
Abstract
Background
This study investigated if cerebral blood flow (CBF) regulation by changes of the arterial partial pressure of carbon dioxide (PaCO2) can be used therapeutically to increase CBF and improve neurological outcome after subarachnoid hemorrhage (SAH).
Methods
In 12 mechanically ventilated poor-grade SAH-patients, a daily trial intervention was performed between day 4 and 14. During this intervention, PaCO2 was decreased to 30 mmHg and then gradually increased to 40, 50, and 60 mmHg in 15-min intervals by modifications of the respiratory minute volume. CBF and brain tissue oxygen saturation (StiO2) were the primary and secondary endpoints. Intracranial pressure was controlled by an external ventricular drainage.
Results
CBF reproducibly decreased during hyperventilation and increased to a maximum of 141 ± 53 % of baseline during hypercapnia (PaCO2 60 mmHg) on all days between day 4 and 14 after SAH. Similarly, StiO2 increased during hypercapnia. CBF remained elevated within the first hour after resetting ventilation to baseline parameters and no rebound effect was observed within this time-span. PaCO2-reactivities of CBF and StiO2 were highest between 30 and 50 mmHg and slightly decreased at higher levels.
Conclusion
CBF and StiO2 reproducibly increased by controlled hypercapnia of up to 60 mmHg even during the period of the maximum expected vasospasm. The absence of a rebound effect within the first hour after hypercapnia indicates that an improvement of the protocol is possible. The intervention may yield a therapeutic potential to prevent ischemic deficits after aneurysmal SAH.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Under physiological conditions, cerebral blood flow (CBF) is determined by cerebral autoregulation, a self-regulating mechanism which adapts to changes of arterial wall tension and keeps CBF constant over a large mean arterial blood pressure (MABP) range, by an equilibrium of dilating and constricting local paracrine factors, and by the arterial partial pressure of carbon dioxide (PaCO2). After aneurysmal subarachnoid hemorrhage (SAH), autoregulation is deranged and the equilibrium of local factors is shifted towards contraction. In contrast, the reactivity of CBF upon changes of PaCO2 is still intact [1, 2]. There is experimental and clinical evidence that in cases of marginal CBF, such as during delayed vasospasm after SAH, hyperventilation induces ischemia and infarction due to an additional vessel constriction and is associated with poor outcome [3–6]. Recently, poor reactivity of CBF to changes of PaCO2 has been identified as a prognosticator of delayed ischemia after aneurysmal SAH [7]. However, in spite of the observation that CBF can be enhanced by higher PaCO2 in experimental studies and its clinical confirmation, it has not been investigated whether a manipulation of arterial carbon dioxide values in terms of hypercapnia may be used therapeutically to increase CBF and prevent ischemia after SAH. In a previous publication, we reported preliminary data of the first patients included in this study, indicating that an increase of CBF may in fact be achieved by transient hypercapnia [8]. The current article reports about a completed Phase 1 trial of hypercapnia for the treatment of poor-grade SAH patients focussing on changes of vessel reactivity and their relation to alterations of PaCO2 during the course of the disease.
Materials and Methods
The study was approved by the ethics committee of the medical faculty of the University of Wuerzburg, Germany (Wue112/12). Based on experimental data of Diringer et al. (increase of CBF from baseline to 265 ± 50 % of baseline by a PaCO2 increase of 20 mmHg) and clinical data of Schmieder et al. (increase of MFV in MCA of 30 cm/s by a PaCO2 increase of 12.7 %), a desired test-power of 0.8 and a level of significance of p < 0.05 (one-way repeated measurement ANOVA with 5 points of measurements), a sample size of 6 and 10 patients, respectively, were calculated. The predefined primary endpoint was an increase of CBF upon an elevation of PaCO2 from 40 to 60 mmHg for each individual day [1, 2]. For calculation of the sample size, G-Power 3.1 statistical software was used. Accordingly, the ethics vote covered 10 patients. Two further patients were included and treated for compassionate use according to the study protocol. Preliminary data of the first patients included in this trial have been reported previously [8]. This Phase 1 clinical trial was registered at ClinicalTrials.gov. (Trial-ID: NCT01799525, “Hypercapnia to Prevent Secondary Ischemia in SAH”).
Inclusion Criteria
Patients were eligible for inclusion if they had suffered aneurysmal SAH of a poor clinical (Hunt/Hess° 3–5) and radiological grade (Fisher° 3) within the last 96 h, had received an external ventricular drainage (EVD) and had undergone surgical or endovascular occlusion of the aneurysm within the first 72 h after SAH. In all patients, the EVD was placed into the right lateral ventricle via a burr hole at Kocher’s point. The written consent of a legal guardian was obtained before inclusion into the study.
