, R. H., & Greenamyre, J. T. (1996). Glutamate and Parkinson’s disease. Molecular Neurobiology, 12(1), 73–94.
Blum, D., Torch, S., Lambeng, N., Nissou, M., Benabid, A. L., Sadoul, R., et al. (2001). Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: Contribution to the apoptotic theory in Parkinson’s disease. Progress in Neurobiology, 65(2), 135–172.
Camps, M., Nichols, A., & Arkinstall, S. (2000). Dual specificity phosphatases: A gene family for control of MAP kinase function. FASEB journal, 14(1), 6–16.
Chambers, J. W., Pachori, A., Howard, S., Iqbal, S., & Lograsso, P. V. (2013). Inhibition of JNK mitochondrial localization and signaling is protective against ischemia/reperfusion injury in rats. The Journal of biological chemistry, 288(6), 4000–4011.
Che, W., Manetsch, M., Quante, T., Rahman, M. M., Patel, B. S., Ge, Q., et al. (2012). Sphingosine 1-phosphate induces MKP-1 expression via p38 MAPK- and CREB-mediated pathways in airway smooth muscle cells. Biochimica et Biophysica Acta, 1823(10), 1658–1665.
Choi, W. S., Eom, D. S., Han, B. S., Kim, W. K., Han, B. H., Choi, E. J., et al. (2004). Phosphorylation of p38 MAPK induced by oxidative stress is linked to activation of both caspase-8- and -9-mediated apoptotic pathways in dopaminergic neurons. The Journal of biological chemistry, 279(19), 20451–20460.
Choi, B. H., Hur, E. M., Lee, J. H., Jun, D. J., & Kim, K. T. (2006). Protein kinase Cdelta-mediated proteasomal degradation of MAP kinase phosphatase-1 contributes to glutamate-induced neuronal cell death. Journal of Cell Science, 119(Pt 7), 1329–1340.
Chu, J. M., Chan, Y. S., Chen, L. W., & Yung, K. K. (2012). Neurokinin receptor 3 peptide exacerbates 6-hydroxydopamine-induced dopaminergic degeneration in rats through JNK pathway. Journal of Neurochemistry, 123(3), 417–427.
Coleman, M. (2005). Axon degeneration mechanisms: Commonality amid diversity. Nature Reviews Neuroscience, 6(11), 889–898.
Collins, L. M., Toulouse, A., Connor, T. J., & Nolan, Y. M. (2012). Contributions of central and systemic inflammation to the pathophysiology of Parkinson’s disease. Neuropharmacology, 62(7), 2154–2168.
Crampton, S. J., Collins, L. M., Toulouse, A., Nolan, Y. M., & O’Keeffe, G. W. (2012). Exposure of foetal neural progenitor cells to IL-1beta impairs their proliferation and alters their differentiation—a role for maternal inflammation? Journal of Neurochemistry, 120(6), 964–973.
Crocker, C. E., Khan, S., Cameron, M. D., Robertson, H. A., Robertson, G. S., & Lograsso, P. (2011). JNK inhibition protects dopamine neurons and provides behavioral improvement in a rat 6-hydroxydopamine model of Parkinson’s disease. ACS chemical neuroscience, 2(4), 207–212.
Crotty, S., Fitzgerald, P., Tuohy, E., Harris, D. M., Fisher, A., Mandel, A., et al. (2008). Neuroprotective effects of novel phosphatidylglycerol-based phospholipids in the 6-hydroxydopamine model of Parkinson’s disease. European Journal of Neuroscience, 27(2), 294–300.
Davis, S., Vanhoutte, P., Pages, C., Caboche, J., & Laroche, S. (2000). The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. The Journal of neuroscience, 20(12), 4563–4572.
Ding, Y. M., Jaumotte, J. D., Signore, A. P., & Zigmond, M. J. (2004). Effects of 6-hydroxydopamine on primary cultures of substantia nigra: Specific damage to dopamine neurons and the impact of glial cell line-derived neurotrophic factor. Journal of Neurochemistry, 89(3), 776–787.
Doddareddy, M., Rawling, T., & Ammit, A. J. (2012). Targeting mitogen-activated protein kinase phosphatase-1 (MKP-1): structure-based design of MKP-1 inhibitors and upregulators. Current Medicinal Chemistry, 19(2), 163–173.
Eljaschewitsch, E., Witting, A., Mawrin, C., Lee, T., Schmidt, P. M., Wolf, S., et al. (2006). The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron, 49(1), 67–79.
Farooq, A., & Zhou, M.-M. (2004). Structure and regulation of MAPK phosphatases. Cellular Signalling, 16(7), 769–779.
Franklin, C. C., & Kraft, A. S. (1997). Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. The Journal of biological chemistry, 272(27), 16917–16923.
Gallo, G. (2011). The cytoskeletal and signaling mechanisms of axon collateral branching. Developmental neurobiology, 71(3), 201–220.
