
Abstract
Endophytic diazotrophic plant growth-promoting bacteria Herbaspirillum rubrisubalbicans (HCC103), Herbaspirillum seropedicae (HRC54), Paraburkholderia tropica (Ppe8T), Gluconacetobacter diazotrophicus (Pal5T), and Nitrospirillum amazonense (CBAmC) have been used as inoculants for sugarcane. The genome sequences of these strains were used to design a set of specific primers for the real-time PCR (qPCR) assay. Primer specificity was confirmed by conventional PCR using the genomic DNAs of 25 related bacterial species and the five target strains. The qPCR assays were conducted using root and shoot samples from two sugarcane varieties (RB867515 and RB92579). These samples were collected both with and without inoculation, using the target strains specified in this study. The sugarcane plants were grown in a greenhouse, utilizing a substrate composed of sterile sand and vermiculite in a 2:1 ratio, for a duration of 55 days. The primers designed for this study successfully amplified target DNA fragments from each of the bacterial species, enabling their differentiation at the species level. The total bacterial population present in the sugarcane quantified using qPCR was on average 105.2 cells g−1 of fresh tissue. Across both evaluated varieties, it was observed that the population of inoculated bacteria tended to decrease over time and became more concentrated in the sugarcane roots compared to the aerial parts. The qPCR results suggest that both the host and the microbes influence the endophytic population and the bacterial number decreases with plant age.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sugarcane (Saccharum officinarum) is the main energy crop in Brazil and is used to produce sugar and ethanol. Bagasse, dry fibrous material that remains after crushing sugarcane stalks, is also used to generate electricity. Brazil continues to be the world's largest producer of sugarcane, with an estimated yield of approximately 642.7 million tons, from a cultivated area of 8,481 thousand hectares, with an average yield of 75,783 kg ha−1 in the year 2019–20 [1]. However, the average national yield of sugarcane corresponds to only 20% of the potential productivity (380 Mg ha−1 year−1) of Brazil [2]. The low productivity of Brazilian sugarcane is related to several edaphoclimatic factors, including management practices such as lack of irrigation and use of nitrogen fertilizers. Some of the nitrogen requirements of sugarcane crops can be met using inoculants with selected diazotrophic plant growth-promoting bacteria (DPGPB) [3,4,5], thereby promoting an increasing interest in developing potential biotechnological applications using endophytes for the sustainable production of sugarcane for biomass and biofuel generation [6, 7].
Based on the results of Oliveira et al. [3,4,5,6,7,8] a sugarcane inoculant was developed comprising a mixture of five bacterial species isolated from different plant tissues: Herbaspirillum rubrisubalbicans (HCC103), Herbaspirillum seropedicae (HRC54), Paraburkholderia tropica (Ppe8T), Gluconacetobacter diazotrophicus (Pal5T), and Nitrospirillum amazonense (CBAmC). The use of this inoculant in sugarcane plants does not fully replace the need for additional nitrogen fertilizers, but may provide up to 30% of the nitrogen accumulated by plant tissues depending on the soil type, nitrogen fertilizer dose, and plant variety [9,10,11]. In addition to supplying nitrogen, DPGPB also helps with the supply of phosphorus (P), potassium (K), and sulfur (S), which are macronutrients, and iron (Fe) and manganese (Mn), which are micronutrients [12]. DPGPBs also promote plant growth by synthesizing phytohormones, such as indole-3-acetic acid (IAA), cytokinins, and gibberellins, and regulating internal plant hormone levels, facilitating water access, increasing resistance to drought stress [13,14,15], and affecting the industrial characteristics of sugarcane [16].
Since its conception, several research groups have tested the efficacy of sugarcane inoculants [5, 17,18,19,20]. However, the lack of information concerning the number of inoculated bacteria and the “natural” diazotrophicus bacteria population makes it difficult to establish the beneficial effects of the inoculants. One way to show the benefits of inoculation is to ensure that the bacteria in the inoculant are associated with sugarcane tissue. Therefore, a specific methodology should be able to quantify and distinguish between inoculated bacteria and “natural” bacterial populations. Additionally, it is essential that this methodology can monitor the inoculant bacterial population and provide a quick and accurate diagnostic method.
Currently, several culture-dependent, microscopic, and molecular methods have been developed to monitor bacteria in the soil and plants [21,22,23]. These include the use of reporter genes, immunoassays, and nucleic acids [24]. However, most of these methodologies have low precision, are time-intensive and laborious, and quantify microorganisms that are not of interest [25,26,27]. To ensure that DPGPB of the inoculant is present in the plant for a long period, detection and quantification techniques must be sensitive, specific, and reproducible. Quantitative real-time PCR (qPCR) possesses all these features and is considered the most robust diagnostic microbial assay, especially for samples with a complex mix of organisms [24]. The qPCR is a well-established method for detecting and quantifying different organisms related to growth promotion in plants or the pathology such as nematode, and fungi [28] bacteria, and viruses [29]. This technique has been successfully used to quantify several bacteria associated with plants, including Azospirillum lipoferum (CRT1) [30] and Azospirillum brasilense (FP2) in maize [31, 32] and wheat [33], endophytic diazotrophic bacteria H. seropedicae (SmR1) in maize roots [34], and G. diazotrophicus in sugarcane [35]. Despite the ubiquity of qPCR, it is essential to design and use specific PCR primers that target particular microbes. Well-designed primers play a critical role in ensuring the specificity, sensitivity, and robustness of a PCR assay. [36,37,38]. Although the design of strain-specific primers for closely related microbes poses a challenge, the recent increase in the availability of many sequenced microbial genomes has made this task more feasible. Thus, the present study aimed to design specific primers and establish ideal reaction conditions for qPCR for the detection and quantification of the endophytic DPGPB that constitute the sugarcane inoculant.
Materials and methods
Bacteria strains and growth conditions
The bacteria H. rubrisubalbicans (strain HCC103), H. seropedicae (strain HRC54), Paraburkholderia tropica (strain Ppe8T), G. diazotrophicus (strain Pal5T), and N. amazonense (strain CBAmC) used as inoculants for sugarcane were grown in modified DYGS liquid medium [39] containing (g L−1): glucose 2.0, malic acid 2.0; peptone 1.5; yeast extract 2.0, K2HPO4 0.5, MgSO4.7H2O 0.5, and glutamic acid 1.5, with the final pH adjusted to 6.0. The bacteria were cultivated in a 250 mL Erlenmeyer, in a final volume of 100 mL, incubated in biological triplicates under aeration on a stirring table at 200 rpm, 30 °C for 24 h, except for the strain Pal5T, which was grown for up to 36 h under the same conditions of rotation and temperature. The optical density (OD) at 600 nm of each bacterial suspension was determined in triplicate and then serially diluted (dilution factor = 10). Dilutions were spread on solid modified DYGS medium (agar 15 g L−1) and incubated for 72 h at 30 °C; next, the bacteria in the plates were counted. Aliquots containing 1 mL of the bacterial suspension were used for the extraction of genomic DNA. The genomic DNA of another 25 strains (Table 1) was obtained from the Johanna Döbereiner Biological Resource Center (CRB-JD) Embrapa Agrobiologia and was used to evaluate primer specificity.
