
Abstract
Background
Delayed cerebral ischemia (DCI) is among the most dreaded complications following aneurysmal subarachnoid hemorrhage (SAH). Despite advances in neurocritical care, DCI remains a significant cause of morbidity and mortality, prolonged intensive care unit and hospital stay, and high healthcare costs. Large artery vasospasm has classically been thought to lead to DCI. However, recent failure of clinical trials targeting vasospasm to improve outcomes has underscored the disconnect between large artery vasospasm and DCI. Therefore, interest has shifted onto other potential mechanisms such as microvascular dysfunction and spreading depolarizations. Animal models can be instrumental in dissecting pathophysiology, but clinical relevance can be difficult to establish.
Methods
Here, we performed a systematic review of the literature on animal models of SAH, focusing specifically on DCI and neurological deficits.
Results
We find that dog, rabbit and rodent models do not consistently lead to DCI, although some degree of delayed vascular dysfunction is common. Primate models reliably recapitulate delayed neurological deficits and ischemic brain injury; however, ethical issues and cost limit their translational utility.
Conclusions
To facilitate translation, clinically relevant animal models that reproduce the pathophysiology and cardinal features of DCI after SAH are urgently needed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
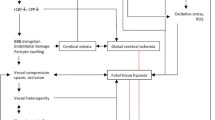
Delayed cerebral ischemia (DCI) is one of the most feared complications of aneurysmal subarachnoid hemorrhage (SAH). It occurs in approximately 20% of survivors of initial aneurysmal rupture and is a major contributor to subsequent death, poor functional outcome, and prolonged intensive care unit (ICU) and hospital stay [1]. The calcium channel blocker nimodipine is the only drug that is the U.S. food and drug administration (FDA)-approved for the prevention of DCI. The Rho-associated, coiled-coil containing protein kinase (ROCK) inhibitor, fasudil, is also used as standard of care for the prevention of DCI in Japan. However, all other investigational therapies have failed in randomized controlled trials [2], including the endothelin receptor antagonist, clazosentan, which effectively reversed angiographic vasospasm in humans but failed to improve the overall clinical outcome [3]. Although clinical trial design and flawed preclinical studies may have played some role in the failure to translate preclinical findings [4], it is more likely that attention was misdirected on reducing large artery vasospasm instead of other potential causes of DCI such as microvascular dysfunction, inflammation, thromboembolism and spreading depolarizations [1, 5,6,7]. There are numerous models of experimental SAH, but it is unclear to what extent DCI is recapitulated in each model. This has been a limiting factor in the pathophysiological investigations and therapeutic testing. Here, we review the available animal models of SAH and their propensity to develop DCI, in search of pathophysiologically and clinically relevant experimental models.
Methods
We conducted a PubMed search inclusive of November 2017 using keywords “subarachnoid hemorrhage” AND “delayed cerebral ischemia” AND [“animal” OR “experimental”]. We excluded clinical studies and review articles, articles in a language other than English, studies that only assessed extracranial vasculature, and studies that employed hemolyzed blood or blood products other than whole blood. We also excluded one study that only used a transgenic animal model without a wild-type control [8], one study that did not assess any delayed endpoint relevant for DCI [9], one study that artificially induced spreading depolarizations [10], and one study that induced focal ischemia after induction of SAH [11]. Overall, our DCI-focused search strategy was representative of the rest of the literature on experimental SAH [12]. We recorded SAH outcomes only in control groups without any physiological, pharmacological, or genetic intervention. We defined DCI as a worsening in tissue perfusion over time after the recovery of the initial global ischemia at the time of SAH induction. Delayed neurological deficits (DND) were defined as sensory, motor or cognitive deterioration after a clearly discernible period of stability or improvement. Tissue injury was noted if cell death was confirmed using histological markers such as Fluoro-Jade staining or TdT-mediated dUTP nick end labeling, or reduced cell counts demonstrated histologically. Frank infarction was also recorded if examined histologically, or by computed tomography (CT) or magnetic resonance imaging (MRI). We used a general linear random intercept mixed effects model to construct a multivariable linear prediction model of vasospasm as measured by arterial diameter which incorporated species, SAH induction model, time of vessel measurement, artery measured, and measurement method.
Results
Overall Characteristics of Experimental SAH Models
The initial search strategy identified 324 studies. After applying the exclusion criteria outlined above, 119 studies describing a total of 121 experimental cohorts were analyzed (Table 1). Seven species were studied, most commonly rats (32%) and dogs (22%), followed by mice (17%), primates (16%), rabbits (12%), cats (1%), and pigs (1%). In two reports, two different species were used (dogs and primates, and rats and rabbits). SAH induction methods included prechiasmatic cistern, cisterna magna, or intrathecal blood injection (61%), endovascular perforation of the internal carotid terminus or external perforation of the basilar artery (24%), direct clot placement around an intracranial artery (14%), or transection of a subarachnoid vein (1%). Blood injection was used in all species, endovascular perforation was only used in rodents, and peri-arterial clot placement was only used in dogs and primates. Cisterna magna blood injection was repeated 2–3 times in some models (9 single, 7 double injection studies in rats; 11 single, 3 double injection studies in rabbits; 25 double, 1 triple injection studies in dogs). Large artery vasospasm was the most often studied endpoint (57%), particularly in larger animals. In contrast, microvascular abnormalities were assessed in only a small fraction of studies (8%), mostly in mice, and relatively recently. DND was assessed in less than half and tissue outcome in less than a fourth of all studies. Endpoint assessments were limited to the first week in most studies, although some continued for up to 48 days.
Large Artery Spasm
Using our search criteria, large artery spasm was reported most often in primates (37% of all cohorts), followed by dogs (25%), rats (19%), rabbits (9%) and mice (8%) (Table 2). The most common SAH induction methods to examine large artery spasm were cisterna magna injection (46%), perivascular clot (35%) and endovascular puncture (11%) models. Vasospasm was examined in the basilar (52%), middle cerebral (19%), anterior cerebral (11%), and internal carotid arteries (10%). Although present in the majority of cohorts, the degree of vasospasm was generally mild, corresponding to a median of 70% (58–86% interquartile range) of control vessel diameters (i.e., only a 30% reduction) in the pooled dataset. A general linear random intercept mixed effects model showed that species [F (4, 109) = 7.00; p < 0.0001], SAH induction method [F (3, 109) = 3.28; p = 0.0237], time after SAH [F (2, 109) = 6.71; p = 0.0018], arteries involved [F (4, 109) = 10.77; p < 0.0001], and method of vasospasm assessment [F (3, 109) = 2.77; p = 0.0449] were independent predictors of the degree of vasospasm. Post hoc analyses suggested that vasospasm was most severe in dogs, followed in decreasing order by mice, rabbits and rats, and least severe in primates. Cisterna magna injection and perivascular clot placement led to the most severe large artery vasospasm, followed by endovascular puncture and prechiasmatic injection. Vasospasm tended to be more severe in middle cerebral arteries, followed by internal carotid and anterior cerebral arteries, and less severe in basilar and posterior cerebral arteries. Although the few studies examining multiple time points after SAH did not reveal a clear temporal progression of vasospasm, in the pooled dataset of all studies vasospasm was worse when measured 4–7 days after SAH compared with 1–3 days or 8–21 days. Data also suggested that while some degree of large artery vasospasm was detectable in most cases, even in worst cases it appeared too mild to precipitate DCI, DND and infarction.
