
Abstract
The plant growth promoting bacteria Herbaspirillum seropedicae SmR1 is an endophytic diazotroph found in several economically important crops. Considering that methods to monitor the plant–bacteria interaction are required, our objective was to develop a real-time PCR method for quantification of PGPB H. seropedicae in the rhizosphere of maize seedlings. Primer pairs were designed, and their specificity was verified using DNA from 12 different bacterial species. Ten standard curves of qPCR assay using HERBAS1 primers and tenfold serial dilutions of H. seropedicae SmR1 DNA were performed, and PCR efficiency of 91 % and correlation coefficient of 0.99 were obtained. H. seropedicae SmR1 limit of detection was 101 copies (corresponding to 60.3 fg of bacterial DNA). qPCR assay using HERBAS1 was used to detect and quantify H. seropedicae strain SmR1 in inoculated maize roots, cultivated in vitro and in pots, harvested 1, 4, 7, and 10 days after inoculation. The estimated bacterial DNA copy number per gram of root was in the range 107–109 for plants grown in vitro and it was around 106 for plants grown in pots. Primer pair HERBAS1 was able to quantify H. seropedicae SmR1, and this assay can be useful for monitoring plant–bacteria interaction.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Plant growth promoting bacteria (PGPB) are a heterogeneous group of free-living soil bacteria associated with the plant rhizosphere, that can contribute, direct or indirectly, to plant growth and can increase the productivity of crops, vegetables, and other plants of economic importance [2, 18]. Indirectly PGPB can promote the plant growth through antagonisms against phytopathogenic microorganisms [18, 26]. Directly PGPB can promote plant growth by phosphate solubilization, providing nitrogen fixation and phytohormones production [2, 26, 32].
The PGPB Herbaspirillum seropedicae is a Gram-negative diazotroph that belongs to the class of the β-proteobacteria [3]. The genus Herbaspirillum comprises 13 species, most of which are plant colonizers, but can occasionally be found in other habitats [27]. H. seropedicae strain SmR1 is a spontaneous streptomycin resistant mutant of the strain Z78 (ATCC 35893) and its complete genome was published recently, GenBank accession number NC_014323.1 [30]. H. seropedicae has been known for establishing interaction with important economic crops such as rice, maize, wheat, and sugarcane [16, 20, 22, 42]. The association of the bacteria with plants probably starts with the attraction of the bacteria to the roots, as they provide carbon sources to the bacteria, followed by attachment and infection on the roots, and subsequent colonization of the plant tissues [5, 28]. Evidence showed that H. seropedicae promote differential plant growth by fixing nitrogen under conditions of ammonium and oxygen limitation [4, 21], and promote the beneficial regulation of phytohormones, like auxins, gibberellins, and ethylene [6, 8, 28]. Nevertheless, the mechanisms of infection and colonization are not completely understood [5, 29], and the identification of endophytic bacteria within the host plant is a necessary step to monitor the colonization. Despite the need for techniques allowing identification of the bacteria on internal plant tissues, few methods are available to identify rapidly and reliably the PGPB within the host plant [12, 29].
In order to monitor the establishment of the presence and colonization of the PGPB on field conditions or in association with crops, molecular methods based on PCR have been successful in tracking and identifying isolates of the genus Azospirillum [39], and the inoculant Azospirillum lipoferum strain CRT1 [7]. Also, the use of BOX PCR was employed for the evaluation of plant growth promoting potential of diazotrophs on deep water rice [44] and to determine the Azospirillum genetic diversity in Pennisetum purpureum [45]. Real-time PCR method has been successfully applied for bacterial DNA quantification of the PGPB A. lipoferum strain CRT1 in maize seedlings and rhizosphere [12], Azospirillum brasilense strain FP2 in inoculated maize seedlings [17], and Paenibacillus polymyxa in the rhizosphere of wild barley [43]. PCR method was employed to monitor phytopathogens in crops. For example, anthracnose caused by Colletotrichum gloeosporioides was detect in infected plant material of yam using nested PCR [31], and the progression of rice blast disease, caused by Magnaporthe oryzae, was quantified using real-time PCR [41]. The aim of this study was to develop a real-time PCR method for quantification of the PGPB H. seropedicae strain SmR1 in maize roots.
Materials and Methods
Bacterial Strains
For the primer specificity test, 12 bacterial strains were used: five different Herbaspirillum species (H. hiltneri, H. huttiense, H. lusitanum, H. rubrisubalbicans, and H. frisingense) and seven other bacterial species (A. brasilense, Bacillus cereus, Bacillus subtilis, E.coli, Rhizobium sp., Microbacterium sp., and Pseudomonas sp.). H. seropedicae strain SmR1 (strain Z78 ATCC 35893 SmR) and other Herbaspirillum strains used in this work were routinely grown in orbital shaker (120 rpm) at 30 °C in 30 mL NFbHPN medium supplemented with 5 mg/L malic acid [25]. Optical density (OD) of bacterial cell culture was measured at 600 nm using Hitachi U2910 Spectrophotometer. A correlation was obtained between OD and the number of colony forming unit (CFU) by plating serially diluted cell cultures in NFbHPN agar plates (Table S1). A. brasilense strain FP2 was grown under the same conditions, except that NFbHPN medium was supplemented with 5 mg/L sodium lactate. Rhizobium [13], Microbacterium, and Pseudomonas were grown in Luria–Bertani medium (USB) at 28 °C. E. coli (ATCC 25922) was grown in Brain–Heart Infusion broth (Himedia), while B. subtilis (ATCC 6051) and B. cereus (ATCC 14579) were grown in nutrient broth (Himedia) at 37 °C.