Plant and bacterial DNA extraction
The extraction of total DNA from plant tissues (root and basal stalks) and bacteria were conducted using the cetyltrimethylammonium bromide (CTAB) buffer [48] modified with the addition of 5 mM ascorbic acid and 4 mM diethyldithiocarbamate sodium (DIECA), 4% polyvinylpyrrolidone (PVP) 40 in substitution with polyvinylpolypyrrolidone (PVPP), and withdrawal of β-mercaptoethanol. For total DNA extraction from the roots, the buffer was supplemented with 5% of β-mercaptoethanol (v:v), proteinase K (1 mg mL−1), lysozyme (25 mg mL−1), 20% sodium dodecyl sulfate (SDS) (w:v), and phenol. Approximately 300 mg of powdered plant tissue was incubated for 45 min at 65 °C with 800 µL of preheated CTAB buffer at 65 °C. In the case of bacteria, a pellet of 1 mL of bacteria grown in DYGS culture medium was incubated for 5 min at 65 °C with 750 μL of CTAB preheated at 65 °C and homogenized twice during the incubation period. The samples of plant tissues and bacterial pellets were then left on the lab bench until they reached room temperature. Subsequently, 5 μL of RNase (20 mg mL-1) and 5 μL of proteinase K (1 mg mL-1) were added to the solution, which was homogenized using a vortex shaker and then incubated at 37 °C for 30 min. Next, 700 μL of chloroform-isoamyl alcohol (24:1, v:v) was added, and after homogenization, the mixture was centrifuged at maximum rotation (16,000 × g) for 10 min. The supernatant was collected and 70% of isopropanol corresponding to the volume recovered was added. The samples were gently inverted 10 times, placed on ice for 15 min, and centrifuged at 10,000 rpm for 5 min. The supernatant was discarded and 500 μL of 70% ethanol was added to the pellet, followed by centrifugation at 10,000 rpm for 5 min. The supernatant was discarded, and the pellet was dried in an exhaust hood under air flow for 2 h. The DNA, thus obtained, was rehydrated with 50 μL Tris-ethylene diamine tetraacetic acid (EDTA) (TE) buffer [10 mM Tris, 1 mM EDTA, pH 8.0]. The yield of extracted DNA (ng μL-1) and its purity (accessed by the ratio A260 nm/A280 nm) were determined using a NanoDrop® 2000 spectrophotometer.
Genomic typing of bacterial strains
BOX-PCR was performed using DNA from the five target strains and 25 taxonomically related strains (Table 1). The PCR reactions were prepared using the primer BOX A1R (5'-CTACGGCAAGGCGACGCTGACG-3') [49] and the following reagents: 2.5 µL of buffer 10 x; 1.5 µL MgCl2 50 mM; 0.75 µL dNTP 10 mM; 2.5 µL of primer 10,000 nM; 0.25 µL of Taq polymerase enzyme 5 U; 2.0 µL of 2% Tween 20; 14.25 µL of water for PCR and 1 µL of bacterial DNA (10 ng µL−1). The conditions used for amplification were initial denaturation at 95 °C for 5 min, followed by 35 cycles (94 °C for 1 min, annealing temperature, 53 °C for 1 min, then 65 °C for 8 min, and final extension at 65 °C for 16 min). The product of BOX-PCR (13 μL) was used for electrophoresis (70 V 300 min−1) on a 2% agarose gel (w:v), stained with ethidium bromide, and visualized on Kodak Logic 100 KODAK® photo documentation. The 1 kb Plus Ladder marker (Invitrogen™, USA) was used as a molecular weight marker. The pattern of bands observed in the gel was used to construct a dendrogram to evaluate the degree of similarity between the different strains using the program BioNumerics v.7.6 (Applied Maths, Belgium). The similarity matrices used for dendrogram construction were calculated using the Jaccard correlation coefficient and grouping of similarity matrices by the unweighted pair group method with arithmetic mean (UPGMA) algorithm and optimization levels and tolerances adjusted to 1.0.
Primer design
To design specific primers for the Pal5T, Ppe8T, and CBAmc strains, we used the genome sequences available in the GenBank database under the accession numbers NC_011365.1 [50], MSDZ00000000.1 [51], and NZ_CP022110.1 [52], respectively. For the HRC54 strain, primers were designed based on the sequence of the M15 family metallopeptidase gene (accession number ACP92_01130/WP_013232277.1). The genomic sequence of the HCC103 strain, which was used for primer design, was extracted from the Genome Assembly and Annotation Tool (GAAT) database at Embrapa Agrobiology [53] and is available upon request. The genomic sequences of Pal5T, Ppe8T, CBAmC, and HCC103 were fragmented in silico into contigs of 500 bp and were subjected to a similarity analysis against sequences from other microorganisms available on the National Center for Biotechnology Information (NCBI) using the BLAST program (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Contigs with no similarity to any other bacteria were selected for primer design. None of the primers designed from random 500 bp contigs of the HRC54 genome proved to be specific. Therefore, in order to design primers specific to the HRC54 strain, we had to identify genes that are unique to the Herbaspirillum seropedicae species. The primers were designed using the Primer3plus program [54] and Oligo Explorer v.1.0 (http://www.softpedia.com/get/Science-CAD/Oligo-Explorer.shtml). Care was taken to avoid the formation of dimers and hairpin structures in the primer sequences. The following conditions were used: i) length between 19 and 21 nucleotides, (ii) GC content between 40 and 60%, iii) amplicons with sizes less than 200 bp, and iii) melting temperature close to 60 °C with differences between the primers (ΔTm) < 2 °C. The designed primers were prefixed with Hr103, Hs54, Pt8, Gd5, and NaCb to represent HCC103, HRC54, Ppe8T, Pal5T, and CBAmc strains, respectively. Initially, all 39 primer pairs were assessed in conventional PCR to confirm their specificity for each respective strain.
Primer test using conventional PCR
The designed primers were initially used for conventional PCR (cPCR) to verify the specificity of each primer pair. cPCR reactions were performed at a final volume of 25 μL with the following regents: 2.5 μL of 10 × buffer, 2.25 μL of MgCl2 50 mM, 0.5 μL of dNTP 10 mM, 1.0 μL of each primer (10 nM), Taq 0.2 μL of polymerase 5 U; 1 μL of bacterial DNA (10 ng μL−1), and water 17.55 μL. The amplification conditions were as follows: initial denaturation at 95 °C for 5 min, followed by 35 cycles of denaturation at 95 °C for 15 s, annealing at the specific temperature and time (refer to Table 2), extension at 72 °C for 30 s, and a final extension step at 72 °C for 5 min. The PCR product was subjected to agarose gel electrophoresis 2% (w:v), stained with ethidium bromide, and photo documented.