Microvascular Abnormalities
Given the disconnect between large artery spasm and clinical outcomes [3], attention in recent years has turned to microvascular abnormalities that may affect tissue perfusion and lead to DCI and DND (Table 3). Microvascular abnormalities in pial or parenchymal vessels were detected in all cohorts where this endpoint was examined, most often in mice, but also in dogs and rats. Microthrombi involving cerebral cortical, hippocampal and cerebellar parenchymal arterioles were commonly observed on routine histology and immunohistochemistry. However, absence of quantitative comparisons to sham controls in some studies made it difficult to assess the magnitude of this finding. Microthrombi peaked 2 days after SAH in mice in the only study that examined the time course. Reduced caliber of pial and parenchymal arterioles, and even capillaries, was also commonly observed on electron microscopy, optical microangiography, and two-photon microscopy. Smaller caliber vessels appeared to be more severely affected. The extent of constriction was sufficient to diminish perfusion when examined using two-photon microscopy, in vivo. There was also evidence of inflammation (e.g., increased P selectin). These microvascular abnormalities are all predicted to significantly increase cerebrovascular resistance and may be sufficient to cause tissue ischemia, especially when coupled to mild or moderate large artery spasm. However, in most cases, they were present at the first time point examined after SAH and did not worsen over time. Therefore, whether they are relevant for the development of DCI is unclear.
Perfusion Abnormalities
Cerebral blood flow (CBF) was examined in 17 studies within our search parameters. The species, SAH method, and the timing and techniques of perfusion assessment after SAH are summarized in Table 4. Although about half of all studies revealed perfusion abnormalities, hypoperfusion was only moderate (~ 40–80% of baseline), regardless of the cerebral blood flow (CBF) measurement technique. Moreover, only about half of the studies had more than 1 time point of assessment after SAH. Among those, worsening of perfusion (i.e., DCI) was detected in only three studies. One study in dogs found significantly lower cerebral, cerebellar and brain stem blood flow on day 8 (CBF ~ 60–70% of baseline) compared with day 1 using the microsphere technique after cisterna magna blood injection [13]. Interestingly, large artery spasm did not correlate with CBF in this study. In a more recent study, mice displayed worse perfusion on day 3 than day 1 after endovascular puncture of the intracranial carotid artery, when examined using optical coherence tomography [14]. However, the perfusion deficit was only mild (CBF 83% of controls). Finally, in a rat prechiasmatic blood injection model, hypoperfusion was significantly worse on day 2 compared with 1 h after SAH (CBF 40% of controls) [15]. Altogether, the data suggest that the majority of animal models of SAH do not faithfully reproduce DCI that is severe enough to cause infarction, regardless of the method and species.
Delayed Neurological Deficits
DND was detected in only ~ 20% of all studies where they were examined using grading systems or qualitatively based on consciousness, motor function and/or appetite (Table 5). DND was not detected in mice (0/9 cohorts) or dogs (0/8 cohorts) and was uncommon in rats (1/16 cohorts) and rabbits (1/5 cohorts). Signs of DND developed more commonly in primates (detected in 7/13 cohorts), albeit in only a small subset of animals in each cohort (10 out of 83 animals in total). Nevertheless, the proportion of animals that developed DND was similar to the incidence of DND in humans after aneurysmal SAH, and majority of these animals did develop ischemic infarcts demonstrated on imaging or histopathology. The most common signs of DND in primates were hemiparesis or reduced level of alertness observed between 3 and 8 days after SAH. Unfortunately, all but one primate study used perivascular clot placement, precluding any conclusion on the propensity of different SAH models for DND. These data show that, except for primates, animal models do not recapitulate the DND observed in aneurysmal SAH in humans.
Tissue Injury
Scattered cell death was detected in 15 out of 18 cohorts where it was assessed, mostly in mice after cisterna magna or prechiasmatic blood injection, and in rats after endovascular puncture (Table 6). The small number of studies precluded any conclusions on the propensity of different species and SAH models to develop scattered cell death. Cortex and hippocampus were most commonly involved. Evidence for delayed injury, however, was sparse. In a mouse model of endovascular perforation, hippocampal neuronal counts appeared more severely reduced compared with cell death detected on day 1 (fluoro-Jade). In a rat model of cisterna magna blood injection, neuronal counts significantly decreased in both hippocampus and cortex at day 5 compared with day 3 [16]. Both studies detected large artery vasospasm (50% reduction in middle cerebral artery caliber), as well as DND. Other studies examined only a single time point or did not compare early and late time points side by side using the same readouts. For example, in a mouse endovascular puncture model hippocampal and cortical neuronal loss (NeuN staining) was more severe at 14 days compared to the histological evidence of cell death present on day 1 (fluoro-Jade staining) [17]. Such studies could not distinguish delayed cell death due to DCI from acute cell death triggered by the SAH induction method, such as global ischemia due to high intracranial pressure, or direct focal ischemia due to endovascular puncture. Indeed supporting the latter, cell death was often ipsilateral in endovascular puncture studies.
Frank infarction was assessed more often than scattered cell death (27 studies) and was detected in half of all cohorts, often using neuroimaging. It was most commonly observed in primate perivascular clot placement followed by mouse rodent puncture models (Table 7). The small number of studies again precluded any conclusions on the propensity of different species and SAH models to develop frank infarcts. As with DND, infarction appeared more common in primates (75%), in most cases involved the cortex ipsilateral to the SAH procedure, and was associated with DND in the same animal. Direct evidence for delayed emergence of infarcts using serial imaging, however, was once again missing. Using serial MRIs, four mouse studies from one lab and two rat studies from another reported infarcts 2–3 days after SAH [18,19,20,21,22,23]. All six studies employed the endovascular puncture model and defined DCI as new infarcts that were either not present or smaller at the earliest time point of imaging. This definition, however, did not eliminate the possibility that infarcts developed or grew subacutely after the initial hours. Moreover, the distribution of infarcts was not described and might have been directly in the territory of the punctured artery. Hence, the data did not provide incontrovertible evidence that infarcts were caused by DCI after SAH, rather than the endovascular puncture procedure itself disrupting the structural integrity of the artery.
Mortality
Nearly 80% of all studies captured by our search strategy reported mortality, ranging between 0 and 67% among all species and SAH methods. It was generally higher in rodents and after endovascular perforation, possibly reflecting inadvertent direct morbidity associated with the procedure.
Discussion
This focused literature review suggests that animal models, with the exception of primates, do not fully recapitulate the progression and outcome of aneurysmal SAH in patients. The disconnect stems from large artery vasospasm, microvascular, and perfusion abnormalities that are too mild in most animal models, and from the lack of delayed emergence of cerebral ischemia, neurological deficits, and infarction. The latter is one of the most feared subacute consequences of SAH in surviving patients, and one that should be most amenable to treatment by virtue of the delayed therapeutic window of opportunity, in contrast to the global or focal ischemic injury suffered at the time of rupture. Therefore, there is an unmet need to develop new animal models of SAH, or improve upon existing models, by optimizing the choice of species, SAH induction method, experimental design and clinically relevant readouts.
Failure to recapitulate DCI after SAH in animal models in part stems from species differences. Brain size and morphology, white matter content, vascular anatomy and physiology all differ between human brain and other species. Notably, the clearance rate of subarachnoid blood varies among species [24] and is especially rapid in rodents after cisterna magna and to a lesser extent prechiasmatic injections. Indeed, rapid clearance of subarachnoid blood is the rationale for double injections in many models of SAH in rats as well as in dogs. Furthermore, abundant collaterals in animal brain may compensate for any regional vascular dysfunction [25]. Consequently, other delayed processes that may be important in human pathophysiology, such as cortical spreading depolarizations, microthrombosis, and microembolism [2, 5, 26] might not develop or have the same impact in experimental animals. Alternatively, DCI and DND may have been missed in studies without longitudinal examinations in the same animal, or reporting bias may have led to underrepresentation of experimental studies showing the absence of DND or DCI. Hence, the true incidence of DND and DCI in experimental SAH is difficult to discern from the literature.