Germination, Inoculation, and Growth of Seedlings
Seeds of Zea mays (variety Dekalb240) were surface sterilized in laminar flow cabinet by washing three times with autoclaved ultrapure water, followed by 70 % ethanol for 3 min and shaken in 2 % sodium hypochlorite and 2.5 % Tween-20 for 30 min [34]. Seeds were then washed three times with autoclaved ultrapure water by gentle shaking, transferred to plates containing 0.8 % agar-water, and maintained for 3 days in growth chamber at a temperature of 25 °C, in the dark, for germination.
For inoculation of the seedlings, a bacterial culture, OD600 0.8 corresponding to ~108 CFU/mL, was diluted in NFb malate medium (1:1,000) [25]. After preparing the cell suspension, thirty germinated seedlings were incubated in 30 mL of ~105 bacterial cells/mL of H. seropedicae SmR1 in NFb malate medium without nitrogen source in an orbital shaker for 30 min at 30 °C and 80 rpm [5]. Control seedlings were Mock-inoculated under the same conditions. For seedlings grown in vitro, inoculated and control seedlings were washed in autoclaved 0.9 % saline buffer for 1 min and placed in the glass tubes containing plant medium solution [15]. Seedlings from each treatment were grown in a controlled-environmental chamber, placed side-by-side for 10 days (16 h photoperiod at 150 μmol/(m2 s), 25 °C light/23 °C dark, and 40 % humidity). For seedlings grown in pots, inoculated and control seedlings were washed with 0.9 % saline solution, and then transferred to pots containing sterilized sand with Hoagland’s solution [15] without nitrogen. They were maintained in a controlled-growth chamber adjusted to 12 h photoperiod, photosynthetic active radiation of 150 μmol/(m2 s), temperature of 25 °C, and watered daily. Hoagland’s solution without nitrogen was added over again 7 days after inoculation.
The results of in vitro and in pots experiments reported represent three independent experiments (biological replicates) performed in different periods of time. Maize seedlings from each treatment were randomly collected on the 1st, 4th, 7th, and 10th day after inoculation (DAI).
For seedlings grown in natural soil, inoculated and control seedlings were transferred to 2-L pots containing 1.5 kg natural soil previously supplemented with 100 mL nutrient solution [33] with reduced nitrogen (0.5 mM KNO3). Pots were placed in a greenhouse without temperature control and under natural light. Watering was performed everyday, and 100 mL of nutrient solution was added every two days. Maize seedlings were cultivated for 28 days when roots were collected and stored in -80 °C freezer until the DNA extraction.
DNA Extraction Protocols
Maize genomic DNA was extracted and purified from frozen roots using the DNeasy Plant Mini Kit (Qiagen) according to the manufacturer’s protocol, with a previous step of lysis treatment; samples were incubated for 15 min at 65 °C with a CTAB buffer (20 g/L CTAB, 1.4 M NaCl, 0.1 M Tris buffer, 20 mM Na2EDTA, and pH 8.0) instead of incubation with the AP1 buffer supplied in the kit. Thus, 20 μL of proteinase K (20 mg/mL) was added to each sample, and samples were incubated for 15 min at 65 °C [14]. For bacterial genomic DNA extraction, 2 mL medium aliquots containing 108 CFU/mL were centrifuged (6,000 g, 3 min, 4 °C), and pellets were stored at −80 °C until DNA extraction. Pellets were suspended in 100 μL of ultrapure water, frozen at −80 °C for 20 min, and immediately heated in boiling water (100 °C) for 10 min. Cell suspension was cooled to room temperature and centrifuged (13,000 g, 10 s), and supernatant was used for DNA extraction using Wizard® Genomic DNA purification kit (Promega™, Madison, WI, USA) according to the manufacturer’s protocol. DNA concentration was determined on a Thermo Scientific NanoDrop™ 2000 spectrophotometer (Wilmington, DE, USA) with measurements at 260 and 280 nm.
Primer Design
In order to select a specific DNA sequence for a H. seropedicae-specific qPCR assay, an extensive BLAST search was performed using the complete genome of the microorganism (GenBank accession number NC_014323.1 [30] ). Two regions, found as a single copy in the H. seropedicae genome, were selected and their specificity was confirmed by in silico alignment analyses: Bacteriophage Tail Fiber Protein (GenBank accession CP002039.1|216307-217431) and Outer Membrane Protein (Porin) (GenBank accession CP002039.1|194590-195762). Sets of primers were designed using Primer Express 3.0 (Applied Biosystems) and two primer pairs were selected, HERBAS1 and HERBAS2 (Table 1), each one targeting one specific sequence. Primers were synthesized by IDT (Coralville, IA, USA).
Real-Time PCR Quantification
In order to detect and quantify the presence of H. seropedicae SmR1 in inoculated maize roots, primers HERBAS1 and HERBAS2, which amplify fragments of 76 bp (theoretical T m = 85 °C) and 63 bp (theoretical T m = 82 °C), respectively, were tested by qPCR. Also, primer ZM1 (hmg), which amplifies a fragment of 79 bp, was used to quantify maize DNA in the tested samples [14]. Quantitative real-time PCR was performed in ABI PRISM 7500 Detection System (Applied Biosystems, Foster City, CA, USA). Amplification reactions were carried out in a final volume of 25 μL containing 12.5 μL of 2X SYBR Green Master Mix (Applied Biosystems), 100 nM of each primer (HERBAS1, HERBAS2, and ZM1), water and template DNA. The amplification protocol consisted of an initial incubation at 50 °C for 2 min, 95 °C incubation for 10 min, followed by 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. All real-time PCR runs were analyzed using automatic software settings. Maize inoculated samples were tested in triplicate and control samples in duplicate. All maize samples were tested by qPCR at a final concentration of 50 ng DNA per reaction.