Real time PCR quantification
qPCR reactions were performed on a 7500 Fast Real-Time PCR System (Applied Biosystems, USA, Cat. No. 275013373) in a final volume of 15 µL containing 6.5 µL of SYBR ™ Green (Promega ™, USA), primers (Table 2), 5 μL of DNA samples, final concentration of 15 to 30 ng of bacterial DNA or 50 ng of plant tissue DNA, and water to complete the reaction volume. The amplification conditions were as follows: initial denaturation at denaturation at 95 °C for 2 min, followed by 40 cycles of 95 °C for 30 s, and annealing temperature, as shown in Table 2.
Construction of standard curves
The genomic DNA from the HCC103, HRC54, Ppe8T, Pal5T, and CBAmC strains was quantified using the Qubit dsDNA HS (high sensitivity) assay kit with the Qubit fluorometer. The concentrations of the samples were standardized to 5.0, 5.0, 6.0, 4.0, and 5.0 ng μL−1, respectively, and they were serially diluted for use in qPCR and for constructing the calibration curve. To calculate the copy number of each target DNA molecule, the genome size of the bacteria (Table S1), Avogadro constant (6.023 × 1023), and molecular weight of the DNA (660 Da bp−1) were used. The curves were plotted using log10 of the target DNA copy number corresponding to the cycle threshold value (Ct) generated by dilution points ranging from 106.5 to 101.5 copies in the qPCR reaction. Primer amplification efficiencies for the qPCR was determined using Eq. 1, where E is the amplification efficiency (%) and S is the slope value of the standard curve.
Bacterial quantification
Each strain of the sugarcane inoculant was quantified by qPCR using the genomic DNA extracted from 1 mL of the culture medium or 300 mg of plant tissue and bacterial counting in plates using dilutions enough to observe 30 to 300 colony-forming units (CFU). Bacterial quantification by qPCR from DYGS culture medium was performed by calculating the number of cells mL−1 and the quantification of sugarcane plant tissues was performed by calculating the number of bacterial cells g−1 tissue. The cell number was calculated according to Eq. 2 [55].
where A is the amount of target DNA fragments or cells calculated using the Ct value of qPCR, B is the concentration of genomic DNA (ng μL−1), C is the volume of extracted DNA (μL), D is the mass of total DNA (ng) used in the qPCR reaction, and E is the volume (mL) or mass (g) value when the total DNA is isolated from the culture medium and plant tissue, respectively.
qPCR quantification of sugarcane inoculant from in vitro cultivated tissues
The quantification of the five bacterial strains present in the sugarcane inoculant was conducted using qPCR with the most suitable primers. Sugarcane plants, which were 45 days old and cultivated in vitro in the growth chamber, were divided into two tissue types: roots and shoots. Each tissue was inoculated with an equal amount of the five bacteria from the inoculant. To achieve this, the initial inoculum solution for each bacterium was adjusted to approximately 109 cells per mL and then combined. Subsequently, 100 mg of each tissue was inoculated with 100 µL of the bacterial suspension, which contained the five bacteria from the inoculant and was prepared in serial dilutions ranging from 101 to 109, with a 10 × dilution factor. The mixture was gently homogenized, incubated at room temperature, and after 3 h, DNA extraction was carried out to quantify the bacteria using the qPCR technique, following the previously described procedure.
Greenhouse experiment to test primer specific
The experiments were carried out at Embrapa Agrobiology, located at the BR 465 highway, km 7, in the Seropédica, RJ, Brazil (22°44′38″S, 43°42′28″W, and 26 m altitude). The experiment was conducted in a greenhouse with two commercial sugarcane varieties (RB867515 and RB92579). The mini-stalks (mini-sett) containing a single node were used for inoculation, as described by Schultz et al. [4]. The standard size mini-stalks selected for planting were packed in raffia bags according to the number required per plant row (15 buds per meter) and immersed for 30 min in the inoculum suspension in 200 L containers. Before bacterial inoculation, the mini-stalks were subjected to heat treatment at 52 °C for 30 min [56] and treated with a 0.1% fungicide solution methyl N-(2-{[1-(4-chlorophenyl)-1H-pyrazol-3-yl] oxymethyl} phenyl) N-methoxycarbamate (Piraclostrobin 250 g L−1) for 3 min and maintained for 2 h at room temperature (~ 25 °C). After inoculation, the seedlings were dried in the shade for 30 min and immediately planted in plastic boxes containing 12 kg of sterile substrate (sand and vermiculite 2:1, w:w) and grown at greenhouse. The experiment had a completely randomized block design with three replications that had roots and shoots treated as subplots and collected at 25, 40, and 55 days after inoculation (DAI). During this period, the plants were irrigated every 2 days with the nutritional Hoagland’s solution [0.1% KH2PO4 1 M (v:v); 0.1% K2HPO4 1 M (v:v); 0.2% MgSO4.7H2O 1 M (v:v); 0.172 g L−1 CaSO4.2H2O; 0.1%; minor elements (v:v); 0.1% of Fe (1.21 g Na2H2EDTA in 100 mL of distilled water and 0.6 g FeCl3.6H2O) (v:v)] [57] and supplemented with 1% nitrogen source (urea) at 40 days after planting.
Statistical analysis
All DNA samples were analyzed in three biological replicates, each with three technical replicates. The quantification data were log transformed and analyzed for normal distribution by the Shapiro–Wilk test, submitted to analysis of variance and Tukey's average test, all with p < 0.05, using the statistical program SISVAR™ (Lavras, Brazil) version 5.6 [58].
Results
PCR reaction optimization and primer specificity in conventional PCR (cPCR)
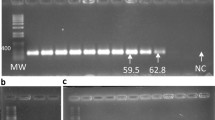
To select the best primer pair for each of the five target strains, 39 primer pairs, including 13 for HRC54, 10 for HCC103, 8 for CBAmC, 4 for Ppe8T, and 4 for Pal5T, were tested. To determine the specificity and optimize the reaction conditions, all primers were evaluated by cPCR using the genomic DNA of 30 related bacterial strains (Table 1). MgCl2 concentrations for cPCR reactions ranged from 1.0 to 2.5 mM, depending on the primers (Table 2). In addition, the best concentrations of primers for cPCR were between 100 and 400 nM, and for qPCR, it ware 200 to 500 nM. The cPCR reactions with the Hs54CpepM15 and Gd5C4525 primers showed the best amplification when 0.02 and 0.05% Tween 20 were added, respectively. Primer specificity was evaluated by cPCR using genomic DNA isolated from 30 taxonomically related bacterial strains, including the five bacteria used as sugarcane inoculants. The cPCR reactions were carried out using either DNA from each strain or a mix of DNA from the five strains. Even when cPCR was carried out using a mix of DNA of the five strains, specific amplification was observed for each primer pair selected (Fig. 1). However, initial analysis of primer specificity suggested that the strains used within each species (Table 1) could be closely related; therefore, we decided to study the phylogenetic relationships between the strains. For this, we carried out BOX-PCR analyses of the five strains used as inoculants, as well as 25 taxonomically related strains. These data revealed that Pt8C14 primers, designed to be specific to the Ppe8T strain of the P. tropica species, showed some degree of cross-reactivity with closely related strains (Table S2 and Fig. S1). In contrast, when using Gd5C4525 primers for the Pal5T strain of the G. diazotrophicus species and NaCbgyrA primers for the CBAmC strain of the N. amazonense species, the expected DNA fragment size was consistently amplified in all evaluated strains of their respective species (Table S2). The primers Hr103C1780, designed for H. rubrisubalbicans strain HCC103, also amplified a fragment of ~ 112 bp from strain M6 (Table S2). In the case of the primer Hs54CpepM15 designed for H. seropedicae strain HRC54, five out of the six strains tested (HRC54, ZAE76T, HR C80, H S4, and ZAE94) had a specific fragment (~ 186 bp) amplified by the cPCR (Table S2). However, it is important to note that phylogenetic analyses showed that most of these strains are phylogenetically remarkably close to those included in the sugarcane inoculant (Fig. S1). Therefore, all selected primers are species-specific and, in the case of the primers for the HCC103 and Ppe8T strains, the specificity was very high because the PCR reactions showed only one cross-reaction for HCC103 and two for Ppe8T.