Exclusion Criteria
Pregnancy, chronic obstructive lung disease, an intracranial pressure (ICP) of more than 20 mmHg on day 4 after SAH, and age under 18 years were predefined exclusion criteria. An arterial pH under 7.25, an ICP over 25 mmHg for more than 2 min, and spontaneous breathing during the intervention were the predefined interruption criteria.
Standard Care
If patients met the inclusion criteria, had received an EVD, and the aneurysm was occluded, sedative and analgesic medications were reduced. If the patient did not wake up appropriately or ICP increased to continuous values above 20 mmHg, sedative and analgesic medication were readministered. A wake-up attempt was not conducted in patients who had developed pneumonia from aspiration after aneurysm rupture. For this purpose, Midazolam (10–30 mg/h) and Fentanyl (60–180 µg/h) were administered and the patients were mechanically ventilated. If further pharmacological therapy of elevated ICP was required, Thiopental was administered under continuous electroencephalographic surveillance (100–350 mg/h). EVDs were leveled at 15 cm above the external auditory canal and continuously drained CSF. Temperature was continuously measured via a probe included in the indwelling catheter. Aggressive temperature control was performed using external cooling, antiphlogistic medication, and targeted antibiotic therapy, if indicated. During the trial interventions, pharmacological therapy was continued and no other drugs or relaxants were administered.
Respirator Settings
Patient underwent one daily trial intervention between day 4 and 14. For the trial intervention, all patients were mechanically ventilated in a volume-controlled mode (pressure trigger −5 cm H2O). After baseline measurements of arterial blood gases (PaCO2, pH, PaO2) and outcome parameters (see below), the respiratory minute volume (RMV) was, at first, increased from baseline values (PaCO2 36–44 mmHg) to reach a target PaCO2 of 30 mmHg. Subsequently, the RMV was reduced in 15-min intervals to reach PaCO2 values of 40, 50, and 60 mmHg. Thereafter, the respirator settings were reset to baseline values.
Primary Endpoint
CBF during the trial intervention was the primary endpoint. CBF was measured by an intracerebral thermodilution probe (Q-Flow 500®, Hemedex™, Cambridge, MA, USA). The probe was fixed in a bolt screw (BOLT KIT, Raumedic, Helmbrechts, Germany) which was tightly screwed into a burr hole in the right frontal bone 1.5 cm anterior to the entry point of the EVD (Kocher’s point). The probe was inserted 5 cm from the surface of the bolt screw and fixed in the screw. CBF was monitored by a Bowman Perfusion Monitor® (Hemedex™, Cambridge, MA, USA) and recorded throughout the 1-h trial intervention. After the end of the trial intervention, observation and CBF recording were continued for another hour to exclude a rebound effect.
Secondary Endpoints
StiO2 was measured transcutaneously by near infrared spectroscopy (NIRS) (INVOS®, Covidien, Neustadt/Donau, Germany) attached bilaterally on the forehead skin. Mean flow velocities (MFV) in both middle cerebral artery (MCA) trunks were determined by transcranial Doppler sonography (TCD) before and at each step of the trial intervention. TCD measurements were not continued after the end of the trial intervention. Heart rate (HR), MABP, and peripheral oxygen saturation were continuously monitored throughout the intervention. Cardiac Output (CO) was continuously measured using a FloTrac® sensor and monitored by a Vigileo® monitor (Edwards Life Science, Unterschleissheim, Germany).
Calculation of PaCO2-Reactivity of CBF and StiO2
The HEMEDEX™ thermodilution probe displays absolute values of CBF in the right frontal cortex (in ml/100 mg brain tissue/min). In each patient, CBF values at baseline and at PaCO2 target values of 30, 40, 50, and 60 mmHg were recorded. At the same time points, the actual PaCO2 values were determined by arterial blood gas analysis. The differences of CBF- and the actual PaCO2-values between the respective target points (40–30, 50–40, and 60–50) were calculated to determine the change of CBF and PaCO2 in the respective interval. Thereafter, the change of CBF was divided by the change of PaCO2 for each interval to calculate the PaCO2 reactivity of CBF (ml/100 mg/min/mmHg). Mean values ± standard deviations for the intervals 30–40, 50–40, and 60–50 mmHg are presented in Fig. 3. In analogy, the PaCO2-reactivity of StiO2 was calculated over both hemispheres. StiO2-values, displayed in % of maximum values, were recorded. The changes of StiO2 in the above-mentioned intervals were divided by the changes of PaCO2 in the respective intervals for each patient to obtain the PaCO2-reactivity of StiO2 (%/mmHg).