Gass, P., Eckhardt, A., Schroder, H., Bravo, R., & Herdegen, T. (1996). Transient expression of the mitogen-activated protein kinase phosphatase MKP-1 (3CH134/ERP1) in the rat brain after limbic epilepsy. Brain Research. Molecular Brain Research, 41(1–2), 74–80.
Gutierrez, H., & Davies, A. (2007). A fast and accurate procedure for deriving the Sholl profile in quantitative studies of neuronal morphology. Journal of Neuroscience Methods, 163(1), 24–30.
Gutierrez, H., O'Keeffe, G. W., Gavalda, N., Gallagher, D., & Davies, A. M. (2008). Nuclear factor kappa B signaling either stimulates or inhibits neurite growth depending on the phosphorylation status of p65/RelA. Journal of Neuroscience, 28(33), 8246–8256.
Hutter, D., Chen, P., Barnes, J., & Liu, Y. (2000). Catalytic activation of mitogen-activated protein (MAP) kinase phosphatase-1 by binding to p38 MAP kinase: Critical role of the p38 C-terminal domain in its negative regulation. Biochemical journal, 352(Pt 1), 155.
Jeanneteau, F., & Deinhardt, K. (2011). Fine-tuning MAPK signaling in the brain: The role of MKP-1. Communicative and Integrative Biology, 4(3), 281–283.
Jeanneteau, F., Deinhardt, K., Miyoshi, G., Bennett, A. M., & Chao, M. V. (2010). The MAP kinase phosphatase MKP-1 regulates BDNF-induced axon branching. Nature Neuroscience, 13(11), 1373–1379.
Kim, H. J. (2011). Stem cell potential in Parkinson’s disease and molecular factors for the generation of dopamine neurons. Biochimica et Biophysica Acta, 1812(1), 1–11.
Lawan, A., Shi, H., Gatzke, F., & Bennett, A. M. (2012). Diversity and specificity of the mitogen-activated protein kinase phosphatase-1 functions. Cellular and Molecular Life Sciences, 70(2), 223–237.
Long-Smith, C. M., Collins, L., Toulouse, A., Sullivan, A. M., & Nolan, Y. M. (2010). Interleukin-1β contributes to dopaminergic neuronal death induced by lipopolysaccharide-stimulated rat glia in vitro. Journal of Neuroimmunology, 226(1–2), 20–26.
Mandel, S., Grunblatt, E., Riederer, P., Gerlach, M., Levites, Y., & Youdim, M. B. (2003). Neuroprotective strategies in Parkinson’s disease: An update on progress. CNS drugs, 17(10), 729–762.
Manetsch, M., Che, W., Seidel, P., Chen, Y., & Ammit, A. J. (2012). MKP-1: A negative feedback effector that represses MAPK-mediated pro-inflammatory signaling pathways and cytokine secretion in human airway smooth muscle cells. Cellular Signalling, 24(4), 907–913.
McKelvey, L., Gutierrez, H., Nocentini, G., Crampton, S. J., Davies, A. M., Riccardi, C. R., et al. (2012). The intracellular portion of GITR enhances NGF-promoted neurite growth through an inverse modulation of Erk and NF-kappaB signalling. Biology open, 1(10), 1016–1023.
Michel, P. P., & Hefti, F. (1990). Toxicity of 6-hydroxydopamine and dopamine for dopaminergic neurons in culture. Journal of Neuroscience Research, 26(4), 428–435.
Mishra, O. P., & Delivoria-Papadopoulos, M. (2004). Effect of hypoxia on the expression and activity of mitogen-activated protein (MAP) kinase-phosphatase-1 (MKP-1) and MKP-3 in neuronal nuclei of newborn piglets: The role of nitric oxide. Neuroscience, 129(3), 665–673.
Nolan, A. M., Nolan, Y. M., & O’Keeffe, G. W. (2011). IL-1beta inhibits axonal growth of developing sympathetic neurons. Molecular and cellular neurosciences, 48(2), 142–150.
Nolan, Y., Vereker, E., Lynch, A. M., & Lynch, M. A. (2003). Evidence that lipopolysaccharide-induced cell death is mediated by accumulation of reactive oxygen species and activation of p38 in rat cortex and hippocampus. Experimental Neurology, 184(2), 794–804.
O’Keeffe, G. W., Dockery, P., & Sullivan, A. M. (2004). Effects of growth/differentiation factor 5 on the survival and morphology of embryonic rat midbrain dopaminergic neurones. Journal of Neurocytology, 33(5), 479–488.
O’Keeffe, G. W., Gutierrez, H., Pandolfi, P. P., Riccardi, C., & Davies, A. M. (2008). NGF-promoted axon growth and target innervation requires GITRL-GITR signaling. Nature Neuroscience, 11(2), 135–142.
Owens, D. M., & Keyse, S. M. (2007). Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene, 26(22), 3203–3213.
Peinado-Ramon, P., Wallen, A., & Hallbook, F. (1998). MAP kinase phosphatase-1 mRNA is expressed in embryonic sympathetic neurons and is upregulated after NGF stimulation. Brain Research. Molecular Brain Research, 56(1–2), 256–267.