Construction of Standard Curves
Standard curves were prepared with serial dilutions of genomic DNA isolated from H. seropedicae SmR1 and maize roots. The number of genome copies was calculated on the basis of the size of the H. seropedicae SmR1 (GenBank accession number NC_014323.1) [30] genome (5.51 Mbp) using the Avogadro’s constant (6.023 × 1023) and the molecular weight of DNA (660 Da/bp). Genomic DNA was tenfold serially diluted in ultrapure water to final concentrations ranging from 106 to 100 genome copies per 2 μL, equivalent to concentrations of 6.03 ng to 6.03 fg. Each standard curve run was analyzed in three different PCR runs in duplicate. The C t versus log CFU of H. seropedicae SmR1 was estimated using genomic DNA extracted from the bacteria culture. Ten times serial dilutions of DNA extracted from H. seropedicae SmR1, grown until stationary growth phase (OD 0.8) was performed, and the corresponding CFU was calculated based on plate counting. The maize copy number was calculated as described above considering the size of 2.3 Gbp [38] and one diploid genome, 2C value of 5.6 pg [19]. Ten-fold serial dilutions (56–0.0056 ng per reaction) of maize genomic DNA were used for the preparation of standard curves, ranging from 104 to 100 genome copies per reaction. Bacterial DNA standard curves were also prepared in the presence of a background DNA of maize roots (6 ng per reaction) isolated from control seedlings. Genomic DNA isolated from inoculated maize root was also tenfold serially diluted in ultrapure water to final concentrations equivalent to 106–100 bacterial genome copies per reaction. Standard curves for each DNA dilution were generated by the plot cycle threshold (C t) values versus logarithm of bacterial genome copy number. Amplification efficiencies were determined using the equation E = 10(−1/s) − 1, where E is the efficiency and s is the slope obtained from the standard curve.
Results
Primer Specificity
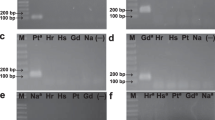
Primer pairs HERBAS1 and HERBAS2 were designed based on the genome of H. seropedicae strain SmR1 (Table 1). In silico, analyses using BLAST alignment tools showed no match for both primer pairs, except for the expected H. seropedicae sequences. Specificity tests were conducted using DNA extracted from H. seropedicae SmR1, other Herbaspirillum species, and other bacterial genera to verify if the primer pairs HERBAS1 and HERBAS2 were able to detect exclusively H. seropedicae SmR1. When qPCR was conducted using HERBAS2 primers, amplicons with similar Tm were observed for all Herbaspirillum species (Table 2). Because HERBAS2 presented positive detection with similar Tm for species other than H. seropedicae, it was not used for the quantification experiments.
Regarding qPCR using HERBAS1 primers and annealing temperature of 60 °C, DNA samples from H. seropedicae presented an amplicon with calculated Tm of 85 °C and observed Tm of 83.28 °C at a C t value of 13.37 (6 ng of template DNA). DNA samples from other Herbaspirillum species presented non-specific amplifications with different Tm values at higher C t values (Table 2, Figures S1, S2, and S3). DNA samples from the other bacterial genera presented amplifications at late C t values (C t of 25 for H. rubrisubalbicans and C t > 30 for other species). DNA from Bacillus cereus and Bacillus subtilis did not present amplification (Table 2). qPCR using HERBAS1 primers was also performed with high-annealing temperatures, 61 and 63 °C, and similar results were obtained (Table S2).
qPCR Reaction Parameters for H. seropedicae SmR1 Quantification
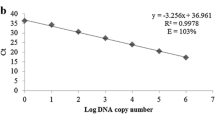
The reaction parameters (efficiency, slope, and correlation coefficient) of the qPCR assay using HERBAS1 and annealing temperature of 60 °C were determined based on standard curves obtained from tenfold serial dilution of three different extracts of bacterial DNA isolated from H. seropedicae SmR1. The reaction parameters were calculated by plotting the C t values against the log10 of the genome copy number (Fig. 1a) and log10 corresponding CFU of H. seropedicae SmR1 (Fig. 1b). Ten independent standard curves were determined for HERBAS1 (Table 3). The standard curves presented a suitable linear correlation (R 2 = 0.99) and a mean slope value of −3.54, corresponding to a PCR efficiency of 91 %. The reaction parameters (efficiency, slope, and correlation coefficient) of the qPCR assay using HERBAS1 and annealing temperature of 61 or 63 °C were also determined based on bacterial DNA serial dilution curves. For both conditions, the efficiency decreased to unacceptable values, lower than 90 % (Table S3); thus, all following qPCR runs were performed at annealing temperature of 60 °C.

qPCR standard curves for Herbaspirillum seropedicae quantification generated using 3 DNA extractions from Herbaspirillum seropedicae strain SmR1 as template DNA and HERBAS1 primers. a C t versus log DNA copy number. Average C t ± SD (n = 21). b C t versus log CFU (n = 6)
The limit of detection (LOD) for H. seropedicae SmR1 was 101 genome copies (corresponding to 60.3 fg), although 100 genome copies were detected in 18 out of 21 reactions (mean C t = 35.50). This quantity of one copy was not detected in all reactions, so it was not used for the construction of the standard curve.
Repeatability standard deviation (%RSDr) of the C t values from ten real-time PCR runs was calculated and all values presented below 10 %. The highest %RSDr value was 9.19 % for 106 genome copies, and the lowest was 5.34 % for 101 genome copies (Table 4). The relative repeatability standard deviation (%RSDr) of PCR run was determined for three DNA extracts (A, B, and C) of H. seropedicae SmR1 using mean C t of different PCR runs of the same DNA extract (Table 5), and the %RSDr values were below 10 %. In addition, the %RSDr of DNA extract was determined for three independent PCR runs (R1, R2, and R3) using mean C t of three different DNA extracts from H. seropedicae SmR1 of the same PCR run (Table 6), and %RSDr values were below 15 %.