PCR product of primers genomic DNA individual of each strain (a) Hr103C1870 (112 bp) (b) Hs54CpepM15 (186 bp) (c) Pt8C14 (111 bp) (d) Gd5C4525 (143 bp) (e) NaCbCgyrA (68 bp) and (f) Mix of the five bacterial strains. M: Molecular marker 1 Kb Plus DNA Ladder (Invitrogen®, USA). a target strain, Hr: H. rubrisubalbicans; Hs: H. seropedicae; Pt: P. tropica; Gd: G. diazotrophicus and Na: N. amazonense
Performance of primers in qPCR
For quantification of the target bacteria, a standard curve (linear equation) was generated from the qPCR with known DNA concentrations (Fig. S2). The averages of the Ct values of three independent qPCR reactions were used to determine the amplification efficiency (E) and the coefficient of determination (R2) of each pair of primers. Most of the values observed for these parameters are in accordance with the recommendations for amplification by primers in qPCR, with E between 90 and 105% and R2 higher than 0.985 (Table S3). Melting curves of the amplified products for all selected primers showed only a single peak above 80 °C that indicating the amplification of only one DNA fragment. The sensitivity or limit of detection (LOD) of the primers was obtained using the sample with the lowest mass of genomic DNA, which demonstrated amplification of 100% of the replicates used in the three qPCR assays. All selected primers presented LOD in the order of 102 femtograms (fg) of DNA (Table S4).
Quantification of bacteria in the sugarcane tissues by qPCR
The five bacteria used were individually quantified by qPCR, after inoculation of the mixture of the strains under serial dilutions in sugarcane tissues grown in vitro. As expected, there was a reduction in the number of bacterial cells quantified according to the dilution (Fig. 2). The plant tissue present in the DNA extraction mixture did not affect primer efficiency during the qPCR reactions. The number of bacteria quantified was remarkably similar to that quantified using the calibration curve prepared using only bacterial genomic DNA. The best primers selected for each strain were then used to quantify the five bacteria in sugarcane grown in a greenhouse for up to 55 days after inoculation.

Quantification of bacteria from in vitro sugarcane cultivation inoculated under serial dilution (a) root (R) and (b) shoot (S)
Quantification of sugarcane bacterial inoculant in the greenhouse-growing plants
The roots and shoots of the sugarcane plants harvested on the 25th, 40th, and 55th day after inoculation were used for total DNA extraction and bacterial quantification by qPCR with specific primers. The overall average of the number of bacteria that make up the sugarcane inoculant indicated a sharp decline over the collection period. In addition, it was observed that the number of bacteria of the five strains was the same in the inoculated and non-inoculated plants (Table 3). This effect was observed in the evaluated plant tissues, shoots, and roots (Fig. 3). When we calculated the bacterial numbers for each strain, significant differences were observed among bacterial species, and these differences were influenced by the plant variety (Table 4). The species P. tropica was quantified, but in relatively low numbers. However, inoculation significantly increased the abundance of these bacteria, particularly in the shoots and roots. N. amazonense, H. seropedicae, and G. diazotrophicus were detected at the species level because the primers designed for the target strains CBAmC, HRC54, and Pal5T amplified DNA fragments from the genomes of all strains evaluated within their respective species (Table S2). Among the five bacteria evaluated, N. amazonense was the only one in which the detected population was significantly higher in the roots and shoots of inoculated plants (Table 4).

Monitoring of bacteria populations of five bacterial inoculated in sugarcane throughout the experiment conducted up to 55 DAI: (a) Aerial part and (b) Root. Hr: Herbaspirillum rubrisubalbicans, Hs: H. seropedicae, Pt: P. tropica, Gd: G. diazotrophicus and Na: N. amazonense. Mean statistically different from the others by the Tukey test (p < 0.05)*
In the case of the two strains HCC103 and HRC54 Herbaspirillum sp. there was also a significant difference in the number of cells quantified in plant tissues between 25, 40, and 55 DAI-extracted samples (Fig. 3). The total number of H. seropedicae (HRC54 strain) detected during the three different periods was the highest. However, a significant increase was observed only in the shoots of plants inoculated with the HCC103 strain (Table 4). In the case of G. diazotrophicus, qPCR quantification showed that, similar to most inoculant bacteria, its population was reduced at 40 DAI (Fig. 3). In addition, the number of bacteria detected of this species was significantly higher in the roots and shoots of the control plants (Table 4).
In general, the population of sugarcane bacterial inoculants in variety RB867515 was significantly higher than that for variety RB92579 (Table 3). In addition, when each bacterial species was evaluated, most were found in higher numbers in the shoots of the RB867515 variety compared to those of the RB92579 variety (Table 4).
Discussion
Primer validation
Primer design is a fundamental step in the quantitative accuracy of qPCR [36,37,38]. Specific regions from the genomic sequence of each strain in the sugarcane inoculant were used to design strain-specific primers. However, even when an exhaustive sequence analysis was performed in silico, finding specific primers for a given strain is not an easy task. The AOAC [59] recommended the ISO 16140 guideline, which suggests that a number of strains, between 20 and 50, should be evaluated to ascertain primer exclusivity. To test primer specificity, we used DNA from 30 different closely related bacterial strains (Table 1), as shown by BOX-PCR phylogenetic analysis (Fig. S1); however, an additional challenge was the absence of genomic sequences for several strains of the same species. Studies have also shown that most of the designed primers are not used, mainly because they are not specific to the target strain [31, 33, 35]. We designed and validated a set of bacterial species-specific primers for the sugarcane inoculant (Fig. 1). The primers designed for HRC54, CBAmC, and Pal5T could be considered species-specific because they could amplify a DNA fragment from several strains within the species (Table S2). We must consider that most of these strains are closely related and do not have any genomic information available to clearly define their phylogenetic position and aid in the identification of strain-specific sequence regions. Furthermore, qPCRs with strain-specific primers can be difficult to run using samples obtained from the field because closely related indigenous bacteria may interfere with the amplification and quantification [33]. Increasing the number of sequenced genomes from different strains may help solve this problem and help in designing a set of strain-specific primers.