Computed tomography (CT) was obtained in 3-day intervals to monitor for newly appearing hypodensities [9] representing “delayed cerebral infarction.” In case of suspected vasospasm, angiography and, if possible, vasospasmolysis were performed. A final neurological examination was conducted after 6 months to evaluate “neurological recovery” including the assessment of the Glasgow Outcome Scale (GOS) score.
Documentation and Statistical Analysis
Values were documented in written protocols and transferred to electronic datasheets. Testing for normal distribution was performed by a D’Agostino and Pearson normality test. If normality testing was passed for all measurement points, a one-way analysis of variance for repeated measurements (one-way RM ANOVA) was applied, followed by a Dunnett’s multiple comparisons test comparing measurements at the respective time-points to baseline values. Statistical analyses were performed using GraphPad Prism 4.0 Statistical Software.
Results
Following the inclusion criteria, all patients had suffered from SAH of poor clinical and radiological grades (Table 1). All aneurysms were occluded within the first 2 days after SAH by surgical clipping (5 patients) or endovascular coiling (7 patients).
Ventilation and Arterial Blood Gases
From a baseline of 7.5 ± 1.3 l/min, the RMV was changed to 11.8 ± 2.3, 5.9 ± 1.4, 4.1 ± 1.0, and 2.9 ± 0.7 l/min to obtain a PaCO2 of 38.7 ± 3.8, 31.8 ± 2.5, 41.0 ± 2.6, 49.8 ± 2.5, and 60.7 ± 4.0 mmHg, respectively. Changes of pH, PaO2, and cardiocirculatory parameters are depicted in Table 2.
Cerebral Blood Flow
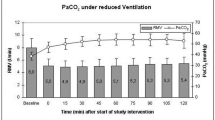
After reducing PaCO2 to 30 mmHg, CBF declined to a mean of 83 ± 35 % of baseline. Increasing PaCO2 to 40, 50, and 60 mmHg resulted in mean CBF values of 98 ± 41, 123 ± 48, and 141 ± 53 % of baseline, respectively. 15, 30, 45, and 60 min after resetting ventilation to baseline parameters, CBF gradually declined to 137 ± 39, 134 ± 47, 123 ± 35, and 114 ± 23 % of baseline values, respectively (Fig. 1).

Course of cerebral blood flow (CBF) during the hypercapnic trial intervention and 15, 30, 45, and 60 min after hypercapnia. CBF remained elevated after resetting the respirator to baseline parameters and slowly returned to pre-hypercapnia levels without a negative rebound effect. The gray columns indicate the respiratory minute volume (mean ± SD) before, at the different stages during, and after the trial intervention. Numbers within the columns represent the mean respiratory minute volume during the respective tome interval. After the end of the trial intervention, the respirator settings were returned to baseline, and pre-hypercapnia settings were maintained there during the following hour. (hyp + 15 = 15 min after resetting the ventilator to baseline, pre-hypercapnia, parameters, RMV respiratory minute volume, *p < 0.05, **p < 0.01 compared to baseline values, one-way RM ANOVA, Dunnett’s test for multiple comparisons)
Brain Tissue Oxygenation Saturation (StiO2)
StiO2, measured by NIRS over the right forebrain, decreased to 94 ± 4 % of baseline values at a PaCO2 of 30 mmHg and increased to 99.9 ± 4, 105 ± 6, and 110 ± 7 % at PaCO2 values of 40, 50, and 60 mmHg, respectively. 15, 30, 45, and 60 min after resetting ventilation to baseline parameters, StiO2 gradually declined to 109 ± 7, 106 ± 6, 103 ± 6, and 101 ± 5 % of baseline. Over the left forebrain, StiO2 decreased to 94 ± 5 % of baseline values at a PaCO2 of 30 mmHg and increased to 101 ± 6, 107 ± 7, and 113 ± 9 % at PaCO2 values of 40, 50, and 60 mmHg, respectively. 15, 30, 45, and 60 min after resetting ventilation to baseline parameters, StiO2 gradually declined to 113 ± 10, 108 ± 8, 104 ± 7, and 102 ± 6 % of baseline values. On both sides, the increase of StiO2 was statistically significant at PaCO2 values of 50 and 60 mmHg and 15, 30, 45, and 60 min after resetting the respirator to baseline values.