In order to evaluate the effect of background maize DNA in qPCR quantification of H. seropedicae SmR1, amplification efficiencies were determined by the construction of standard curves of serial dilution of bacterial DNA in the presence of a constant amount of DNA extracted from maize roots (6 ng per well). In addition, a tenfold serially diluted standard curve of DNA extracted from inoculated maize roots was analyzed (Table 7). When amplification efficiencies for H. seropedicae detection were determined using DNA samples extracted from inoculated maize roots, values ranged from 108 to 118 % (Table 7). When serial dilutions of the 3 different bacterial DNA extracts were prepared in the presence of background maize DNA isolated from control maize root (non-inoculated), amplification efficiencies were, respectively, 95, 99, and 103 % (Table 7). In the presence of background maize DNA, the limit of detection of H. seropedicae DNA was near 100 copy (mean C t = 33.46). Using maize DNA as template, no amplification was detected.
qPCR Reaction Parameters for Maize Quantification
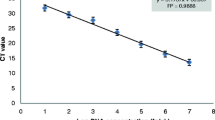
Reaction parameters for maize DNA quantification were determined using primers ZM1 [14] and standard curves prepared from tenfold serial dilutions ranging from 104 to 100 genome copies. Four DNA samples isolated from control maize root samples were used for construction of the standard curves. The reaction parameters (efficiency, slope, and correlation coefficient) were calculated by plotting the C t values against the log10 DNA copy number (Fig. 2). The obtained standard curve presented an efficiency of 98.67 % and a correlation coefficient of 0.99.

qPCR standard curve for Zea mays quantification generated using 4 DNA extractions from maize (Zea mays) as template DNA and ZM1 (hmg) primers. Average C t ± SD (n = 10)
Quantification of H. seropedicae DNA in Maize Root Samples
Maize germinated seeds were inoculated with H. seropedicae (105 bacterial cells per seed), and seedlings were harvest 1, 4, 7, and 10 DAI (maize seedlings grown in vitro) and 4, 7, and 10 DAI (maize seedlings grown in pots). DNA samples isolated from inoculated and non-inoculated maize roots were used as template in qPCR in order to quantify H. seropedicae DNA. The amount of H. seropedicae copy number was estimated using the mean standard curve previously calculated (Fig. 1a). Regarding DNA samples from inoculated maize roots, PCR was performed in triplicate and C t values ranged from 25.84 (1 DAI) to 13.94 (10 DAI). Melting curves of inoculated root samples were similar to melting curve of H. seropedicae (Figures S4 and S5). For the DNA samples isolated from control maize samples, PCR was performed in duplicate, and only late C t values (>35) were observed. These late C ts correspond to unspecific amplification under the established experimental conditions. For the in vitro experiment, quantification results showed that bacterial DNA copy number per gram of root fresh weight increased from 5.16 × 107 (1 DAI) to 1.42 × 109 (10 DAI) (Fig. 3). For the plants grown in pots, bacterial DNA copy number per gram of root fresh weight ranged from 3.25 × 106 (4 DAI) to 3.5 × 106 (10 DAI) (Fig. 4).

Bacterial DNA copy number/g of root (fresh weight) of maize seedlings grown in vitro after inoculation with Herbaspirillum seropedicae strain SmR1 DKB240 variety. Inoculated samples collected 1, 4, 7, and 10 DAI. Data are presented as mean ± SD (n = 9)

Bacterial DNA copy number/g of root (fresh weight) of maize seedlings grown in pots after inoculation with Herbaspirillum seropedicae strain SmR1 DKB240 variety. Inoculated samples collected 4, 7, and 10 days after inoculation (DAI). Data are presented as mean ± SD (n = 9)
Quantification of Maize DNA in Maize Root Samples
DNA samples isolated from inoculated and non-inoculated maize roots grown in vitro were used as template in qPCR reactions targeting hmg gene (primers ZM1) in order to quantify maize DNA. The amount of maize DNA copy number was estimated using the mean standard curve previously calculated (Fig. 2). Results for all samples showed a mean C t value of 22.51 ± 0.29, corresponding to an average value of 4000 diploid maize genome copies per reaction.
Ratio Between H. seropedicae SmR1 and Maize Copy Number
Using the copy number obtained by qPCR from bacterial DNA quantification and maize DNA quantification, the number of H. seropedicae genome copies in relation to the number of maize DNA copies was determined for plants grown in vitro. The results showed an increase in the number of H. seropedicae SmR1 DNA copies when compared to the number of maize DNA copies. Ratios ranged from 10 (1 DAI) to 275 (10 DAI), as shown in Fig. 5.

Ratio bacterial DNA copy number per maize DNA copy number of maize seedlings grown in vitro after inoculation with Herbaspirillum seropedicae strain SmR1 DKB240 variety. Inoculated samples collected 1, 4, 7, and 10 DAI. Data are presented as mean ± SD (n = 9)
Detection of H. seropedicae DNA in Maize Root Samples Cultivated in Natural Soil
For detection of H. seropedicae in inoculated maize roots cultivated in natural soil, samples were harvested 28 DAI. DNA samples were isolated from inoculated and control maize roots and used as template in real-time PCR. The inoculated root samples presented an amplicon with the same Tm of H. seropedicae DNA, whereas no amplification was detected for non-inoculated root samples (Figure S6).
Discussion
It has been shown that Herbaspirillum spp. are able of entering and rapidly colonizing roots, stems, and leaves of host plants, through cortical intercellular infection and invasion of the xylem vessels [10, 23, 27]. However, the mechanisms involving such interaction remain unclear, and studies attempting to contribute for the understanding of these mechanisms are necessary. Understanding the plant-bacterial associations is important for future biotechnological applications, such as using bacteria species as plant biofertilizers, and it could be an important tool to improve crop production, to conserve biodiversity, and to sustain agro-ecosystems [40]. In order to evaluate the plant–microbe interaction between maize and H. seropedicae SmR1, maize seedlings were inoculated with H. seropedicae SmR1 and analyzed 1, 4, 7, and 10 DAI.