Pratt, P. F., Bokemeyer, D., Foschi, M., Sorokin, A., & Dunn, M. J. (2003). Alterations in subcellular localization of p38 MAPK potentiates endothelin-stimulated COX-2 expression in glomerular mesangial cells. The Journal of biological chemistry, 278(51), 51928–51936.
Raff, M. C., Whitmore, A. V., & Finn, J. T. (2002). Axonal self-destruction and neurodegeneration. Science, 296(5569), 868–871.
Rajadhyaksha, A., Husson, I., Satpute, S. S., Küppenbender, K. D., Ren, J. Q., Guerriero, R. M., et al. (2004). L-Type Ca2 + channels mediate adaptation of extracellular signal-regulated kinase 1/2 phosphorylation in the ventral tegmental area after chronic amphetamine treatment. The Journal of neuroscience, 24(34), 7464–7476.
Raman, M., Chen, W., & Cobb, M. H. (2007). Differential regulation and properties of MAPKs. Oncogene, 26(22), 3100–3112.
Rayport, S., Sulzer, D., Shi, W. X., Sawasdikosol, S., Monaco, J., Batson, D., et al. (1992). Identified postnatal mesolimbic dopamine neurons in culture: Morphology and electrophysiology. The Journal of neuroscience, 12(11), 4264–4280.
Ries, V., Silva, R. M., Oo, T. F., Cheng, H. C., Rzhetskaya, M., Kholodilov, N., et al. (2008). JNK2 and JNK3 combined are essential for apoptosis in dopamine neurons of the substantia nigra, but are not required for axon degeneration. Journal of Neurochemistry, 107(6), 1578–1588.
Slack, D. N., Seternes, O. M., Gabrielsen, M., & Keyse, S. M. (2001). Distinct binding determinants for ERK2/p38alpha and JNK map kinases mediate catalytic activation and substrate selectivity of map kinase phosphatase-1. The Journal of biological chemistry, 276(19), 16491–16500.
Staples, C. J., Owens, D. M., Maier, J. V., Cato, A. C., & Keyse, S. M. (2010). Cross-talk between the p38alpha and JNK MAPK pathways mediated by MAP kinase phosphatase-1 determines cellular sensitivity to UV radiation. The Journal of biological chemistry, 285(34), 25928–25940.
Takaki, M., Ujike, H., Kodama, M., Takehisa, Y., Nakata, K., & Kuroda, S. (2001). Two kinds of mitogen-activated protein kinase phosphatases, MKP-1 and MKP-3, are differentially activated by acute and chronic methamphetamine treatment in the rat brain. Journal of Neurochemistry, 79(3), 679–688.
Toulouse, A., & Sullivan, A. M. (2008). Progress in Parkinson’s disease-where do we stand? Progress in Neurobiology, 85(4), 376–392.
Valjent, E., Caboche, J., & Vanhoutte, P. (2001). Mitogen-activated protein kinase/extracellular signal-regulated kinase induced gene regulation in brain: A molecular substrate for learning and memory? Molecular Neurobiology, 23(2–3), 83–99.
Walsh, S., Finn, D. P., & Dowd, E. (2011). Time-course of nigrostriatal neurodegeneration and neuroinflammation in the 6-hydroxydopamine-induced axonal and terminal lesion models of Parkinson’s disease in the rat. Neuroscience, 175, 251–261.
Wancket, L. M., Frazier, W. J., & Liu, Y. (2012). Mitogen-activated protein kinase phosphatase (MKP)-1 in immunology, physiology, and disease. Life Sciences, 90(7–8), 237–248.
Winter, C., Schenkel, J., Zimmermann, M., & Herdegen, T. (1998). MAP kinase phosphatase 1 is expressed and enhanced by FK506 in surviving mamillary, but not degenerating nigral neurons following axotomy. Brain Research, 801(1–2), 198–205.
Wu, J. J., Zhang, L., & Bennett, A. M. (2005). The noncatalytic amino terminus of mitogen-activated protein kinase phosphatase 1 directs nuclear targeting and serum response element transcriptional regulation. Molecular and Cellular Biology, 25(11), 4792–4803.
Acknowledgments
The authors would like to thank Ms Aisling Gavin for kindly providing us with tissue. This work was supported by the College of Medicine and Health, University College Cork, Science Foundation Ireland Grant No. SFI/RFP/NSC1298 (YN), Science Foundation Ireland Grant No. 10/RFP/NES2786 (GO’K), and the Irish Research Council for Science Engineering and Technology.
Conflict of interest
The authors declare that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Collins, L.M., O’Keeffe, G.W., Long-Smith, C.M. et al. Mitogen-Activated Protein Kinase Phosphatase (MKP)-1 as a Neuroprotective Agent: Promotion of the Morphological Development of Midbrain Dopaminergic Neurons. Neuromol Med 15, 435–446 (2013). https://doi.org/10.1007/s12017-013-8230-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-013-8230-5