TCD
During the intervention, MFV in the left (right) MCA declined from a baseline of 81 ± 41 (81 ± 26)–68 ± 40 (63 ± 22) cm/s at a PaCO2 of 30 mmHg and rose to 95 ± 45 (96 ± 33), 114 ± 47 (117 ± 35), and 131 ± 45 (127 ± 36) mmHg at PaCO2 values of 40, 50, and 60 mmHg, respectively. All differences were significant compared to baseline values (Fig. 2).

Transcranial Doppler sonography during the trial intervention. The course of flow velocities in both middle cerebral arteries during the trial intervention is concordant with the course of CBF and StiO2 and indicates that the increase of CBF is a global phenomenon. (MFV mean flow velocity, MCA middle cerebral artery, *p < 0.05, **p < 0.01 compared to baseline values, one-way RM ANOVA, Dunnett’s test for multiple comparisons)
Side Effects
All patients had received an EVD prior to trial inclusion for continuous cerebrospinal fluid (CSF) drainage. Hence, only a mild decrease and increase of ICP from 11.2 ± 4.5 mmHg at baseline to 9.4 ± 4.3, 12.3 ± 4.3, 13.7 ± 4.9, and 13.9 ± 4.8 mmHg at a PaCO2 of 30, 40, 50, and 60 mmHg was observed during the trial intervention. The decrease and increase of ICP reached the level of significance. A potential further elevation of ICP under increasing PaCO2 was reflected by a statistically significant surplus drainage of CSF from 4.3 ± 4.5 ml over 15 min at baseline to 1.7 ± 1.3, 4.6 ± 4.0, 7.9 ± 6.3, and 7.4 ± 6.2 ml over 15 min at a PaCO2 of 30, 40, 50, and 60 mmHg. ICP and the amount of CSF drained during the trial intervention are depicted in Fig. 3.

CBF, ICP, and CSF collection during the trial intervention. The course of CBF, ICP, and the volume of CSF drained within the 15 min of each trial step are presented. During the increase of CBF, a minor but statistically significant increase of ICP occurred. More excessive elevations of ICP have obviously been prevented by a surplus drainage of CSF during the trial intervention. (*p < 0.05 vs. baseline, one-way RM ANOVA, Dunnett’s test for multiple comparisons)
Reactivity of CBF and StiO2 by PaCO2 Interval
PaCO2-reactivities of CBF and StiO2 were calculated for the intervals between PaCO2-steps during the intervention (between 30–40, 40–50, and 50–60 mmHg, respectively). PaCO2-reactivity of CBF was 0.45 ± 0.74, 0.56 ± 0.83, and 0.28 ± 0.44 ml/100 g/min/mmHg between a PaCO2 of 30–40, 40–50, and 50–60 mmHg, respectively (Fig. 4). PaCO2-reactivity of StiO2 measured over the left forebrain was 0.52 ± 0.43, 0.60 ± 0.73, and 0.46 ± 0.51 %/mmHg between a PaCO2 of 30–40, 40–50, and 50–60 mmHg, respectively. PaCO2-reactivity of StiO2 measured over the right forebrain was 0.54 ± 0.36, 0.48 ± 81, and 0.41 ± 0.47 %/mmHg between a PaCO2 of 30–40, 40–50, and 50–60 mmHg, respectively.

PaCO2-reactivity of CBF in different CO2-intervals. The reactivity of CBF is highest between 40 and 50 mmHg, slightly decreases at higher PaCO2-values. The differences of CBF- and actual PaCO2-values between the respective target points (40–30, 50–40, and 60–50 mmHg) were calculated to determine the change of CBF and PaCO2 in the respective interval. Thereafter, the change of CBF was divided by the change of PaCO2 for each interval to calculate the PaCO2 reactivity of CBF (ml/100 mg/min/mmHg). Mean values ± standard deviations for each interval are presented
Reactivity of CBF and StiO2 by Days After SAH
The increase of CBF by elevating PaCO2 to 60 mmHg was significant on days 4, 5, 7, 8, and 10–14, the increase of StiO2 on the left side on days 4–14, on the right side on days 4–6 and days 8–14 (Fig. 5). The increase of CBF by elevating PaCO2 to 50 mmHg was not on any day. The increase of StiO2 on the left side was significant on days 6–12 and day 14, and the increase of StiO2 on the right side on days 8, 12 and 13. There was a trend to a higher PaCO2-reactivity of CBF before day 5 and after day 9 after SAH which did not reach the level of significance.