Using the H. seropedicae strain SmR1 genome recently published [30], two primer pairs were designed (Table 1) for specific detection and quantification of H. seropedicae. In order to verify the specificity of the primers, a qPCR assay was performed using both primer pairs and DNA samples extracted from different bacterial species. Primer HERBAS1 presented amplification (late C t) for eleven analyzed DNA samples. However, ∆C t observed between H. seropedicae SmR1 (C t = 13.37), and the other bacteria (C t > 25.59) is sufficient to propose the use of this primer pair because the majority of these amplifications are non-specific and they are easily distinguishable by their Tm (Table 2) and melting curves (Figures S1 and S2). Comparing the C t values (Table 2) for different species of Herbaspirillum, C t observed with 6 ng of H. rubrisubalbicans corresponded to C t observed using 0.006 ng of H. seropedicae, and C t observed with 6 ng of H. huttiense corresponded to C t observed using 0.0006 ng of H. seropedicae; therefore, it is possible to quantify H. seropedicae DNA using this assay since the error is negligible. The primers designed in the present work can quantify 1,000 copies of H. seropedicae, and if there is the same amount of 1,000 copies of H. rubrisubalbicans, it will be detected at a C t corresponding to only one copy, so the error will be 1,001 copies instead of 1,000 copies. Moreover, the presence of unspecific amplification was tested in maize root samples cultivated in natural soil. Only the expected Tm of 83 °C for H. seropedicae was detected showing that C t values were not influenced by amplifications other than the H. seropedicae amplification (Figure S6).
In order to test the HERBAS1 primers for quantification of H. seropedicae DNA, the reaction parameters were determined and the repeatability of this assay was evaluated using tenfold serial dilutions of H. seropedicae SmR1 DNA. The standard curves presented suitable reaction parameters (Fig. 1a). A reliable standard curve should present a R 2 value higher than 0.95 and a slope between −3.9 and −3.0, corresponding to PCR efficiencies between 80 and 115 %, respectively [46]. On the other hand, for genetically modified organisms (GMO) analysis, the parameters of the standard curve should be more restrictive, presenting a R2 higher than 0.98 and a slope between −3.6 and −3.1, corresponding to PCR efficiency between 90 and 110 % [11]. The parameters obtained in the present work (R 2 = 0.99 and E = 91 %) fit in the restrictive parameters required for the GMO analysis resulting in an accurate and reliable detection and quantification of H. seropedicae.
According to the European Union Reference Laboratory (EURL) (http://gmo-crl.jrc.ec.europa.eu/) requirements for GMO quantification by qPCR, the repeatability of qPCR expressed as RSDr should be less than 25 %. The results obtained in the present study show that both PCR run (Table 5) and DNA extraction (Table 6) presented low variation, indicating a suitable degree of accuracy of the assay. RSDr values were lower within PCR runs than within DNA extraction, indicating that variation of the DNA extraction method is higher than the variation of the PCR run. Nevertheless, RSDr values obtained for both steps of the analysis are lower than the 25 % suggested by EURL. Also, according to the EURL, the sensitivity of a qPCR quantification method can be determined through the limit of detection (LOD). LOD is the lowest amount of sample that can be reliably detected. The qPCR assay developed in this work ensured the reliably detection of amounts of H. seropedicae SmR1 DNA ranging from 106 genome copies (6.03 ng) to 101 genome copies (60.3 fg), showing the high sensitivity of this method (Table 4). Similar results were obtained in previous qPCR studies. A LOD of 1 pg [43] was detected in the quantification of Paenibacillus polymyxa in the rizhosphere of wild barley. DNA quantification of Xanthomonas oryzae pv. oryzicola presented a LOD of 50 fg [24], corresponding to approximately 101 genome copies, an amount close to the DNA amount detected in the present assay.
PCR amplification of DNA samples extracted from plant tissues or other sources can influence the reliability of the quantification [9]. Furthermore, the presence of large amounts of background DNA could also have some effect on the target DNA amplification [1]. The results obtained for the standard curve constructed with mixed DNA showed that maize background DNA did not seem to have a negative effect on bacterial DNA amplification and efficiency, once the amplification mean efficiency was 99 % in the presence of maize DNA (Table 7). Meanwhile, the amplification efficiency of the standard curve prepared with DNA isolated from inoculated maize roots was 113 % (Table 7). Such differences in efficiency values in the presence of background DNA could be due to compound or structural conformation that makes template DNA copies inaccessible for the polymerase enzyme during the first PCR cycles. Gradual changes in these conformations make template copies more and more accessible in the following cycles [9].
Plant growth-promoting bacteria (PGPB) are becoming an important tool to support plant nutrition in field conditions. Consequently, a fast method to monitor the presence of these bacteria in plant tissues is necessary. In order to detect and quantify the presence of H. seropedicae SmR1, qPCR experiments were performed using primer pair HERBAS1. The results of the quantification experiments obtained in this study showed an increase in the genome copy number of H. seropedicae in inoculated maize roots along the period of evaluation (1, 4, 7, and 10 DAI) for seedlings cultivated in vitro. It was observed that H. seropedicae SmR1 genome copy number in maize root was 5.1 × 107 DNA copy number/g root fresh weight one DAI, increasing to 1.4 × 109 DNA copy number/g root fresh weight ten DAI (Fig. 3). This increase becomes clear when the bacterial genome copy number is estimated in relation to the maize DNA copy number (Fig. 5). These data corroborate the conclusions obtained by Monteiro et al. [29], which report that H. seropedicae colonization and invasion occurs very quickly. This rapid pattern of H. seropedicae colonization was also reported in other studies for different crop cultures such as rice, sugarcane, and common bean [21, 22, 35, 37].