Non-human primate models are attractive in SAH research due to phylogenetic, anatomic, and morphologic similarities to humans, as well as relatively well-established behavioral outcomes. In most primate studies, SAH was induced by perivascular clot placement, which likely leads to lasting presence of blood and breakdown products in subarachnoid space and is critical for DCI [27]. However, perivascular clot placement does not reproduce the sharp increase in intracranial pressure (ICP) and resulting transient global ischemia at the time of aneurysm rupture, or the acute exposure to fresh arterial blood, both of which may also contribute to DCI in humans [1]. Although DND and DCI were more common in primates than other species tested, ethical issues, cost and low throughput make non-human primate research prohibitive except in a few specialized facilities [25]. Therefore, it is not realistic to utilize non-human primates as the main species in SAH research, but perhaps as the gateway to a clinical trial. It should be acknowledged, however, that there is no evidence supporting that therapeutic efficacy in non-human primates are more predictive than phylogenetically lower species for therapeutic efficacy in human, no matter how intuitive it sounds.
Experimental models of SAH, as in most other disease models, range from holistic to mechanistic (i.e., reductionist). Holistic approaches try to approximate all aspects of human aneurysmal SAH with the intent of, for example, examining clinically relevant outcomes for therapeutic testing. Mechanistic models, on the other hand, mimic one particular aspect to better understand the pathophysiology. Clearly, none of the existing SAH models achieve a truly holistic representation of human aneurysmal SAH. The endovascular puncture model is practical, acutely introduces oxygenated arterial blood into the subarachnoid space, and raises intracranial pressure. However, it is also associated with a high rate of acute focal cerebral ischemia and infarcts from destruction of arterial continuity by the endovascular filament, and likely by gross endothelial injury in extended segments of the carotid artery during filament insertion. If acute tissue injury is not examined by a sensitive technique in the endovascular puncture model (e.g., MRI < 24 h after the procedure), any ischemic injury detected subsequently can be erroneously attributed to DCI. As discussed above in the context of primates, perivascular clot placement ensures dense presence of blood and breakdown products for extended periods of time, but the model does not reproduce fresh arterial blood exposure or the intracranial pressure spike at the time of rupture. Conversely, direct prechiasmatic or cisterna magna blood injection models reproduce the oxygenated arterial blood exposure and the intracranial pressure spike, but injected blood is often rapidly cleared without recreating the dense presence of blood and breakdown products for extended periods of time. To the extent that these processes are relevant for DCI and DND, perivascular clot placement and direct subarachnoid blood injection models remain more mechanistic than holistic. A holistic model to examine DCI and DND in rodents is the elastase-hypertension model [28,29,30,31]. Aneurysms are induced by subarachnoid elastase injection on a hypertensive background via mechanisms involving inflammation and focal vessel wall weakening that are implicated in human aneurysm formation as well. The model faithfully recreates spontaneous aneurysmal rupture and SAH with a natural range of severities mimicking human aneurysmal SAH [32, 33]. Mechanistically, however, the unpredictable timing of spontaneous aneurysmal rupture (5–14 days) blurs the timing of DCI and DND, and elastase-induced changes in arterial morphology may, in theory, affect the vascular mechanisms of DCI and DND. Despite these caveats, we believe the elastase-hypertension model is valuable to examine therapeutic interventions on SAH outcomes, when administered after the spontaneous rupture, which is marked by an acute neurological worsening in this model. Therefore, we believe preclinical efficacy screening would benefit from this holistic SAH model.
Outcome endpoints can also be more holistic or more mechanistic. A holistic approach to quantify DND as a clinically relevant (i.e., functional) outcome measure after experimental SAH is not easy due to the poor sensitivity of neurocognitive testing in most species. Moreover, unlike in models of focal ischemia, the severity and distribution of ischemia in SAH models can be highly variable even within a single model and species. As a result, the tissue and neurocognitive outcomes of each SAH model have not been well defined [34], and as noted above, most studies have indeed failed to detect DND using available tests. There may also be differences in the definition of what constitutes DND. Moreover, immediate or early deficits directly caused by SAH induction method may be severe enough to mask subsequent DND. More mechanistic readouts are easier to define. After the realization that large artery spasm is not a good predictor of DCI or DND [27] and that its successful treatment has not improved the outcomes, the focus on large artery spasm as the primary endpoint has shifted to microvascular dysfunction, inflammation, thromboembolism and spreading depolarizations [1, 5, 6]. However, these more mechanistic endpoints have not yet been proven to be a critical contributor or predictor of DCI and DND. Therefore, more studies are needed to establish causality.
Longitudinal outcome assessments, preferably in the same animal, are critical to ascertain whether any ischemia or neurocognitive deficit develops in a delayed manner, to qualify as DCI and DND. This important study design principle, unfortunately, has been missing in all but a handful of experimental SAH studies examining DCI and DND, as summarized above. Moreover, it is important to examine the spatiotemporal correspondence among various endpoints (e.g., CBF, infarction, neurocognitive deficits) to establish causation. This has also been missing except for a few studies. Longitudinal assessments in the same animal, and establishing correlations among various readouts, can best be established using advanced neuroimaging techniques to measure supply–demand mismatch and injury [35]. Finally, it is worth noting that when prioritizing investigational therapies for clinical trials, we must take into account the limitations of preclinical models, and perhaps more importantly the strength of preclinical data, such as proper experimental design and independent replication.
Although developing guidelines on preclinical SAH models for novel therapeutic intervention screening requires an expert consortium, we here offer some recommendations that we feel are critical to enhance the reproducibility and congruity among studies targeting DCI or DND, as well as their predictive value toward clinical translation. Most importantly, DCI must be distinguished from acute ischemia, and DND from acute deficits directly caused at the time of SAH induction. This requires longitudinal examination of the same animal, preferably for more than 7 days. Ideally, neuroimaging should be used to quantify injury and CBF within the first 24 h of SAH induction as a reference point for subsequent changes. Of course, availability and high cost are significant barriers for routine use of these tools. Neurological examination should document acute deficits within the first 24 h of SAH induction, and serial exams should seek evidence for worsening deficits over time. Importantly, severe deficits at onset may mask subsequent DND. For example, certain models that are known to induce a concurrent large vessel ischemic stroke, such as endovascular perforation, should be interpreted with additional caution. Sensorimotor tests can be performed during acute to subacute stages. However, cognitive tests (e.g., learning and memory) are often confounded by acute systemic disturbances and sensorimotor deficits and are thus suitable in later stages. In principle, therapeutic interventions should be tested in more than one species in a stratified manner. For practical reasons one can start with smaller, high-throughput species (e.g., rodents), and escalate to larger and preferably gyrencephalic species (e.g., pigs) if the results are promising. Similarly, interventions should be tested in more than one experimental model. All models have advantages and disadvantages, and none has been shown to be more predictive than the others, precluding an algorithmic approach. When selecting models, one has to keep in mind their strengths and caveats, as well as the relevance of model readouts for the targeted mechanism. Although initial screening in high-throughput models may be justified, we advocate a multimodal approach to provide information about tissue and neurological outcomes. Physiological (e.g., CBF), neurological (e.g., sensorimotor or cognitive deficits) and tissue (e.g., neuroimaging or histological) readouts should be collected and reconciled in each animal as much as possible. Furthermore, rigor and reproducibility are indispensible. Experiments should adhere to principles of good laboratory practice, including randomization and blinding, and predetermined exclusion criteria (or adopting intention-to-treat), primary and secondary endpoints, and sample sizes for sufficient statistical power. We recommend including equal numbers of male and female animals in the initial cohort, but powering the study for the pooled sample size with sex as an independent variable. If a trend for sex differences is observed in outcomes of this initial cohort, sample size can be increased for within-sex comparisons. While knowing the estrus cycle stage of the females is of scientific and biological significance, it is not a relevant parameter for clinical translation, since clinical trials do not include or stratify women based on their estrus cycle stage at the time of the event.