Increase of CBF upon hypercapnia through the course of therapy. The elevation of PaCO2 to 60 mmHg resulted in a marked increase of CBF and StiO2 between days 4 and 14. While the increase of StiO2 was largely constant throughout the observation period, the increase of CBF tended to be lower between days 5 and 10. (*p < 0.05, **p < 0.01 compared to baseline values, one-way RM ANOVA, Dunnett’s test for multiple comparisons)
Clinical and Radiological Outcome
10 of 12 patients were forwarded to re-angiography because of suspected severe vasospasm by TCD and/or Perfusion-CT. Nine received balloon dilatation of spasms and/or pharmacological vasospasmolysis. One patient developed delayed cerebral infarction in the watershed zone between the left ACA and MCA territories in spite of vasospasmolysis of the left internal carotid artery. Patient characteristics and neurological outcome after 6 months are depicted in Table 1.
Discussion
In this Phase 1 study, we investigated the feasibility and safety of therapeutic hypercapnia in patients with poor-grade SAH. We found a reliable and reproducible improvement of the primary and secondary endpoints—CBF and StiO2—by a stepwise increase of PaCO2 without relevant harmful side effects. Considering that the patients included in this study have a particularly high risk for delayed cerebral ischemia and poor outcome, the incidence of secondary infarctions was low and the neurological outcome is comparatively good [10–12].
Side Effects
Known side effects of hypercapnia include ICP elevation and blood acidification. According to the Monro-Kellie doctrine, the intracranial volume is the sum of brain tissue, intracranial CSF, and intracranial blood-volume. As such, an increase of ICP is an inevitable side effect of all CBF enhancing therapies. Hence, the presence of an EVD for continuous CSF drainage was a prerequisite for inclusion into the present study. During the interventions, it was closed for approximately 30 s every 5 min in order to measure ICP. Furthermore, predefined exclusion and interruption criteria were an ICP above 20 mmHg at the time of proposed trial inclusion and an ICP above 25 mmHg for more than 2 min during the intervention. Although ICP increased by a mean of 2.7 mmHg under hypercapnia, neither of the predefined interruption criteria occurred in this series of twelve consecutive poor-grade SAH patients, who met the inclusion criteria and underwent a total of 107 interventions. During the interventions, a surplus drainage of CSF was noted and may have prevented an excessive rise in ICP. The increase of ICP by 2.7 mmHg may be caused by the stepwise PaCO2 change and the 15-min time scale and of the study protocol including a reduction of PaCO2 to 30 mmHg prior to the induction of hypercapnia. During this period, almost no CSF was drained. In the following period of hypercapnia, the drainage of CSF occurred slower than the increase of CBF and resulted in a transient increase of ICP. This is supported by the observation that even during the first step of PaCO2 increase from 30 mmHg back to a target value of 40 mmHg, ICP was slightly higher than baseline values. However, no intervention had to be interrupted with respect to the predefined interruption criteria. This is in accordance with the findings of Petridis and coworkers who reported about the effects of lung-protective ventilation and permissive hypercapnia (PaCO2 50–60 mmHg) in SAH-patients who had developed early ARDS. All of these patients had an EVD for continuous CSF drainage and an intracerebral ICP probe for continuous ICP-measurement. In analogy to our results, the authors did not find a relevant increase of ICP in this setting during hypercapnia [13]. Some patients in this series had moderate early brain swelling as defined by Claassen et al. [14], but none had decompensated brain edema. In patients with even more pronounced brain edema, the surplus drainage of CSF may not be sufficient to prevent ICP elevations during hypercapnia. These patients will not be candidates for a hypercapnic therapy. If decompressive craniectomy is indicated, their eligibility may be reevaluated thereafter.
A pH below 7.25 during the intervention was a further potential interruption criterion in our study in order to exclude a possible cardiocirculatory effect by profound acidosis [15]. However, no intervention in this series had to be interrupted because pH fell below this predefined value. The reduction of PaO2 during hyperventilation and slight increase during hypercapnia might be explained by pulmonary vessel contraction during a lack of CO2 causing a decreased uptake of O2 [16]. On the other hand, hyperventilation has been reported to increase systemic vascular resistance and to reduce perfusion in most tissues while peripheral oxygen extraction may increase [17].
Increase of CBF
Secondary ischemic events are a major cause of in-hospital mortality and morbidity after aneurysmal SAH. Most likely, the cause of delayed ischemic neurological deficit (DIND) is multifactorial [18]. Delayed vasospasm (DV) probably contributes, but cortical electrical hyperactivity, hypermetabolism, a disturbance of local vasorelaxing and vasoconstricting factors, and inflammation may also be of importance. Our therapeutic approach does not focus on a single pathophysiological factor but rather targets the common denominator—the mismatch of oxygen demand and supply. While cerebral autoregulation is disturbed after SAH, there is evidence in animals [1, 19, 20] and humans that the PaCO2-reactivity remains intact after SAH [2, 21, 22].