The sequence targeted by HERBAS1 primer pair occurs as a single copy in the H. seropedicae genome, so we can estimate that each H. seropedicae genome copy corresponds to approximately one cell (Fig. 1b). In this sense, the estimated bacterial cell number in root samples of plants grown in vitro (107–109) and in root samples of plants grown in pots (around 106) are in similar amounts presented by other studies reporting DNA quantification of other PGPB species. Quantification of Enterobacter radicincitans in Brassica oleracea plants was 108 bacterial cells/g root fresh weight [36], and quantification of A. lipoferum CRT1 by qPCR in maize roots was 104–106 CFU equivalents/g of root dry weight [12].
In conclusion, the present study describe a suitable, reliable, and accurate qPCR assay for the detection and quantification of Herbaspirillum seropedicae SmR1 in maize inoculated roots, using primers HERBAS1. This assay presented high specificity to the target and it can be useful for monitoring the colonization of the PGPB H. seropedicae SmR1 in plant roots.
References
Andersen, C. B., Holst-Jensen, A., Berdal, K. G., Thorstensen, T., & Tengs, T. (2006). Equal performance of TaqMan, MGB, molecular beacon, and SYBR green-based detection assays in detection and quantification of roundup ready soybean. Journal of Agricultural and Food Chemistry, 54(26), 9658–9663. doi:10.1021/jf061987c.
Bal, H. B., Nayak, L., Das, S., & Adhya, T. K. (2013). Isolation of ACC deaminase producing PGPR from rice rhizosphere and evaluating their plant growth promoting activity under salt stress. Plant and Soil, 366(1–2), 93–105. doi:10.1007/s11104-012-1402-5.
Baldani, J., Baldani, V., Seldin, L., & Dobereiner, J. (1986). Characterization of Herbaspirillum seropedicae Gen-Nov, Sp-Nov, a root-associated nitrogen-fixing bacterium. International Journal of Systematic Bacteriology, 36(1), 86–93.
Baldani, J. I., Pot, B., Kirchhof, G., Falsen, E., Baldani, V. L. D., Olivares, F. L., et al. (1996). Emended description of Herbaspirillum; Inclusion of Pseudomonas rubrisubalbicans, a mild plant pathogen, as Herbaspirillum rubrisubalbicans comb nov; and classification of a group of clinical isolates (EF group 1) as Herbaspirillum species 3. International Journal of Systematic Bacteriology, 46(3), 802–810.
Balsanelli, E., Serrato, R. V., de Baura, V. A., Sassaki, G., Yates, M. G., Rigo, L. U., et al. (2010). Herbaspirillum seropedicae rfbB and rfbC genes are required for maize colonization. Environmental Microbiology, 12(8), 2233–2244. doi:10.1111/j.1462-2920.2010.02187.x.
Bastian, F., Cohen, A., Piccoli, P., Luna, V., Baraldi, R., & Bottini, R. (1998). Production of indole-3-acetic acid and gibberellins A(1) and A(3) by Acetobacter diazotrophicus and Herbaspirillum seropedicae in chemically defined culture media. Plant Growth Regulation, 24(1), 7–11. doi:10.1023/A:1005964031159.
Baudoin, E., Couillerot, O., Spaepen, S., Moenne-Loccoz, Y., & Nazaret, S. (2010). Applicability of the 16S-23S rDNA internal spacer for PCR detection of the phytostimulatory PGPR inoculant Azospirillum lipoferum CRT1 in field soil. Journal of Applied Microbiology, 108(1), 25–38. doi:10.1111/j.1365-2672.2009.04393.x.
Brusamarello-Santos, L. C. C., Pacheco, F., Aljanabi, S. M. M., Monteiro, R. A., Cruz, L. M., Baura, V. A., et al. (2012). Differential gene expression of rice roots inoculated with the diazotroph Herbaspirillum seropedicae. Plant and Soil, 356(1–2), 113–125. doi:10.1007/s11104-011-1044-z.
Cankar, K., Stebih, D., Dreo, T., Zel, J., & Gruden, K. (2006). Critical points of DNA quantification by real-time PCR: Effects of DNA extraction method and sample matrix on quantification of genetically modified organisms. BMC Biotechnology, 6, 37. doi:10.1186/1472-6750-6-37.
Chubatsu, L. S., Monteiro, R. A., de Souza, E. M., Schuler de Oliveira, M. A., Yates, M. G., Wassem, R., et al. (2012). Nitrogen fixation control in Herbaspirillum seropedicae. Plant and Soil, 356(1–2), 197–207. doi:10.1007/s11104-011-0819-6.
Costa, J., Mafra, I., Kuchta, T., & Oliveira, M. B. P. P. (2012). Single-tube nested real-time PCR as a new highly sensitive approach to trace hazelnut. Journal of Agricultural and Food Chemistry, 60(33), 8103–8110. doi:10.1021/jf302898z.
Couillerot, O., Bouffaud, M.-L., Baudoin, E., Muller, D., Caballero-Mellado, J., & Moenne-Loccoz, Y. (2010). Development of a real-time PCR method to quantify the PGPR strain Azospirillum lipoferum CRT1 on maize seedlings. Soil Biology & Biochemistry, 42(12), 2298–2305. doi:10.1016/j.soilbio.2010.09.003.
de Souza, R., Beneduzi, A., Ambrosini, A., da Costa, P. B., Meyer, J., Vargas, L. K., et al. (2013). The effect of plant growth-promoting rhizobacteria on the growth of rice (Oryza sativa L.) cropped in southern Brazilian fields. Plant and Soil, 366(1–2), 585–603. doi:10.1007/s11104-012-1430-1.
Dinon, A., Prins, T., van Dijk, J., Arisi, A., Scholtens, I., & Kok, E. (2011). Development and validation of real-time PCR screening methods for detection of cry1A.105 and cry2Ab2 genes in genetically modified organisms. Analytical and Bioanalytical Chemistry, 400(5), 1433–1442. doi:10.1007/s00216-011-4875-9.