In conclusion, there are important limitations to each animal model of SAH that contribute to the difficulty in translating preclinical findings to effective therapies. We have also learned that mechanistic readouts such as vasospasm cannot substitute for readouts with direct clinical relevance such as tissue and functional outcomes. In order to enhance clinical relevance and predictive value for DCI and DND, we should dedicate more time and effort to develop better experimental models, rather than continue using previously published models without modification. We must better define the characteristics of existing models in multiple species and perhaps develop a battery of models representing different aspects of aneurysmal SAH as testing grounds. Finally, we must adhere to good study design and reporting principles to enhance the collective effort [36].
References
Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nature reviews. Neurology. 2014;10(1):44–58.
Zoerle T, Ilodigwe DC, Wan H, Lakovic K, Sabri M, Ai J, et al. Pharmacologic reduction of angiographic vasospasm in experimental subarachnoid hemorrhage: systematic review and meta-analysis. J Cereb Blood Flow Metab. 2012;32(9):1645–58.
Macdonald RL, Kassell NF, Mayer S, Ruefenacht D, Schmiedek P, Weidauer S, et al. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1): randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke. 2008;39(11):3015–21.
van der Worp HB, Howells DW, Sena ES, Porritt MJ, Rewell S, O’Collins V, et al. Can animal models of disease reliably inform human studies? PLoS Med. 2010;7(3):e1000245.
Dreier JP, Major S, Manning A, Woitzik J, Drenckhahn C, Steinbrink J, et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain J Neurol. 2009;132(Pt 7):1866–81.
Naraoka M, Matsuda N, Shimamura N, Asano K, Ohkuma H. The role of arterioles and the microcirculation in the development of vasospasm after aneurysmal SAH. Biomed Res Int. 2014;2014:253746.
Chung DY, Oka F, Ayata C. Spreading depolarizations: a therapeutic target against delayed cerebral ischemia after subarachnoid hemorrhage. J Clin Neurophysiol. 2016;33(3):196–202.
Garzon-Muvdi T, Pradilla G, Ruzevick JJ, Bender M, Edwards L, Grossman R, et al. A glutamate receptor antagonist, S-4-carboxyphenylglycine (S-4-CPG), inhibits vasospasm after subarachnoid hemorrhage in haptoglobin 2-2 mice [corrected]. Neurosurgery. 2013;73(4):719–28 (discussion 729).
Nyberg C, Karlsson T, Hillered L, Ronne Engstrom E. Metabolic pattern of the acute phase of subarachnoid hemorrhage in a novel porcine model: studies with cerebral microdialysis with high temporal resolution. PLoS ONE. 2014;9(6):e99904.
Hamming AM, van der Toorn A, Rudrapatna US, Ma L, van Os HJ, Ferrari MD, et al. Valproate reduces delayed brain injury in a rat model of subarachnoid hemorrhage. Stroke. 2017;48(2):452–8.
Oka F, Hoffmann U, Lee JH, Shin HK, Chung DY, Yuzawa I, et al. Requisite ischemia for spreading depolarization occurrence after subarachnoid hemorrhage in rodents. J Cereb Blood Flow Metab. 2017;37(5):1829–40.
Marbacher S, Fandino J, Kitchen ND. Standard intracranial in vivo animal models of delayed cerebral vasospasm. Br J Neurosurg. 2010;24(4):415–34.
Bassiouni H, Schulz R, Dorge H, Stolke D, Heusch G. The impact of subarachnoid hemorrhage on regional cerebral blood flow and large-vessel diameter in the canine model of chronic vasospasm. J Stroke Cerebrovasc Dis. 2007;16(2):45–51.
Siler DA, Martini RP, Ward JP, Nelson JW, Borkar RN, Zuloaga KL, et al. Protective role of p450 epoxyeicosanoids in subarachnoid hemorrhage. Neurocrit Care. 2015;22(2):306–19.
Povlsen GK, Edvinsson L. MEK1/2 inhibitor U0126 but not endothelin receptor antagonist clazosentan reduces upregulation of cerebrovascular contractile receptors and delayed cerebral ischemia, and improves outcome after subarachnoid hemorrhage in rats. J Cereb Blood Flow Metab. 2015;35(2):329–37.
Guresir E, Vasiliadis N, Dias S, Raab P, Seifert V, Vatter H. The effect of common carotid artery occlusion on delayed brain tissue damage in the rat double subarachnoid hemorrhage model. Acta Neurochir. 2012;154(1):11–9.
Wang H, James ML, Venkatraman TN, Wilson LJ, Lyuboslavsky P, Myers SJ, et al. pH-sensitive NMDA inhibitors improve outcome in a murine model of SAH. Neurocrit Care. 2014;20(1):119–31.
Mutoh T, Mutoh T, Sasaki K, Yamamoto Y, Tsuru Y, Tsubone H, et al. Isoflurane postconditioning with cardiac support promotes recovery from early brain injury in mice after severe subarachnoid hemorrhage. Life Sci. 2016;153:35–40.
Mutoh T, Mutoh T, Nakamura K, Yamamoto Y, Tsuru Y, Tsubone H, et al. Acute cardiac support with intravenous milrinone promotes recovery from early brain injury in a murine model of severe subarachnoid haemorrhage. Clin Exp Pharmacol Physiol. 2017;44(4):463–9.
Mutoh T, Mutoh T, Nakamura K, Sasaki K, Tatewaki Y, Ishikawa T, et al. Inotropic support against early brain injury improves cerebral hypoperfusion and outcomes in a murine model of subarachnoid hemorrhage. Brain Res Bull. 2017;130:18–26.
Mutoh T, Mutoh T, Sasaki K, Nakamura K, Tatewaki Y, Ishikawa T, et al. Neurocardiac protection with milrinone for restoring acute cerebral hypoperfusion and delayed ischemic injury after experimental subarachnoid hemorrhage. Neurosci Lett. 2017;640:70–5.
van den Bergh WM, Schepers J, Veldhuis WB, Nicolay K, Tulleken CA, Rinkel GJ. Magnetic resonance imaging in experimental subarachnoid haemorrhage. Acta Neurochir. 2005;147(9):977–83 (discussion 983).
Hamming AM, Wermer MJ, Umesh Rudrapatna S, Lanier C, van Os HJ, van den Bergh WM, et al. Spreading depolarizations increase delayed brain injury in a rat model of subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2016;36(7):1224–31.
Macdonald RL, Weir BK. A review of hemoglobin and the pathogenesis of cerebral vasospasm. Stroke. 1991;22(8):971–82.
Megyesi JF, Vollrath B, Cook DA, Findlay JM. In vivo animal models of cerebral vasospasm: a review. Neurosurgery. 2000;46(2):448–60 (discussion 460–1).
Vergouwen MD, Vermeulen M, Coert BA, Stroes ES, Roos YB. Microthrombosis after aneurysmal subarachnoid hemorrhage: an additional explanation for delayed cerebral ischemia. J Cereb Blood Flow Metab. 2008;28(11):1761–70.
Rowland MJ, Hadjipavlou G, Kelly M, Westbrook J, Pattinson KT. Delayed cerebral ischaemia after subarachnoid haemorrhage: looking beyond vasospasm. Br J Anaesth. 2012;109(3):315–29.