Recently, the loss of cerebrovascular reactivity on changes of PaCO2 was identified as a predictor of DCI and poor outcome [7]. A large number of experimental and clinical studies investigated the principle mechanisms of CBF reactivity on changes of PaCO2 and the harmful effects of prolonged hyperventilation. Nevertheless, prior to this present Phase 1 trial, no study has investigated the therapeutic potential of hypercapnia in SAH patients. In the current study, the elevations of PaCO2 to a target of 60 mmHg led to reproducible increases of CBF and StiO2 in all patients. As previously reported, CO slightly increases under moderate hypercapnia [23, 24] whereas it decreases under pronounced hypercapnia and acidity [23]. In this study, cardiac output moderately increased by approximately 10 %, partly by an increase of HR, partly by an increase of stroke volume. Assuming that the surplus cardiac output is approximately evenly distributed in the systemic circulation, it cannot account for the increase of CBF. Therefore, an immediate vasodilating effect is likely to occur under hypercapnia. The measurement of CBF in this study was only punctual in the right frontal cortex. Rabinstein and coworkers observed a correlation of secondary ischemia with the site of the aneurysm [25]. Due to ethical aspects, however, a non-eloquent area was chosen for this single-point CBF measurement. Hence, the issue of a possible “steal-effect” may be raised with a shift of blood from areas with no reserve capacity into better perfused areas. To account for this, we added bilateral frontal StiO2 measurements and bilateral TCD as secondary endpoints. By this way, four of six supratentorial vascular territories were covered and a “steal-effect” was not observed during the trial interventions. The bilateral symmetric increase of TCD values indicates that the increase of CBF and StiO2 is a global rather than a local phenomenon.
The increase of CBF tended to be slightly less pronounced in case of higher baseline flow velocities in TCD in the major cerebral arteries. This indicates that the increase of CBF upon hypercapnia may be somewhat lower in case of cerebral vasospasm, an observation which is in accordance with the findings of Carrera et al. [7]. However, this correlation was not statistically significant in this present series. In general, the PaCO2 reactivity was preserved in all patients and CBF could reliably be increased by hypercapnia of over 50 mmHg even in patients with signs of severe vasospasm as determined by high baseline TCD values.
The fact that 10 of 12 patients were transferred to reangiography due to suspected vasospasm indicates that temporary hypercapnia does not prevent or treat vasospasm in large vessel trunks but rather affect the cerebral vasculature at the level of small conductive vessel or microcirculation. We based the decision for reangiography and endovascular vasospasm therapy on clinical, ultrasonographic and CT-criteria as described previously resulting in a relatively high number of endovascular spasm therapies [26]. The parallel course of TCD in the MCA trunks and CBF also indicates that there is no relevant dilatory effect of hypercapnia in large vessels. If there was such an effect, the increase of MFV in TCD would be expected to be less pronounced than the increase of CBF during hypercapnia. The high rate of patients transferred to reangiography and angioplasty results from the above-mentioned criteria and may be center-specific and might have helped to protect from DCI. Although the development of DCI was rare and outcome was comparatively good considering the very poor starting condition, this may limit the power of this series to generalize the beneficial effect regarding the secondary endpoints “delayed cerebral infarction” and “neurological recovery.”
Improvement of Study-Protocol
The next step to this phase 1 trial will be a randomized controlled trial comparing patients treated by intermittent hypercapnia to a control group. Prior to starting an RCT, the optimum trial protocol needs to be found. The present data suggests that the current protocol can be optimized in two ways. First, our results indicate that the maximum reactivity of both CBF and StiO2 lies between a PaCO2 of 30–50 mmHg and already decreases between 50 and 60 mmHg. Considering the accompanying changes of pH, we hypothesize that a PaCO2 of 55–60 mmHg may be an effective target to reliably increase CBF while maintaining patient safety over a longer period.
Second, the absence of a rebound effect after resetting the respirator to baseline values suggests that longer periods of hypercapnia may safely be possible and might actually increase CBF for a longer period of time. The study was designed as a Phase 1 trial with the primary target to determine the changes of CBF during temporary iatrogenic induction of hypercapnia in order to evaluate its potential capability for a therapeutic use in SAH patients. The primary observation time was during the 60 min of the trial intervention. Unfortunately, TCD and arterial blood gases were not measured at the end of the trial intervention. In contrast, CBF and StiO2 were further recorded, mainly because a rebound effect had to be excluded after returning to normoventilation. Regarding the dynamics of PaCO2 changes during the trial intervention, it seems likely that PaCO2 values returned to baseline within several minutes after resetting the respirator to baseline parameters. In contrast, CBF and StiO2 had not returned to baseline even 60 min after restoring normoventilation. However, since arterial blood gases were not available after the end of the hypercapnic period, the course of PaCO2 and its correlation with the sustained elevation of CBF and StiO2 cannot be drawn from the present data.