To enhance the performance of both cPCR and qPCR, some primers underwent optimization using 2% Tween 20. According to Bachmann et al. [60], this nonionic detergent can be used at concentrations of up to 2.5% (v:v) without inhibiting PCR. Bustin and Huggett [37] recommended annealing temperatures within the range of 59 °C ± 2 °C for qPCR primers. However, they also demonstrated that well-designed primers could function effectively even when subjected to temperatures as high as 64 °C, without compromising quantification accuracy. It's worth noting that all the primers developed in this study were subjected to annealing temperatures exceeding 60 °C (Table 2).
The amplification efficiency (E) of the primers in this study showed values within the recommended range of 95–105% [37]. However, R2 of the standard curves for Hs54CpepM15 and Gd5C4525 primers in some tests was below 0.98, which is the minimum recommended value [37]. A single peak was observed above 80 °C in the melting curves of most of the primers after qPCR. According to Bustin and Huggett [37], this characterizes the specificity of qPCR assays using the fluorescent dye, SYBR Green. It is important to point out that a second peak was formed in the melting curves when using primers Hr103C1870, Hs54CpepM15, and NaCbCgyrA with heavily diluted genomic DNA samples of strains HCC103, HRC54, and CBAmC (Fig. S3). Nonspecific amplification was observed with the use of the primers when the qPCR product was visualized on an agarose gel, as shown in Fig. S4a, b, and e. According to Azevedo et al. [61], an increase in the annealing time in the qPCR assay can be used to reduce nonspecific amplification. Additionally, an extra touchdown PCR step can also be used in the qPCR assay, or an annealing temperature that is higher than the recommended range (59 °C ± 2 °C) is used which is then successively reduced to reach the best annealing temperature for the primers [62,63,64].
The LOD analysis showed that the primers Pt8C14 and Gd5C4525, designed for the strains Ppe8T and Pal5T, respectively, presented higher sensitivity and were able to amplify target fragments from 100 fg of genomic DNA. However, primers Hr103C1870, HsCpep54M15, and NaCbCgyrA amplified target DNA fragments from 1000 fg of genomic DNA from the bacterial strains HCC103, HRC54, and CBAmC, respectively (Table S4). The LODs were confirmed with more than 95% reproducibility in the amplification stage of the qPCR of the target DNA fragment using standard curves of heavily diluted DNA samples (Fig. S4), as recommended by Kralik and Ricchi [65]. The LOD of the primers used in the present study was higher than the previously reported LOD of primers used for the quantification of other bacteria, such as 1.0, 60.3, and 60.0 fg for Pseudomonas putida BP25 [66], H. seropedicae SmR1 [34], and A. lipoferum CRT1 [30], respectively.
A statistically significant difference (p < 0.05) between the different quantification techniques was observed only for the HCC103 primers (Fig. 3). The higher number of bacteria quantified by qPCR in this experimental assay is dependent on the fact that qPCRs quantify both viable and non-viable cells, whereas plate counts only account for viable cells. As observed by Ilha et al. [62], the number of CFUs per mL of Lactobacillus paracasei was statistically different from the results of the qPCR only at 28 days of incubation, when the number of viable L. paracasei cells in the yogurt samples was reported to be low. Quantification of A. brasilense FP2 from wheat seedlings and roots of corn seedlings also showed that qPCR was more sensitive than plaque quantification [31,32,33]. However, it is important to emphasize that quantification in plaques uses semi-selective media, and there may be an overestimation of the actual population of target bacteria, especially when the analyzed matrix is a plant, as other microorganisms besides the target may also grow. Additionally, when primers have a low LOD or when there is a low concentration of target DNA in the sample due to losses in the isolation process, quantification using qPCR can be underestimated [67].
Bacterial quantification by qPCR using DNA isolated from sugarcane tissues
The primers developed in this study were suitable for quantifying each of the five bacterial species in the sugarcane inoculant. This paves the way for the fast and accurate quantitative investigation of sugarcane microbial communities after inoculation. However, it's essential to emphasize that further research is required to design strain-specific primers for monitoring plant endophytic bacterial populations under field conditions using qPCR assays. The quantification data obtained in our experiment showed that there was an increase in the overall average number of bacteria in the inoculated treatments compared to that in the control; however, this increase was not statistically significant (Table 4).
Plants support a specific population of endophytic microorganisms that can colonize them. Endophytic colonization refers to the entry, growth, and multiplication of endophyte populations within the host plant, and fitness outcomes in these traits reveal intense conflicts of interest between partners [68, 69]. It is reasonable to suggest that this interaction can become pathogenic when the plant loses control of the bacterial population, thereby causing a massive colonization of xylem mesophyll [40]. In our experiments, although the propagules were heat-treated according to the method reported by Landell et al. [56], the high number of bacterial cells quantified in the control suggested that the heat treatment was not sufficient to eliminate the bacteria from the plant matrices collected in the field and used as propagules. The number of cells per gram (105 cells g−1 of plant tissue) observed in non-inoculated plants was remarkably similar to that observed in several studies involving greenhouse and field experiments and was quantified using different techniques. We also observed that on average, the number of target bacteria was significantly higher in the roots than in the shoots (Table 4). Other authors have also reported that species such as G. diazotrophicus and Herbaspirillum sp. are abundant in the roots [25]. The qPCR analysis of each species of the inoculant showed that there was a significant increase in the number of quantified cells of N. amazonense and P. tropica per gram of tissue in the roots, N. amazonense and H. rubrisubalbicans in the shoots, and in the case of bacterial species G. diazotrophicus, the opposite effect was observed in both tissues. However, several studies using qPCR to quantify bacteria associated with plant tissues have shown that population behavior may vary according to the culture, substrate, and evaluation time of the experiments [31, 33, 34, 66].
Our results also showed that on average, the number of inoculant bacteria for sugarcane was significantly higher in the RB867515 variety. Although no phytotechnical parameters were evaluated in our study, several studies have shown that sugarcane genotypes respond differently to inoculation with endophytic diazotrophic bacteria. Additionally, sugarcane variety RB867515 has been previously reported to exhibit a greater response to inoculation compared to other varieties [5, 17, 67, 70]. It is important to note that the number of inoculated bacteria quantified in the sugarcane tissues may be underestimated. Kralik and Ricchi [65] suggested that more than 50% of the total DNA contained in the matrix was lost during the extraction process. Therefore, quantification by qPCR is sensitive only to the fraction of bacterial DNA recovered during the total DNA extraction process.