Tada Y, Wada K, Shimada K, Makino H, Liang EI, Murakami S, et al. Roles of hypertension in the rupture of intracranial aneurysms. Stroke. 2014;45(2):579–86.
Tada Y, Kanematsu Y, Kanematsu M, Nuki Y, Liang EI, Wada K, et al. A mouse model of intracranial aneurysm: technical considerations. Acta Neurochir Suppl. 2011;111:31–5.
Nuki Y, Tsou TL, Kurihara C, Kanematsu M, Kanematsu Y, Hashimoto T. Elastase-induced intracranial aneurysms in hypertensive mice. Hypertension. 2009;54(6):1337–44.
Kanematsu Y, Kanematsu M, Kurihara C, Tada Y, Tsou TL, van Rooijen N, et al. Critical roles of macrophages in the formation of intracranial aneurysm. Stroke. 2011;42(1):173–8.
Zhao J, Lin X, He C, Yang GY, Ling F. Study of cerebral aneurysms in a modified rat model: from real-time imaging to histological analysis. J Clin Neurosci. 2015;22(2):373–7.
Makino H, Tada Y, Wada K, Liang EI, Chang M, Mobashery S, et al. Pharmacological stabilization of intracranial aneurysms in mice: a feasibility study. Stroke. 2012;43(9):2450–6.
Jeon H, Ai J, Sabri M, Tariq A, Shang X, Chen G, et al. Neurological and neurobehavioral assessment of experimental subarachnoid hemorrhage. BMC Neurosci. 2009;10:103.
Mandeville ET, Ayata C, Zheng Y, Mandeville JB. Translational MR neuroimaging of stroke and recovery. Transl Stroke Res. 2017;8(1):22–32.
Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8(6):e1000412.
Luo C, Yao X, Li J, He B, Liu Q, Ren H, et al. Paravascular pathways contribute to vasculitis and neuroinflammation after subarachnoid hemorrhage independently of glymphatic control. Cell Death Dis. 2016;7:e2160.
McConnell ED, Wei HS, Reitz KM, Kang H, Takano T, Vates GE, et al. Cerebral microcirculatory failure after subarachnoid hemorrhage is reversed by hyaluronidase. J Cereb Blood Flow Metab. 2016;36(9):1537–52.
Sabri M, Ai J, Lakovic K, D’Abbondanza J, Ilodigwe D, Macdonald RL. Mechanisms of microthrombi formation after experimental subarachnoid hemorrhage. Neuroscience. 2012;224:26–37.
Sabri M, Ai J, Lakovic K, Macdonald RL. Mechanisms of microthrombosis and microcirculatory constriction after experimental subarachnoid hemorrhage. Acta Neurochir Suppl. 2013;115:185–92.
Sabri M, Ai J, Lass E, D’Abbondanza J, Macdonald RL. Genetic elimination of eNOS reduces secondary complications of experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2013;33(7):1008–14.
Vergouwen MD, Knaup VL, Roelofs JJ, de Boer OJ, Meijers JC. Effect of recombinant ADAMTS-13 on microthrombosis and brain injury after experimental subarachnoid hemorrhage. JTH. 2014;12(6):943–7.
Yagi K, Lidington D, Wan H, Fares JC, Meissner A, Sumiyoshi M, et al. Therapeutically targeting tumor necrosis factor-alpha/sphingosine-1-phosphate signaling corrects myogenic reactivity in subarachnoid hemorrhage. Stroke. 2015;46(8):2260–70.
Parra A, McGirt MJ, Sheng H, Laskowitz DT, Pearlstein RD, Warner DS. Mouse model of subarachnoid hemorrhage associated cerebral vasospasm: methodological analysis. Neurol Res. 2002;24(5):510–6.
Sheng H, Spasojevic I, Tse HM, Jung JY, Hong J, Zhang Z, et al. Neuroprotective efficacy from a lipophilic redox-modulating Mn(III) N-Hexylpyridylporphyrin, MnTnHex-2-PyP: rodent models of ischemic stroke and subarachnoid hemorrhage. J Pharmacol Exp Ther. 2011;338(3):906–16.
Vellimana AK, Milner E, Azad TD, Harries MD, Zhou ML, Gidday JM, et al. Endothelial nitric oxide synthase mediates endogenous protection against subarachnoid hemorrhage-induced cerebral vasospasm. Stroke. 2011;42(3):776–82.
Pisapia JM, Xu X, Kelly J, Yeung J, Carrion G, Tong H, et al. Microthrombosis after experimental subarachnoid hemorrhage: time course and effect of red blood cell-bound thrombin-activated pro-urokinase and clazosentan. Exp Neurol. 2012;233(1):357–63.
Fujimoto M, Shiba M, Kawakita F, Liu L, Shimojo N, Imanaka-Yoshida K, et al. Deficiency of tenascin-C and attenuation of blood-brain barrier disruption following experimental subarachnoid hemorrhage in mice. J Neurosurg. 2016;124(6):1693–702.
Muroi C, Kashiwagi Y, Rokugawa T, Tonomura M, Obata A, Nevzati E, et al. Evaluation of a filament perforation model for mouse subarachnoid hemorrhage using 7.0 Tesla MRI. J Clin Neurosci. 2016;28:141–7.
Provencio JJ, Swank V, Lu H, Brunet S, Baltan S, Khapre RV, et al. Neutrophil depletion after subarachnoid hemorrhage improves memory via NMDA receptors. Brain Behav Immun. 2016;54:233–42.
Solomon RA, McCormack BM, Lovitz RN, Swift DM, Hegemann MT. Elevation of brain norepinephrine concentration after experimental subarachnoid hemorrhage. Neurosurgery. 1986;19(3):363–6.
Swift DM, Solomon RA. Subarachnoid hemorrhage fails to produce vasculopathy or chronic blood flow changes in rats. Stroke. 1988;19(7):878–82.
Jackowski A, Crockard A, Burnstock G, Russell RR, Kristek F. The time course of intracranial pathophysiological changes following experimental subarachnoid haemorrhage in the rat. J Cereb Blood Flow Metab. 1990;10(6):835–49.
Ram Z, Sadeh M, Shacked I, Sahar A, Hadani M. Magnesium sulfate reverses experimental delayed cerebral vasospasm after subarachnoid hemorrhage in rats. Stroke. 1991;22(7):922–7.
Germano A, Imperatore C, d’Avella D, Costa G, Tomasello F. Antivasospastic and brain-protective effects of a hydroxyl radical scavenger (AVS) after experimental subarachnoid hemorrhage. J Neurosurg. 1998;88(6):1075–81.
Tekkok IH, Tekkok S, Ozcan OE, Erbengi T, Erbengi A. Preventive effect of intracisternal heparin for proliferative angiopathy after experimental subarachnoid haemorrhage in rats. Acta Neurochir. 1994;127(1–2):112–7.
Suzuki H, Kanamaru K, Tsunoda H, Inada H, Kuroki M, Sun H, et al. Heme oxygenase-1 gene induction as an intrinsic regulation against delayed cerebral vasospasm in rats. J Clin Investig. 1999;104(1):59–66.
Ryba MS, Gordon-Krajcer W, Walski M, Chalimoniuk M, Chrapusta SJ. Hydroxylamine attenuates the effects of simulated subarachnoid hemorrhage in the rat brain and improves neurological outcome. Brain Res. 1999;850(1–2):225–33.
Germano A, d’Avella D, Imperatore C, Caruso G, Tomasello F. Time-course of blood-brain barrier permeability changes after experimental subarachnoid haemorrhage. Acta Neurochir. 2000;142(5):575–80 (discussion 580–1).