The optimal duration cannot be concluded from the present data. While the presented PaCO2 values were measured at the end of a 15-min interval and constitute steady state values, dependent parameters like the pH or target parameters like CBF or StiO2 may not have reached an equilibrium after 15 min. In addition, there may be significant interindividual variations. It has to be considered that CSF quickly acidifies during respiratory acidosis. The decline of pH in the CSF and the interstitial fluid has been shown to be the immediate factor governing the vasodilatory response of hypercapnia, subsequently increasing CBF [27–29]. However, buffer systems in the blood and CSF start to counteract the acidosis, leading to a decline of CBF after its maximum. At that time, a deleterious rebound effect below ischemic thresholds must be expected if the respirator is reset to baseline parameters [30]. Hence, therapeutic hypercapnia should ideally be terminated shortly before CBF leaves its maximum under constant hypercapnia. During hyperventilation in healthy humans, Raichle et al., for example, found a near-complete recovery of CBF after 4 h of constant hypocapnia [31]. Taking our own data into account and assuming that the adaptation to hyper- and hypocapnia occurs in a similar fashion, the ideal time point to terminate therapeutic hypercapnia will probably be after 30–180 min. This issue will be investigated in an further trial.
Conclusion
Graded temporary hypercapnia was easily applicable and could be safely performed under the prerequisite that a continuous CSF drainage buffered excessive increases of ICP. Therapeutic hypercapnia up to 60 mmHg resulted in a reproducible increase of CBF and StiO2. The present data suggest that the therapeutic approach may be further enhanced via a longer duration of hypercapnia which will be investigated in a further trial. The results of this Phase 1 trial are promising. However, the therapeutic potential of this non-pharmacological treatment to prevent secondary ischemic deficits after poor-grade aneurysmal SAH and improve neurological recovery finally has to be evaluated in a randomized trial.
References
Diringer MN, Kirsch JR, Hanley DF, Traystman RJ. Altered cerebrovascular CO2 reactivity following subarachnoid hemorrhage in cats. J Neurosurg. 1993;78(6):915–21.
Schmieder K, Jarus-Dziedzic K, Wronski J, Harders A. CO2 reactivity in patients after subarachnoid haemorrhage. Acta Neurochir (Wien). 1997;139(11):1038–41.
Hassler W, Chioffi F. CO2 reactivity of cerebral vasospasm after aneurysmal subarachnoid haemorrhage. Acta Neurochir (Wien). 1989;98(3–4):167–75.
Carrera E, Schmidt JM, Fernandez L, et al. Spontaneous hyperventilation and brain tissue hypoxia in patients with severe brain injury. J Neurol Neurosurg Psychiatry. 2010;81(7):793–7.
Yamamoto M, Meyer J, Naritomi H, Sakai F, Yamaguchi F, Shaw T. Noninvasive measurement of cerebral vasospasm in patients with subarachnoid hemorrhage. J Neurol Sci. 1979;43(2):301–15.
Curley G, Kavanagh BP, Laffey JG. Hypocapnia and the injured brain: more harm than benefit. Crit Care Med. 2010;38(5):1348–59.
Carrera E, Kurtz P, Badjatia N, et al. Cerebrovascular carbon dioxide reactivity and delayed cerebral ischemia after subarachnoid hemorrhage. Arch Neurol. 2010;67(4):434–9.
Westermaier T, Stetter C, Kunze E, et al. Controlled transient hypercapnia: a novel approach for the treatment of delayed cerebral ischemia after subarachnoid hemorrhage? J Neurosurg. 2014;121(5):1056–62.
von Kummer R, Holle R, Gizyska U, et al. Interobserver agreement in assessing early CT signs of middle cerebral artery infarction. AJNR Am J Neuroradiol. 1996;17(9):1743–8.
Cross DT, Tirschwell DL, Clark MA, et al. Mortality rates after subarachnoid hemorrhage: variations according to hospital case volume in 18 states. J Neurosurg. 2003;99(5):810–7.
Rivero-Arias O, Gray A, Wolstenholme J. Burden of disease and costs of aneurysmal subarachnoid haemorrhage (aSAH) in the United Kingdom. Cost Eff Resour Alloc. 2010;8(6):6.
Frontera JA, Fernandez A, Schmidt JM, et al. Defining vasospasm after subarachnoid hemorrhage: what is the most clinically relevant definition? Stroke. 2009;40(6):1963–8.