Egener, T., Hurek, T., & Reinhold-Hurek, B. (1999). Endophytic expression of nif genes of Azoarcus sp strain BH72 in rice roots. Molecular Plant–Microbe Interactions, 12(9), 813–819. doi:10.1094/mpmi.1999.12.9.813.
Elbeltagy, A., Nishioka, K., Sato, T., Suzuki, H., Ye, B., Hamada, T., et al. (2001). Endophytic colonization and in planta nitrogen fixation by a Herbaspirillum sp. isolated from wild rice species. Applied and Environment Microbiology, 67(11), 5285–5293. doi:10.1128/AEM.67.11.5285-. 5293.2001.
Faleiro, A., Pereira, T., Espindula, E., Brod, F., & Arisi, A. (2013). Real time PCR detection targeting nifA gene of plant growth promoting bacteria Azospirillum brasilense strain FP2 in maize roots. Symbiosis, 61(3), 125–133. doi:10.1007/s13199-013-0262-y.
Fan, B., Chen, X. H., Budiharjo, A., Bleiss, W., Vater, J., & Borriss, R. (2011). Efficient colonization of plant roots by the plant growth promoting bacterium Bacillus amyloliquefaciens FZB42, engineered to express green fluorescent protein. Journal of Biotechnology, 151(4), 303–311. doi:10.1016/j.jbiotec.2010.12.022.
Gaut, B. S., d’Ennequin, M. L., Peek, A. S., & Sawkins, M. C. (2000). Maize as a model for the evolution of plant nuclear genomes. Proceedings of the National Academy of Sciences of the United States of America, 97(13), 7008–7015. doi:10.1073/pnas.97.13.7008.
Gyaneshwar, P., James, E. K., Reddy, P. M., & Ladha, J. K. (2002). Herbaspirillum colonization increases growth and nitrogen accumulation in aluminium-tolerant rice varieties. New Phytologist, 154(1), 131–145. doi:10.1046/j.1469-8137.2002.00371.x.
James, E. K., Gyaneshwar, P., Mathan, N., Barraquio, Q. L., Reddy, P. M., Iannetta, P. P. M., et al. (2002). Infection and colonization of rice seedlings by the plant growth-promoting bacterium Herbaspirillum seropedicae Z67. Molecular Plant–Microbe Interactions, 15(9), 894–906. doi:10.1094/mpmi.2002.15.9.894.
James, E. K., & Olivares, F. L. (1998). Infection and colonization of sugar cane and other graminaceous plants by endophytic diazotrophs. Critical Reviews in Plant Sciences, 17(1), 77–119. doi:10.1016/s0735-2689(98)00357-8.
James, E. K., Olivares, F. L., Baldani, J. I., & Dobereiner, J. (1997). Herbaspirillum, an endophytic diazotroph colonizing vascular tissue in leaves of Sorghum bicolor L Moench. Journal of Experimental Botany, 48(308), 785–797. doi:10.1093/jxb/48.3.785.
Kang, M., Kim, M., Hwang, D., Cho, M., Seol, Y., Hahn, J., et al. (2012). Quantitative in planta PCR assay for specific detection of Xanthomonas oryzae pv. oryzicola using putative membrane protein based primer set. Crop Protection, 40, 22–27. doi:10.1016/j.cropro.2012.04.014.
Klassen, G., Pedrosa, F. O., Souza, E. M., Funayama, S., & Rigo, L. U. (1997). Effect of nitrogen compounds on nitrogenase activity in Herbaspirillum seropedicae SMR1. Canadian Journal of Microbiology, 43(9), 887–891.
Liu, F. C., Xing, S. J., Ma, H. L., Du, Z. Y., & Ma, B. Y. (2013). Plant growth-promoting rhizobacteria affect the growth and nutrient uptake of Fraxinus americana container seedlings. Applied Microbiology and Biotechnology, 97(10), 4617–4625. doi:10.1007/s00253-012-4255-1.
Monteiro, R. A., Balsanelli, E., Tuleski, T., Faoro, H., Cruz, L. M., Wassem, R., et al. (2012). Genomic comparison of the endophyte Herbaspirillum seropedicae SmR1 and the phytopathogen Herbaspirillum rubrisubalbicans M1 by suppressive subtractive hybridization and partial genome sequencing. FEMS Microbiology Ecology, 80(2), 441–451. doi:10.1111/j.1574-6941.2012.01309.x.
Monteiro, R. A., Balsanelli, E., Wassem, R., Marin, A. M., Brusamarello-Santos, L. C. C., Schmidt, M. A., et al. (2012). Herbaspirillum-plant interactions: microscopical, histological and molecular aspects. Plant and Soil, 356(1–2), 175–196. doi:10.1007/s11104-012-1125-7.
Monteiro, R. A., Schmidt, M. A., de Baura, V. A., Balsanelli, E., Wassem, R., Yates, M. G., et al. (2008). Early colonization pattern of maize (Zea mays L. Poales, Poaceae) roots by Herbaspirillum seropedicae (Burkholderiales, Oxalobacteraceae). Genetics and Molecular Biology, 31(4), 932–937. doi:10.1590/s1415-47572008005000007.
Pedrosa, F. O., Monteiro, R. A., Wassem, R., Cruz, L. M., Ayub, R. A., Colauto, N. B., et al. (2011). Genome of Herbaspirillum seropedicae strain SmR1, a specialized diazotrophic endophyte of tropical grasses. Plos Genetics, 7(5), e1002064. doi:10.1371/journal.pgen.1002064.
Raj, M., Jeeva, M. L., Hegde, V., Vidyadharan, P., Archana, P. V., Senthil alias Sankar, M., et al. (2012). Polymerase chain reaction assay for rapid, sensitive detection, and identification of Colletotrichum gloeosporioides causing greater yam anthracnose. Molecular Biotechnology, 52(3), 277–284. doi:10.1007/s12033-012-9496-9.