Conclusions
The designed primers and the developed qPCR assay proved highly effective for quantifying the bacterial strains HCC103, HRC54, Ppe8T, Pal5T, and CBAmC using total DNA isolated from sugarcane. Moreover, this study revealed that the inoculation with these five bacterial species led to an increase in the population of CBAmC and HRC54 in the shoots, as well as CBAmC and Ppe8T in the roots of sugarcane. These findings were observed over a period of up to 55 days after inoculation in sterile substrates within a greenhouse setting. Furthermore, the results demonstrated that the sugarcane variety RB867515 exhibited a higher colonization rate by bacteria from the inoculant compared to the RB92579 variety, and the bacterial population tended to decrease as the plant aged.
References
CONAB (2019) Acompanhamento da Safra Brasileira de Cana-de-açúcar. ISSN 2318–7921. http://www.conab.gov.br. Accessed 5 December 2019
Cruz CHB, Souza GM, Cantarella H, et al (2016) Universidades e empresas: 40 anos de ciência e tecnologia para o etanol brasileiro. São Paulo, Brasil. https://doi.org/10.5151/9788521210627
Oliveira ALM, Canuto EL, Urquiaga S et al (2006) Yield of micropropagated sugarcane varieties in different soil types following inoculation with diazotrophic bacteria. Plant Soil 284:23–32. https://doi.org/10.1007/s11104-006-0025-0
Schultz N, Silva JA, Sousa JS, Monteiro RC, Oliveira RP, Chaves VA, Pereira W, Silva MF, Baldani JI, Boddey RM, Reis VM, Urquiaga S (2014) Inoculation of sugarcane with diazotrophic bactéria. R Bras Ci Solo 38:407–414. https://doi.org/10.1590/S0100-06832014000200005
Schultz N, Pereira W, Silva PA, Baldani JI, Boddey RM, Alves BJR, Urquiaga S, Reis VM (2017) Yield of sugarcane varieties and their sugar quality grown in different soil types and inoculated with a diazotrophic bacteria consortium. Plant Prod Sci 20:366–374. https://doi.org/10.1080/1343943X.2017.1374869
Ryan RP, Germaine K, Franks A, Ryan DJ, Dowling DN (2008) Bacterial endophytes: Recent developments and applications. FEMS Microbiol Lett 278:1–9. https://doi.org/10.1111/j.1574-6968.2007.00918
Weyens N, Van der Lelie D, Taghavi S, Newman L, Vangronsveld J (2009) Exploiting plant-microbe partnerships to improve biomass production and remediation. Trends Biotechnol 27:591–598. https://doi.org/10.1016/j.tibtech.2009.07.006
Oliveira ALM, Canuto ED, Reis VM, Baldani JI (2003) Response of micropropagated sugarcane varieties to inoculation with endophytic diazotrophic bacteria. Braz J Microbiol 34:59–61. https://doi.org/10.1590/S1517-83822003000500020
Urquiaga S, Cruz KHS, Boddey RM (1992) Contribution of nitrogen fixation to sugarcane: Nitrogen-15 and nitrogen balance estimates. Soil Sci Soc Am J 56:105–114. https://doi.org/10.2136/sssaj1992.03615995005600010017x
Urquiaga S, Xavier GR, Morais RF et al (2012) Evidence from field nitrogen balance and 15N natural abundance data for the contribution of biological N2 fixation to Brazilian sugarcane varieties. Plant and Soil 356:5–21. https://doi.org/10.1007/s11104-011-1016-3
Pereira WB, Sousa JS, Schultz N, Reis VM (2019) Sugarcane productivity as a function of nitrogen fertilization and inoculation with Diazotrophic plant growth-promoting bacteria. Sugar Tech 21:71–82. https://doi.org/10.1007/s12355-018-0638-7
Etesami H, Adl SM (2020) Plant Growth-Promoting Rhizobacteria (PGPR) and their action mechanisms in availability of nutrients to plants. In: Kumar M, Kumar V, Prasad R (eds) Phyto-Microbiome in Stress Regulation. Environmental and Microbial Biotechnology. Springer, Singapore. https://doi.org/10.1007/978-981-15-2576-6_9
Vargas L, Santa Brígida AB, Mota Filho JP, de Carvalho TG, Rojas CA, Vaneechoutte D et al (2014) Drought tolerance conferred to sugarcane by association with Gluconacetobacter diazotrophicus: A transcriptomic view of hormone pathways. PLoS One 9(12):e114744. https://doi.org/10.1371/journal.pone.0114744
Silva R, Filgueiras L, Santos B, Coelho M, Silva M, Estrada-Bonilla G, Vidal M, Baldani JI, Meneses C (2020) Gluconacetobacter diazotrophicus changes the molecular mechanisms of root development in Oryza sativa L. growing under water stress. Int J Mol Sci 21(1):333. https://doi.org/10.3390/ijms21010333
Filgueiras L, Silva R, Almeida I et al (2019) Gluconacetobacter diazotrophicus mitigates drought stress in Oryza sativa L. Plant Soil. https://doi.org/10.1007/s11104-019-04163-1
Antunes JEL, Freitas ADS, Oliveira LMS, Lyra MCCP, Fonseca MAC, Santos CERS, Oliveira JP, Araújo ASF, Figueiredo MVB (2019) Sugarcane inoculated with endophytic diazotrophic bacteria: effects on yield, biological nitrogen fixation and industrial characteristics. An Acad Bras Cienc. https://doi.org/10.1590/0001-3765201920180990
Pereira W, Leite JM, Hipólito GS et al (2013) Acúmulo de biomassa em variedades de cana-de-açúcar inoculadas com diferentes estirpes de bactérias diazotróficas. Rev Ciência Agron 44:363–370
Gírio LAS, Dias FLF, Reis MR et al (2015) Bactérias promotoras de crescimento e adubação nitrogenada no crescimento inicial de cana-de-açúcar proveniente de mudas pré-brotadas. Pesq Agrop Bras. https://doi.org/10.1590/S0100-204X2015000100004
Pedula RO, Schultz N, Monteiro RC et al (2016) Growth analysis of sugarcane inoculated with diazotrophic bacteria and nitrogen fertilization. Afr J Agric Res 30:2786–2795. https://doi.org/10.5897/AJAR2016.11141
Dos Santos SG, Ribeiro FS, Fonseca CS et al (2017) Development and nitrate reductase activity of sugarcane inoculated with five diazotrophic strains. Arch Microbiol. https://doi.org/10.1007/s00203-017-1357-2
Ricchi M, Bertasio C, Boniotti MB, Vicari N, Russo S, Tilola M, Bellotti MA, Bertasi B (2017) Comparison among the Quantification of Bacterial Pathogens by qPCR, dPCR, and Cultural Methods. Front Microbiol. https://doi.org/10.3389/fmicb.2017.01174
Zhang Z, Qu Y, Li S et al (2017) Soil bacterial quantification approaches coupling with relative abundances reflecting the changes of taxa. Sci Rep 7:4837. https://doi.org/10.1038/s41598-017-05260-w