Petridis AK, Doukas A, Kienke S, et al. The effect of lung-protective permissive hypercapnia in intracerebral pressure in patients with subarachnoid haemorrhage and ARDS: a retrospective study. Acta Neurochir (Wien). 2010;152(12):2143–5. doi:10.1007/s00701-010-0761-z.
Claassen J, Carhuapoma JR, Kreiter KT, Du EY, Connolly ES, Mayer SA. Global cerebral edema after subarachnoid hemorrhage: frequency, predictors, and impact on outcome. Stroke. 2002;33(5):1225–32.
Stengl M, Ledvinova L, Chvojka J, et al. Effects of clinically relevant acute hypercapnic and metabolic acidosis on the cardiovascular system: an experimental porcine study. Crit Care. 2013;17(6):R303.
Chuang IC, Dong HP, Yang RC, et al. Effect of carbon dioxide on pulmonary vascular tone at various pulmonary arterial pressure levels induced by endothelin-1. Lung. 2010;188(3):199–207.
Karlsson T, Stjernstrom EL, Stjernstrom H, Norlen K, Wiklund L. Central and regional blood flow during hyperventilation: an experimental study in the pig. Acta Anaesthesiol Scand. 1994;38(2):180–6.
Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nat Rev Neurol. 2014;10(1):44–58.
Diringer MN, Kirsch JR, Traystman RJ. Reduced cerebral blood flow but intact reactivity to hypercarbia and hypoxia following subarachnoid hemorrhage in rabbits. J Cereb Blood Flow Metab. 1994;14(1):59–63.
Ma XD, Hauerberg J, Pedersen DB, Juhler M. Effects of morphine on cerebral blood flow autoregulation CO2-reactivity in experimental subarachnoid hemorrhage. J Neurosurg Anesthesiol. 1999;11(4):264–72.
Dernbach PD, Little JR, Jones SC, Ebrahim ZY. Altered cerebral autoregulation and CO2 reactivity after aneurysmal subarachnoid hemorrhage. Neurosurgery. 1988;22(5):822–6.
Poulin MJ, Liang PJ, Robbins PA. Fast and slow components of cerebral blood flow response to step decreases in end-tidal PCO2 in humans. J Appl Physiol. 1998;85(2):388–97.
Komori M, Takada K, Tomizawa Y, Nishiyama K, Kawamata M, Ozaki M. Permissive range of hypercapnia for improved peripheral microcirculation and cardiac output in rabbits. Crit Care Med. 2007;35(9):2171–5.
Hickling KG, Joyce C. Permissive hypercapnia in ARDS and its effect on tissue oxygenation. Acta Anaesthesiol Scand Suppl. 1995;107:201–8.
Rabinstein AA, Friedman JA, Weigand SD, et al. Predictors of cerebral infarction in aneurysmal subarachnoid hemorrhage. Stroke. 2004;35(8):1862–6.
Westermaier T, Pham M, Stetter C, et al. Value of transcranial Doppler, perfusion-CT and neurological evaluation to forecast secondary ischemia after aneurysmal SAH. Neurocrit Care. 2014;20(3):406–12.
Agnoli A. Adaptation of CBF during induced chronic normooxic respiratory acidosis. Scand J Clin Lab Investig Suppl. 1968;102:VIII.
Fencl V, Vale JR, Broch JA. Respiration and cerebral blood flow in metabolic acidosis and alkalosis in humans. J Appl Physiol. 1969;27(1):67–76.
Skinhoj E. CBF adaption in man to chronic hypo- and hypercapnia and its relation to CSF pH. Scand J Clin Lab Investig Suppl. 1968;102:8.
Jones TH, Morawetz RB, Crowell RM, et al. Thresholds of focal cerebral ischemia in awake monkeys. J Neurosurg. 1981;54(6):773–82.
Raichle ME, Posner JB, Plum F. Cerebral blood flow during and after hyperventilation. Arch Neurol. 1970;23(5):394–403.
Acknowledgments
The study was supported by the Else-Kröner-Fresenius Stiftung (2013_A171).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
None of the authors has a conflict of interest to declare.
Rights and permissions
About this article

Cite this article
Westermaier, T., Stetter, C., Kunze, E. et al. Controlled Hypercapnia Enhances Cerebral Blood Flow and Brain Tissue Oxygenation After Aneurysmal Subarachnoid Hemorrhage: Results of a Phase 1 Study. Neurocrit Care 25, 205–214 (2016). https://doi.org/10.1007/s12028-016-0246-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-016-0246-x