Yamamoto S, Teng W, Nishizawa S, Kakiuchi T, Tsukada H. Improvement in cerebral blood flow and metabolism following subarachnoid hemorrhage in response to prophylactic administration of the hydroxyl radical scavenger, AVS, (±)-N, N’-propylenedinicotinamide: a positron emission tomography study in rats. J Neurosurg. 2000;92(6):1009–15.
Aladag MA, Turkoz Y, Sahna E, Parlakpinar H, Gul M. The attenuation of vasospasm by using a sod mimetic after experimental subarachnoidal haemorrhage in rats. Acta Neurochir. 2003;145(8):673–7.
Yilmaz C, Cansever T, Kircelli A, Isiksacan Ozen O, Aydemir F, Akar A, et al. The effects of proanthocyanidins on vasospasm after experimental subarachnoid hemorrhage in rats. Turk Neurosurg. 2015;28(4):667–74.
Pappas AC, Koide M, Wellman GC. Astrocyte Ca2+ signaling drives inversion of neurovascular coupling after subarachnoid hemorrhage. J Neurosci. 2015;35(39):13375–84.
Sun Y, Shen Q, Watts LT, Muir ER, Huang S, Yang GY, et al. Multimodal MRI characterization of experimental subarachnoid hemorrhage. Neuroscience. 2016;316:53–62.
Konczalla J, Wanderer S, Mrosek J, Gueresir E, Schuss P, Platz J, et al. Levosimendan, a new therapeutic approach to prevent delayed cerebral vasospasm after subarachnoid hemorrhage? Acta Neurochir. 2016;158(11):2075–83.
Wang Z, Chen G, Zhu WW, Bian JY, Shen XO, Zhou D. Influence of simvastatin on microthrombosis in the brain after subarachnoid hemorrhage in rats: a preliminary study. Ann Clin Lab Sci. 2010;40(1):32–42.
Larsen CC, Povlsen GK, Rasmussen MN, Edvinsson L. Improvement in neurological outcome and abolition of cerebrovascular endothelin B and 5-hydroxytryptamine 1B receptor upregulation through mitogen-activated protein kinase kinase 1/2 inhibition after subarachnoid hemorrhage in rats. J Neurosurg. 2011;114(4):1143–53.
Maddahi A, Povlsen GK, Edvinsson L. Regulation of enhanced cerebrovascular expression of proinflammatory mediators in experimental subarachnoid hemorrhage via the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase pathway. J Neuroinflammation. 2012;9:274.
Povlsen GK, Johansson SE, Larsen CC, Samraj AK, Edvinsson L. Early events triggering delayed vasoconstrictor receptor upregulation and cerebral ischemia after subarachnoid hemorrhage. BMC Neurosci. 2013;14:34.
Zhang XS, Zhang X, Zhou ML, Zhou XM, Li N, Li W, et al. Amelioration of oxidative stress and protection against early brain injury by astaxanthin after experimental subarachnoid hemorrhage. J Neurosurg. 2014;121(1):42–54.
Muller AH, Edwards AVG, Larsen MR, Nielsen J, Warfvinge K, Povlsen GK, et al. Proteomic expression changes in large cerebral arteries after experimental subarachnoid hemorrhage in rat are regulated by the MEK-ERK1/2 pathway. J Mol Neurosci. 2017;62(3–4):380–94.
Matz PG, Sundaresan S, Sharp FR, Weinstein PR. Induction of HSP70 in rat brain following subarachnoid hemorrhage produced by endovascular perforation. J Neurosurg. 1996;85(1):138–45.
Sun BL, Xia ZL, Yang MF, Qiu PM. Effects of Ginkgo biloba extract on somatosensory evoked potential, nitric oxide levels in serum and brain tissue in rats with cerebral vasospasm after subarachnoid hemorrhage. Clin Hemorheol Microcirc. 2000;23(2–4):139–44.
Sun BL, Zhang SM, Xia ZL, Yang MF, Yuan H, Zhang J, et al. The effects of nimodipine on regional cerebral blood flow, brain water and electrolyte contents in rats with subarachnoid hemorrhage. Clin Hemorheol Microcirc. 2003;29(3–4):337–44.
Yang MF, Sun BL, Xia ZL, Zhu LZ, Qiu PM, Zhang SM. Alleviation of brain edema by l-arginine following experimental subarachnoid hemorrhage in a rat model. Clin Hemorheol Microcirc. 2003;29(3–4):437–43.
Sehba FA, Mostafa G, Knopman J, Friedrich V Jr, Bederson JB. Acute alterations in microvascular basal lamina after subarachnoid hemorrhage. J Neurosurg. 2004;101(4):633–40.
Bendel O, Prunell G, Stenqvist A, Mathiesen T, Holmin S, Svendgaard NA, et al. Experimental subarachnoid hemorrhage induces changes in the levels of hippocampal NMDA receptor subunit mRNA. Brain Res Mol Brain Res. 2005;137(1–2):119–25.
Torok E, Klopotowski M, Trabold R, Thal SC, Plesnila N, Scholler K. Mild hypothermia (33 degrees C) reduces intracranial hypertension and improves functional outcome after subarachnoid hemorrhage in rats. Neurosurgery. 2009;65(2):352–9 (discussion 359).
Suzuki H, Ayer R, Sugawara T, Chen W, Sozen T, Hasegawa Y, et al. Role of osteopontin in early brain injury after subarachnoid hemorrhage in rats. Acta Neurochir Suppl. 2011;110(Pt 1):75–9.
Westermaier T, Jauss A, Eriskat J, Kunze E. Roosen K. The temporal profile of cerebral blood flow and tissue metabolites indicates sustained metabolic depression after experimental subarachnoid hemorrhage in rats. Neurosurgery. 2011;68(1):223–9 (discussion 229–30).
Li Q, Chen Y, Zhang X, Zuo S, Ge H, Chen Y, et al. Scutellarin attenuates vasospasm through the Erk5-KLF2-eNOS pathway after subarachnoid hemorrhage in rats. J Clin Neurosci. 2016;34:264–70.
Thomas C, Vercouillie J, Domene A, Tauber C, Kassiou M, Guilloteau D et al. Detection of neuroinflammation in a rat model of subarachnoid hemorrhage using [18F]DPA-714 PET imaging. Mol Imaging 2016;15:1-8.
Wang L, Li M, Xie Y, Xu L, Ye R, Liu X. Preclinical efficacy of human Albumin in subarachnoid hemorrhage. Neuroscience. 2017;344:255–64.
Acikgoz B, Ozgen T, Ozdogan F, Sungur A, Tekkok IH. Angiotensin II receptor content within the subfornical organ and organum vasculosum lamina terminalis increases after experimental subarachnoid haemorrhage in rats. Acta Neurochir. 1996;138(4):460–5.
Alkan T, Tureyen K, Ulutas M, Kahveci N, Goren B, Korfali E, et al. Acute and delayed vasoconstriction after subarachnoid hemorrhage: local cerebral blood flow, histopathology, and morphology in the rat basilar artery. Arch Physiol Biochem. 2001;109(2):145–53.
Vollmer DG, Kassell NF, Hongo K, Ogawa H, Tsukahara T. Effect of the nonglucocorticoid 21-aminosteroid U74006F experimental cerebral vasospasm. Surg Neurol. 1989;31(3):190–4.
Nelson RJ, Perry S, Burns AC, Roberts J, Pickard JD. The effects of hyponatraemia and subarachnoid haemorrhage on the cerebral vasomotor responses of the rabbit. J Cereb Blood Flow Metab. 1991;11(4):661–6.
Ryba M, Grieb P, Walski M, Sawicki J, Pastuszko M. 2-Chloro-2′ deoxyadenosine prevents angiopathic changes in cerebral arteries in experimental SAH in rabbits. Acta Neurochir. 1993;122(1–2):118–21.