Romano I, Ventorino V, Pepe O (2020) Effectiveness of plant beneficial microbes: Overview of the methodological approaches for the assessment of root colonization and persistence. Front Plant Sci. https://doi.org/10.3389/fpls.2020.00006
Rilling JI, Acuña JJ, Nannipieri P, Cassan F, Maruyama F, Jorquera MA (2019) Current opinion and perspectives on the methods for tracking and monitoring plant growth-promoting bacteria. Soil Biol Biochem 130:205–219. https://doi.org/10.1016/j.soilbio.2018.12.012
Reis Junior FB, Silva LG, Reis VM, Döbereiner J (2000) Ocorrência de Bactérias Diazotróficas em Diferentes Genótipos de Cana-de-açúcar. Pesq Agrop Brasileira 35:985–994. https://doi.org/10.1590/S0100-204X2000000500016
Oliveira ALM, Urquiaga S, Döbereiner J, Baldani JI (2002) The effect of inoculating endophytic N2-fixing bacteria on micropropagated sugarcane plantlets. Plant Soil 242:205–215. https://doi.org/10.1023/A:1016249704336
Magnani GS, Cruz LM, Weber H, Bespalhok JC, Daros E, Baura V, Yates MG, Monteiro RA, Faoro H, Pedrosa FO, Souza EM (2013) Culture-independent analysis of endophytic bacterial communities associated with Brazilian sugarcane. Genet Mol Res. https://doi.org/10.4238/2013.October.15.3
Li W, Yan Z, Nakhla MK (2015) Real-Time for Detection and Identification of Anguina funesta, A. agrostis, A. tritici, and A. pacificae. Am Phytopathol Soc https://doi.org/10.1094/PDIS-09-14-0959-RE
Luchi N, Capretti P, Pazzagli M, Pinzani P (2016) Powerful qPCR assays for the early detection of latent invaders: interdisciplinary approaches in clinical cancer research and plant pathology. Appl Microbiol Biotechnol. https://doi.org/10.1007/s00253-016-7541-5
Couillerot O, Bouffaud M, Baudoin E et al (2010) Development of a real-time PCR method to quantify the PGPR strain Azospirillum lipoferum CRT1 on maize seedlings. Soil Biol Biochem. https://doi.org/10.1016/j.soilbio.2010.09.003
Faleiro AC, Pereira TP, Espindula E et al (2013) Real Time PCR detection targeting nifA gene of plant growth promoting bacteria Azospirillum brasilense strain FP2 in maize roots. Symbiosis. https://doi.org/10.1007/s13199-013-0262-y
Da Cunha ET, Pedrolo AM, Paludo F et al (2020) Azospirillum brasilense viable cells enumeration using propidium monoazide-quantitative PCR. Arch Microbiol. https://doi.org/10.1007/s00203-020-01877-0
Stets MI, Alqueres SMC, Souza EM et al (2015) Quantification of Azospirillum brasilense FP2 bacteria in wheat roots by strain-specific quantitative PCR. Appl Environ Microbiol 81:6700–6709. https://doi.org/10.1128/AEM.01351-15
Pereira TP, do Amaral FP, Dall’Asta P, Brod FCA, Arisi ACM (2014) Real-Time PCR quantification of the plant growth promoting bacteria herbaspirillum seropedicae strain SmR1 in maize roots. Mol Biotechnol 56:660–670. https://doi.org/10.1007/s12033-014-9742-4
Boa Sorte PMF, Simões-Araújo JL, Melo LHV et al (2014) Development of a real-time PCR assay for the detection and quantification of Gluconacetobacter diazotrophicus in sugarcane grown under field conditions. Afr J Microbiol Res. https://doi.org/10.5897/AJMR2014.6779
Robertson JM, Walsh-Weller J (1998) An introduction to PCR primer design and optimization of amplification reactions. Methods Mol Biol 98:121–154. https://doi.org/10.1385/0-89603-443-7:121
Bustin S, Huggett J (2017) qPCR primer design revisited. Biomol Detect Quantif. https://doi.org/10.1016/j.bdq.2017.11.001
Dreier M, Berthoud H, Shani N, Wechsler D, Junier P (2020) SpeciesPrimer: a bioinformatics pipeline dedicated to the design of qPCR primers for the quantification of bacterial species. PeerJ 8:e8544. https://doi.org/10.7717/peerj.8544
Baldani JI, Reis VM, Videira SS et al (2014) The art of isolating nitrogen-fixing bacteria from non-leguminous plants using N-free semi-solid media: a practical guide for microbiologists. Plant Soil. https://doi.org/10.1007/s11104-014-2186-6
Olivares FL, James EK, Baldani JI, Döbereiner J (1997) Infection of mottled stripe disease susceptible and resistant sugarcane varieties by the endophytic diazotroph Herbaspirilium. New Phytol. https://doi.org/10.1046/j.1469-8137.1997.00684.x
Baldani JI, Baldani VLD, Seldin L, Döbereiner J (1986) Characterization of Herbaspirillum seropedicae gen. nov.,sp. A root-associated nitrogen-fixing bacterium. Int J Syst Bacteriol 36(1):86–93. https://doi.org/10.1099/00207713-36-1-86
Reis VM, Santos PEL, Tenorio-Salgado S, Vogel J, Stoffels M, Guyon S, Mavingui P, Baldani VLD, Schmid M, Baldani JI, Balandreau J, Hartmann A, Caballero-Mellado J (2004) Burkholderia tropica sp. nov., a novel nitrogenfixing, plant-associated bacterium. Int J Syst Evol Microbiol 54(Pt 6):2155–2162. https://doi.org/10.1099/ijs.0.02879-0
Rodrigues L da S, Baldani VLD, Reis VM, Baldani JI (2006) Diversidade de bactérias diazotróficas endofíticas dos gêneros Herbaspirillum e Burkholderia na cultura do arroz inundado. Pesq agropec bras [Internet] 41(2):275–84. https://doi.org/10.1590/S0100-204X2006000200012
Reis VM, Olivares FL, Döbereiner J (1994) Improved methodology for isolation of Acetobacter diazotrophicus and confirmation of its endophytic habitat. World J Microbiol Biotechnol 10:401–405. https://doi.org/10.1007/BF00144460
Guedes HV, Santos ST dos, Perin L, Teixeira KR dos S, Reis VM, Baldani JI (2008) Polyphasic characterization of Gluconacetobacterdiazotrophicus isolates obtained from different sugarcane varieties. Braz J Microbiol [Internet] 39(4):718–23. https://doi.org/10.1590/S1517-83822008000400023
Azevedo MS, Teixeira KTS, Kirchhof G, Hartmann A, Baldani JI (2005) Influence of soil and host plant crop on the genetic diversity of Azospirillum amazonense isolates. Elsevier GmbH 49:565–576. https://doi.org/10.1016/j.pedobi.06.008
Magalhães FM, Baldani JI, Souto SM, Kuykendall JR, Döbereiner J (1983) A new acid-tolerant Azospirillum species. Anais da Academia Brasileira de Ciências, Rio de Rio 55:417–4430
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Versalovic J, Schneider M, De Bruijn FJ, Lupski JR (1994) Genomic fingerprinting of bacteria using repetitive sequence-based polymerase chain reaction. Methods Mol Cell Biol 5:25–40