Rashid, S., Charles, T. C., & Glick, B. R. (2012). Isolation and characterization of new plant growth-promoting bacterial endophytes. Applied Soil Ecology, 61, 217–224. doi:10.1016/j.apsoi1.2011.09.011.
Rodriguez-Salazar, J., Suarez, R., Caballero-Mellado, J., & Iturriaga, G. (2009). Trehalose accumulation in Azospirillum brasilense improves drought tolerance and biomass in maize plants. FEMS Microbiology Letters, 296(1), 52–59. doi:10.1111/j.1574-6968.2009.01614.x.
Roncato-Maccari, L. D. B., Ramos, H. J. O., Pedrosa, F. O., Alquini, Y., Chubatsu, L. S., Yates, M. G., et al. (2003). Endophytic Herbaspirillum seropedicae expresses nif genes in gramineous plants. FEMS Microbiology Ecology, 45(1), 39–47. doi:10.1016/s0168-6496(03)00108-9.
Roncato-Maccari, L. D. B., Ramos, H. J. O., Pedrosa, F. O., Alquini, Y., Chubatsu, L. S., Yates, M. G., et al. (2003). Root colonization, systemic spreading and contribution of Herbaspirillum seropedicae to growth of rice seedling. Symbiosis, 35(1–3), 261–270.
Ruppel, S., Ruhlmann, J., & Merbach, W. (2006). Quantification and localization of bacteria in plant tissues using quantitative real-time PCR and online emission fingerprinting. Plant and Soil, 286(1–2), 21–35. doi:10.1007/s11104-006-9023-5.
Schmidt, M., Souza, E., Baura, V., Wassem, R., Yates, M., Pedrosa, F., et al. (2011). Evidence for the endophytic colonization of Phaseolus vulgaris (common bean) roots by the diazotroph Herbaspirillum seropedicae. Brazilian Journal of Medical and Biological Research, 44(3), 182–185. doi:10.1590/S0100-879X2011007500004.
Schnable, P. S. (2012). The B73 maize genome: Complexity, diversity, and dynamics (November, pg 1112, 2009). Science, 337(6098), 1040.
Shime-Hattori, A., Kobayashi, S., Ikeda, S., Asano, R., Shime, H., & Shinano, T. (2011). A rapid and simple PCR method for identifying isolates of the genus Azospirillum within populations of rhizosphere bacteria. Journal of Applied Microbiology, 111(4), 915–924. doi:10.1111/j.1365-2672.2011.05115.x.
Stets, M. I., Pinto, A. S, Jr, Huergo, L. F., de Souza, E. M., Guimaraes, V. F., Alves, A. C., et al. (2013). Rapid identification of bacterial isolates from wheat roots by high resolution whole cell MALDI-TOF MS analysis. Journal of Biotechnology, 165(3–4), 167–174. doi:10.1016/j.jbiotec.2013.04.001.
Su’udi, M., Kim, J., Park, J.-M., Bae, S.-C., Kim, D., Kim, Y.-H., et al. (2013). Quantification of rice blast disease progressions through Taqman real-time PCR. Molecular Biotechnology, 55(1), 43–48. doi:10.1007/s12033-012-9632-6.
Taule, C., Mareque, C., Barlocco, C., Hackembruch, F., Reis, V. M., Sicardi, M., et al. (2012). The contribution of nitrogen fixation to sugarcane (Saccharum officinarum L.), and the identification and characterization of part of the associated diazotrophic bacterial community. Plant and Soil, 356(1–2), 35–49. doi:10.1007/s11104-011-1023-4.
Timmusk, S., Paalme, V., Lagercrantz, U., & Nevo, E. (2009). Detection and quantification of Paenibacillus polymyxa in the rhizosphere of wild barley (Hordeum spontaneum) with real-time PCR. Journal of Applied Microbiology, 107(3), 736–745. doi:10.1111/j.1365-2672.2009.04265.x.
Verma, S. C., Ladha, J. K., & Tripathi, A. K. (2001). Evaluation of plant growth promoting and colonization ability of endophytic diazotrophs from deep water rice. Journal of Biotechnology, 91(2–3), 127–141. doi:10.1016/s0168-1656(01)00333-9.
Videira, S. S., de Oliveira, D. M., de Morais, R. F., Borges, W. L., Divan Baldani, V. L., & Baldani, J. I. (2012). Genetic diversity and plant growth promoting traits of diazotrophic bacteria isolated from two Pennisetum purpureum Schum. genotypes grown in the field. Plant and Soil, 356(1–2), 51–66. doi:10.1007/s11104-011-1082-6.
Zhang, T., & Fang, H. H. (2006). Applications of real-time polymerase chain reaction for quantification of microorganisms in environmental samples. Applied Microbiology and Biotechnology, 70(3), 281–289. doi:10.1007/s00253-006-0333-6.
Acknowledgments
We would like to express our gratitude to Fábio de Oliveira Pedrosa, Leda Chubatsu, and Michelle Tadra-Sfeir, Universidade Federal do Paraná, for providing A. brasilense strain FP2, H. seropedicae strain SmR1, and other Herbaspirillum, to Luciane Passaglia for providing Rhizobium, Microbacterium, and Pseudomonas. This work was financially supported by the National Institute of Science and Technology—Biological Nitrogen Fixation, INCT-FBN, CNPq, Ministry of Science and Technology, Brazil. TPP, FPA, and PD were recipients of Master and PhD fellowships from CAPES, Ministry of Education, Brazil. FCAB is recipient of post-doctoral fellowship from CAPES PNPD, and ACMA is recipient of research fellowship from CNPq (PQ-2).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Pereira, T.P., do Amaral, F.P., Dall’Asta, P. et al. Real-Time PCR Quantification of the Plant Growth Promoting Bacteria Herbaspirillum seropedicae Strain SmR1 in Maize Roots. Mol Biotechnol 56, 660–670 (2014). https://doi.org/10.1007/s12033-014-9742-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-014-9742-4