Imaizumi S, Shimizu H, Ahmad I, Kaminuma T, Tajima M, Yoshimoto T. Effect of calcitonin gene-related peptide on delayed cerebral vasospasm after experimental subarachnoid hemorrhage in rabbits. Surg Neurol. 1996;46(3):263–70 (discussion 270–1).
Ahmad I, Imaizumi S, Shimizu H, Kaminuma T, Ochiai N, Tajima M, et al. Development of calcitonin gene-related peptide slow-release tablet implanted in CSF space for prevention of cerebral vasospasm after experimental subarachnoid haemorrhage. Acta Neurochir. 1996;138(10):1230–40.
Caner HH, Kwan AL, Arthur A, Jeng AY, Lappe RW, Kassell NF, et al. Systemic administration of an inhibitor of endothelin-converting enzyme for attenuation of cerebral vasospasm following experimental subarachnoid hemorrhage. J Neurosurg. 1996;85(5):917–22.
Zuccarello M, Soattin GB, Lewis AI, Breu V, Hallak H, Rapoport RM. Prevention of subarachnoid hemorrhage-induced cerebral vasospasm by oral administration of endothelin receptor antagonists. J Neurosurg. 1996;84(3):503–7.
Kwan AL, Bavbek M, Jeng AY, Maniara W, Toyoda T, Lappe RW, et al. Prevention and reversal of cerebral vasospasm by an endothelin-converting enzyme inhibitor, CGS 26303, in an experimental model of subarachnoid hemorrhage. J Neurosurg. 1997;87(2):281–6.
Kaminuma T, Shimizu H, Ahmad I, Ochiai N, Ehama R, Ohnuma M, et al. Prevention of cerebral vasospasm by vasodilatory peptide maxadilan following subarachnoid hemorrhage in rabbits. J Control Release. 1998;52(1–2):71–80.
Ishiguro M, Wellman TL, Honda A, Russell SR, Tranmer BI, Wellman GC. Emergence of a R-type Ca2+ channel (CaV 2.3) contributes to cerebral artery constriction after subarachnoid hemorrhage. Circ Res. 2005;96(4):419–26.
Miller CA, Lombard FW, Wu CT, Hubbard CJ, Silbajoris L, Borel CO, et al. Role of vascular mitogens in subarachnoid hemorrhage-associated cerebral vasculopathy. Neurocrit Care. 2006;5(3):215–21.
Zheng R, Qin L, Li S, Xu K, Geng H. CT perfusion-derived mean transit time of cortical brain has a negative correlation with the plasma level of Nitric Oxide after subarachnoid hemorrhage. Acta Neurochir. 2014;156(3):527–33.
Egemen N, Sanlidilek U, Zorlutuna A, Baskaya M, Bilgic S, Caglar S, et al. Transclival approach to rabbit basilar artery for experimental induction of chronic vasospasm. Acta Neurochir. 1992;115(3–4):123–6.
Morooka H. Cerebral aterial spasm. I. Adrenergic mechanism in experimental cerebral vasospasm. Acta Med Okayama. 1978;32(1):23–37.
Gavras H, Andrews P, Papadakis N. Reversal of experimental delayed cerebral vasospasm by angiotensin-converting enzyme inhibition. J Neurosurg. 1981;55(6):884–8.
Varsos VG, Liszczak TM, Han DH, Kistler JP, Vielma J, Black PM, et al. Delayed cerebral vasospasm is not reversible by aminophylline, nifedipine, or papaverine in a “two-hemorrhage” canine model. J Neurosurg. 1983;58(1):11–7.
Alexander E 3rd, Black PM, Liszczak TM, Zervas NT. Delayed CSF lavage for arteriographic and morphological vasospasm after experimental SAH. J Neurosurg. 1985;63(6):949–58.
Takayasu M, Suzuki Y, Shibuya M, Asano T, Kanamori M, Okada T, et al. The effects of HA compound calcium antagonists on delayed cerebral vasospasm in dogs. J Neurosurg. 1986;65(1):80–5.
Uemura Y, Okamoto S, Handa Y, Handa H. Disturbance in the intramural circulation of the major cerebro-pial arteries after experimental subarachnoid haemorrhage. Acta Neurochir. 1987;89(1–2):71–6.
Watanabe T, Asano T, Shimizu T, Seyama Y, Takakura K. Participation of lipoxygenase products from arachidonic acid in the pathogenesis of cerebral vasospasm. J Neurochem. 1988;50(4):1145–50.
Shibuya M, Suzuki Y, Takayasu M, Asano T, Harada T, Ikegaki I, et al. The effects of an intracellular calcium antagonist HA 1077 on delayed cerebral vasospasm in dogs. Acta Neurochir. 1988;90(1–2):53–9.
Seifert V, Eisert WG, Stolke D, Goetz C. Efficacy of single intracisternal bolus injection of recombinant tissue plasminogen activator to prevent delayed cerebral vasospasm after experimental subarachnoid hemorrhage. Neurosurgery. 1989;25(4):590–8.
Yokota M, Tani E, Fukumori T, Maeda Y, Yamaura I. Effects of subarachnoid hemorrhage and a thromboxane A2 synthetase inhibitor on intracranial prostaglandins. Surg Neurol. 1991;35(5):345–9.
Diringer MN, Heffez DS, Monsein L, Kirsch JR, Hanley DF, Traystman RJ. Cerebrovascular CO2 reactivity during delayed vasospasm in a canine model of subarachnoid hemorrhage. Stroke. 1991;22(3):367–72.
Matsumura Y, Ikegawa R, Suzuki Y, Takaoka M, Uchida T, Kido H, et al. Phosphoramidon prevents cerebral vasospasm following subarachnoid hemorrhage in dogs: the relationship to endothelin-1 levels in the cerebrospinal fluid. Life Sci. 1991;49(11):841–8.
Kobayashi H, Ide H, Handa Y, Aradachi H, Arai Y, Kubota T. Effect of leukotriene antagonist on experimental delayed cerebral vasospasm. Neurosurgery. 1992;31(3):550–5 (discussion 555–6).
Itoh S, Sasaki T, Ide K, Ishikawa K, Nishikibe M, Yano M. A novel endothelin ETA receptor antagonist, BQ-485, and its preventive effect on experimental cerebral vasospasm in dogs. Biochem Biophys Res Commun. 1993;195(2):969–75.
Willette RN, Zhang H, Mitchell MP, Sauermelch CF, Ohlstein EH, Sulpizio AC. Nonpeptide endothelin antagonist. Cerebrovascular characterization and effects on delayed cerebral vasospasm. Stroke. 1994;25(12):2450–5 (discussion 2456).
Bulter WE, Peterson JW, Zervas NT, Morgan KG. Intracellular calcium, myosin light chain phosphorylation, and contractile force in experimental cerebral vasospasm. Neurosurgery. 1996;38(4):781–7 (discussion 787–8).
Khajavi K, Ayzman I, Shearer D, Jones SC, Levy JH, Prayson RA, et al. Prevention of chronic cerebral vasospasm in dogs with milrinone. Neurosurgery. 1997;40(2):354–62 (discussion 362–3).
Ohkuma H, Itoh K, Shibata S, Suzuki S. Morphological changes of intraparenchymal arterioles after experimental subarachnoid hemorrhage in dogs. Neurosurgery. 1997;41(1):230–5 (discussion 235–6).
Roux S, Breu V, Giller T, Neidhart W, Ramuz H, Coassolo P, et al. Ro 61-1790, a new hydrosoluble endothelin antagonist: general pharmacology and effects on experimental cerebral vasospasm. J Pharmacol Exp Ther. 1997;283(3):1110–8.