Bertalan M, Albano R, Pádua V et al (2009) Complete genome sequence of the sugarcane nitrogen-fixing endophyte Gluconacetobacter diazotrophicus Pal5T. BMC Genomics. https://doi.org/10.1186/1471-2164-10-450
Bertalan PRA, Simões-Araújo JL, Vidal MS et al (2018) Draft genome sequence of Paraburkholderia tropica Ppe8 strain, a sugarcane endophytic diazotrophic bacterium. Braz J Microbiol. https://doi.org/10.1016/j.bjm.2017.07.005
Schwab S, Terra LA, Baldani JI (2018) Genomic characterization of Nitrospirillum amazonense strain CBAmC, a nitrogen-fixing bacterium isolated from surface-sterilized sugarcane stems. Mol Genet Genomics. https://doi.org/10.1007/s00438-018-1439-0
Baldani JI, Guedes HV, Vidal MS, et al (2011) Base de dados genômica de estirpes que compõem o inoculante de cana-de-açúcar e milho. Ed. Embrapa Agrobiologia. Documentos 282. Seropédica, Rio de Janeiro. 1517–8498. http://www.infoteca.cnptia.embrapa.br/infoteca/handle/doc/950789. Accessed 5 Oct 2023
Untergasser A, Cutcutache I, Koressaar T et al (2012) Primer3—new capabilities and interfaces. Nucleic Acids Res 40:115. https://doi.org/10.1093/nar/gks596
Ilha EC, Scariot MC, Treml D et al (2016) Comparison of real-time PCR assay and plate count for Lactobacillus paracasei enumeration in yoghurt. Ann Microbiol. https://doi.org/10.1007/s13213-015-1137-7
Landell MGA, Campana MP, Figueiredo P et al (2012) Sistema de multiplicação de cana-de-açúcar com uso de mudas pré-brotadas (MPB), oriundas de gemas individualizadas. https://www.udop.com.br/ebiblio/pagina/arquivos/2013_sistema_multiplicacao_cana_com_mudas_pre_brotadas.pdf. Accessed 15 September 2020
Hoagland DR, Arnon DI (1938) The water culture method for growing plants without soil. Calif Agr Exp STA Cir 347:32. https://hdl.handle.net/2027/uc2.ark:/13960/t51g1sb8j
Ferreira DF (2014) Sisvar: A Guide for its Bootstrap procedures in multiple comparisons. Ciência Agrotecnologia. https://doi.org/10.1590/S1413-70542014000200001
Environmental Surfaces'. In: Latimer GW Jr. (ed) Official Methods of Analysis of AOAC INTERNATIONAL, 22 (New York, 2023; online edn, AOAC Publications, 4 Jan. 2023). https://doi.org/10.1093/9780197610145.005.010
Bachmann B, Luke W, Hunsmann G (1990) Improvement of PCR amplified DNA sequencing with the aid of detergents. Nucleic Acids Res 18:1309
Azevedo F, Pereira H, Johansson B (2017) Colony PCR. In: Domingues L (ed) PCR, Methods in Molecular Biology, vol 1620. Springer, New York, pp 129–139. https://doi.org/10.1007/978-1-4939-7060-5_8
Henry S, Baudoin E, López-Gutiérrez JC, Martin-Laurent F, Brauman A, Philippot L (2004) Quantification of denitrifying bacteria in soils by nirK gene targeted real-time PCR. J Microbiol Methods. https://doi.org/10.1016/j.mimet.2004.07.002
Korbie D, Mattick J (2008) Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nat Protoc. https://doi.org/10.1038/nprot.2008.133
Zhang Q, Wang J, Deng F, Yan Z, Xia Y et al (2015) TqPCR: A touchdown qPCR assay with significantly improved detection sensitivity and amplification efficiency of SYBR green qPCR. PLoS One 10(7):e0132666. https://doi.org/10.1371/journal.pone.013266
Kralik P and Ricchi M (2017) A basic guide to real time PCR in microbial diagnostics: Definitions, parameters, and everything. Front Microbiol. https://doi.org/10.3389/fmicb.2017.00108
Agisha VN, Eapen SJ, Bhai RS et al (2017) Detecting and monitoring endophytic colonization by Pseudomonas putida BP25 in black pepper (Piper nigrum L.) using quantitative real-time PCR. J Spices Aromat Crops 26(1):1–7. https://doi.org/10.25081/josac.2017.v26.i1.812
Reis VM, Baldani JI, Urquiaga S (2009) Recomendação de uma mistura de estirpes de cinco bactérias fixadoras de nitrogênio para inoculação de cana-de-açúcar: Gluconacetobacter diazotrophicus (BR 11281), Herbaspirillum seropedicae (BR 11335), Herbaspirillum rubrisubalbicans (BR 11504), Azospirillum amazonense (BR 11145) e Burkholderia tropica (BR 11366). Embrapa Agrobiologia/Circular técnica 30. Seropédica, Rio de Janeiro. pp 1519–7328. https://ainfo.cnptia.embrapa.br/digital/bitstream/item/39137/1/CIT30-09. Accessed 05 Oct 2023
Kandel SL, Joubert PM, Doty SL (2017) Bacterial endophyte colonization and distribution within plants. Microorganisms. https://doi.org/10.3390/microorganisms5040077
Ibáñez F, Tonelli ML, Muñoz V, Figueredo MS, Fabra A (2017) Bacterial endophytes of plants: diversity, invasion mechanisms and effects on the host. In: Maheshwari D (ed) Endophytes: Biology and Biotechnology. Sustainable Development and Biodiversity, vol 15. Springer, Cham. https://doi.org/10.1007/978-3-319-66541-2_2
Schultz N, Morais RF, Silva JA et al (2012) Avaliação agronômica de variedades de cana-de-açúcar inoculadas com bactérias diazotróficas e adubadas com nitrogênio. Pesq Agrop Brasileira 47:261–268. https://doi.org/10.1590/S0100-204X2012000200015
Acknowledgements
To the Graduate Program in Crop Science (PPGF) of Federal Rural Rio de Janeiro University (UFRRJ), to Embrapa Agrobiologia, for its infrastructure and technical support; the Coordination for the Improvement of Higher Education Personnel (CAPES), for granting a scholarship to the first author and other collaborators. This work was partially funded by grants from the Carlos Chagas Research Support Foundation (FAPERJ) the National Council for Scientific Development (CNPq), project number INCT 46/33/2014-2 and
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Consent to participate
All authors gave their consent to participate.
Consent for publication
All authors gave their consent for the publication of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Responsible Editor: Luiz Henrique Rosa
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Da Silva, C.G.N., Monteiro, E.d. ., Diniz, P.P. et al. Designing and validation of specific primers for the quantitative detection of bacteria in sugarcane inoculant. Braz J Microbiol 54, 2627–2640 (2023). https://doi.org/10.1007/s42770-023-01144-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42770-023-01144-6