Watanabe T, Nishiyama M, Hori T, Asano T, Shimizu T, Masayasu H. Ebselen (DR3305) ameliorates delayed cerebral vasospasm in a canine two-hemorrhage model. Neurol Res. 1997;19(5):563–5.
Kita T, Kubo K, Hiramatsu K, Sakaki T, Yonetani Y, Sato S, et al. Profiles of an intravenously available endothelin A-receptor antagonist, S-0139, for preventing cerebral vasospasm in a canine two-hemorrhage model. Life Sci. 1998;63(4):305–15.
Wolf EW, Banerjee A, Soble-Smith J, Dohan FC Jr, White RP, Robertson JT. Reversal of cerebral vasospasm using an intrathecally administered nitric oxide donor. J Neurosurg. 1998;89(2):279–88.
Onoue H, Katusic ZS. The effect of subarachnoid hemorrhage on mechanisms of vasodilation mediated by cyclic adenosine monophosphate. J Neurosurg. 1998;89(1):111–7.
Cook DJ, Kan S, Ai J, Kasuya H, Macdonald RL. Cisternal sustained release dihydropyridines for subarachnoid hemorrhage. Curr Neurovasc Res. 2012;9(2):139–48.
Mori K. Double cisterna magna blood injection model of experimental subarachnoid hemorrhage in dogs. Transl Stroke Res. 2014;5(6):647–52.
Hanggi D, Etminan N, Steiger HJ, Johnson M, Peet MM, Tice T, et al. A site-specific, sustained-release drug delivery system for aneurysmal subarachnoid hemorrhage. Neurotherapeutics. 2016;13(2):439–49.
Shiokawa K, Kasuya H, Miyajima M, Izawa M, Takakura K. Prophylactic effect of papaverine prolonged-release pellets on cerebral vasospasm in dogs. Neurosurgery. 1998;42(1):109–15 (discussion 115–6).
Zibly Z, Fein L, Sharma M, Assaf Y, Wohl A, Harnof S. A novel swine model of subarachnoid hemorrhage-induced cerebral vasospasm. Neurol India. 2017;65(5):1035–42.
Espinosa F, Weir B, Overton T, Castor W, Grace M, Boisvert D. A randomized placebo-controlled double-blind trial of nimodipine after SAH in monkeys. Part 1: clinical and radiological findings. J Neurosurg. 1984;60(6):1167–75.
Nosko M, Weir B, Krueger C, Cook D, Norris S, Overton T, et al. Nimodipine and chronic vasospasm in monkeys: part 1. Clinical and radiological findings. Neurosurgery. 1985;16(2):129–36.
Nosko M, Weir BK, Lunt A, Grace M, Allen P, Mielke B. Effect of clot removal at 24 hours on chronic vasospasm after SAH in the primate model. J Neurosurg. 1987;66(3):416–22.
Findlay JM, Weir BK, Steinke D, Tanabe T, Gordon P, Grace M. Effect of intrathecal thrombolytic therapy on subarachnoid clot and chronic vasospasm in a primate model of SAH. J Neurosurg. 1988;69(5):723–35.
Findlay JM, Weir BK, Gordon P, Grace M, Baughman R. Safety and efficacy of intrathecal thrombolytic therapy in a primate model of cerebral vasospasm. Neurosurgery. 1989;24(4):491–8.
Findlay JM, Weir BK, Kanamaru K, Grace M, Baughman R. The effect of timing of intrathecal fibrinolytic therapy on cerebral vasospasm in a primate model of subarachnoid hemorrhage. Neurosurgery. 1990;26(2):201–6.
Pluta RM, Deka-Starosta A, Zauner A, Morgan JK, Muraszko KM, Oldfield EH. Neuropeptide Y in the primate model of subarachnoid hemorrhage. J Neurosurg. 1992;77(3):417–23.
Handa Y, Kubota T, Tsuchida A, Kaneko M, Caner H, Kobayashi H, et al. Effect of systemic hypotension on cerebral energy metabolism during chronic cerebral vasospasm in primates. J Neurosurg. 1993;78(1):112–9.
Hariton GB, Findlay JM, Weir BK, Kasuya H, Grace MG, Mielke BW. Comparison of intrathecal administration of urokinase and tissue plasminogen activator on subarachnoid clot and chronic vasospasm in a primate model. Neurosurgery. 1993;33(4):691–6 (discussion 696–7).
Inoue T, Shimizu H, Kaminuma T, Tajima M, Watabe K, Yoshimoto T. Prevention of cerebral vasospasm by calcitonin gene-related peptide slow-release tablet after subarachnoid hemorrhage in monkeys. Neurosurgery. 1996;39(5):984–90.
Pluta RM, Boock RJ, Afshar JK, Clouse K, Bacic M, Ehrenreich H, et al. Source and cause of endothelin-1 release into cerebrospinal fluid after subarachnoid hemorrhage. J Neurosurg. 1997;87(2):287–93.
Horky LL, Pluta RM, Boock RJ, Oldfield EH. Role of ferrous iron chelator 2,2′-dipyridyl in preventing delayed vasospasm in a primate model of subarachnoid hemorrhage. J Neurosurg. 1998;88(2):298–303.
Pluta RM, Afshar JK, Boock RJ, Oldfield EH. Temporal changes in perivascular concentrations of oxyhemoglobin, deoxyhemoglobin, and methemoglobin after subarachnoid hemorrhage. J Neurosurg. 1998;88(3):557–61.
Pluta RM, Dejam A, Grimes G, Gladwin MT, Oldfield EH. Nitrite infusions to prevent delayed cerebral vasospasm in a primate model of subarachnoid hemorrhage. JAMA. 2005;293(12):1477–84.
Pluta RM, Butman JA, Schatlo B, Johnson DL, Oldfield EH. Subarachnoid hemorrhage and the distribution of drugs delivered into the cerebrospinal fluid. Laboratory investigation. J Neurosurg. 2009;111(5):1001–7.
Norwood CW, Poole J, Moody D. Treatment of experiment delayed cerebral arterial spasm with a beta2-adrenergic stimulator and a phosphodiesterase inhibitor. J Neurosurg. 1976;45(5):491–7.
Boisvert DP, Pickard JD, Graham DI, Fitch W. Delayed effects of subarachnoid haemorrhage on cerebral metabolism and the cerebrovascular response to hypercapnia in the primate. J Neurol Neurosurg Psychiatry. 1979;42(10):892–8.
Espinosa F, Weir B, Boisvert D, Overton T, Castor W. Chronic cerebral vasospasm after large subarachnoid hemorrhage in monkeys. J Neurosurg. 1982;57(2):224–32.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Supported by Grants from the Japanese Heart Foundation/Bayer Yakuhin Research Grant Abroad, NIH (P01NS055104, R01NS102969, R25NS065743, and KL2TR002542), the Foundation Leducq, the Heitman Foundation, the Ellison Foundation, the Brain Aneurysm Foundation’s Timothy P. Susco and Andrew David Heitman Foundation Chairs of Research, the Aneurysm and AVM Foundation, and the American Heart Association (18POST34030369).
Author information
Authors and Affiliations
Contributions
FO collected, analyzed the data, and wrote manuscript; DYC analyzed the data and wrote manuscript; MS edited the manuscript; CA conceived the study, analyzed the data, and wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Rights and permissions
About this article

Cite this article
Oka, F., Chung, D.Y., Suzuki, M. et al. Delayed Cerebral Ischemia After Subarachnoid Hemorrhage: Experimental-Clinical Disconnect and the Unmet Need. Neurocrit Care 32, 238–251 (2020). https://doi.org/10.1007/s12028-018-0650-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-018-0650-5