
Abstract
Objective: Brain arterial critical closing pressure (CrCP) has been studied in several diseases such as traumatic brain injury (TBI), subarachnoid haemorrhage, hydrocephalus, and in various physiological scenarios: intracranial hypertension, decreased cerebral perfusion pressure, hypercapnia, etc. Little or nothing so far has been demonstrated to characterise change in CrCP during mild hypocapnia.
Method: We retrospectively analysed recordings of intracranial pressure (ICP), arterial blood pressure (ABP) and blood flow velocity from 27 severe TBI patients (mean 39.5 ± 3.4 years, 6 women) in whom a ventilation increase (20% increase in respiratory minute volume) was performed over 50 min as part of a standard clinical CO2 reactivity test. CrCP was calculated using the Windkessel model of cerebral arterial flow. Arteriolar wall tension (WT) was calculated as a difference between CrCP and ICP. The compartmental compliances arterial (C a ) and cerebrospinal fluid space (C i ) were also evaluated.
Results: During hypocapnia, ICP decreased from 17±6.8 to 13.2±6.6 mmHg (p < 0.000001). Wall tension increased from 14.5 ± 9.9 to 21.7±9.1 mmHg (p < 0.0002). CrCP, being a sum of WT + ICP, changed significantly from 31.5 ± 11.9 mmHg to 34.9±11.1 mmHg (p < 0.002), and the closing margin (ABP-CrCP) remained constant at an average value of 60 mmHg. C a decreased significantly during hypocapnia by 30% (p < 0.00001) and C i increased by 26% (p < 0.003).
Conclusion: During hypocapnia in TBI patients, ICP decreases and WT increases. CrCP increases slightly as the rise in wall tension outweighs the decrease in ICP. The closing margin remained unchanged, suggesting that the risk of hypocapnia-induced ischemia might not be increased.
Access provided by CONRICYT-eBooks. Download conference paper PDF
Similar content being viewed by others


Keywords
- Traumatic brain injury
- Hypocapnia
- Intracranial pressure
- Critical closing pressure
- Arterial wall tension
- Cerebrovascular compliance
- Brain monitoring
- ICM+
Introduction
Critical closing pressure (CrCP) is the lowest threshold of arterial blood pressure (ABP) below which the cerebral blood flow ceases. The flow cessation happens when the transmural pressure in the cerebral arteries, expressed as the difference between ABP and intracranial pressure (ICP), becomes too low to oppose the tension of the arterial walls leading to collapse of the arteries. Therefore, CrCP is defined as the sum of intracranial pressure (ICP) and the wall tension (WT) pressure. The phenomenon of critical closing pressure in the brain was first described with the appearance of transcranial Doppler (TCD) measurements of blood flow velocity in the middle cerebral artery when the intercept of the regression line through the systolic and diastolic values of flow velocity and arterial blood pressure was studied. Since then, CrCP has been studied in many conditions, including traumatic brain injury, subarachnoid haemorrhage, and hydrocephalus under various haemodynamic conditions such as increased intracranial pressure, decreased cerebral perfusion, and moderate hyper- and hypocapnia.
Moderate hypocapnia has at one point been proposed as a means of controlling elevated intracranial pressure in severe brain trauma patients because of its vasoconstrictive properties, leading to a reduction in cerebral blood volume, and thus ICP. However, subsequent studies showed that despite lowering ICP and improving global brain perfusion, the direct vasoconstriction effect often leads to worsening deficits in local cerebral perfusion in particularly vulnerable regions of the brain. This has led to changes in the management strategies of severe head trauma patients, allowing only for mild hypocapnic treatment to be applied in those patients. However, little is currently known of the effects of such mild hypocapnia on the properties of cerebral arteries.
In this study, we set out to examine what happens to the arterial compliances, the WT and the critical closing pressure in TBI patients with elevated ICP during mild hypocapnic challenges.
Materials and Methods
We retrospectively analysed waveform recordings of ICP, arterial blood pressure (ABP) and TCD blood flow velocity (FV) taken from 27 sedated and ventilated severe TBI patients (mean age 39.5 ± 3.4 years, 6 were women) admitted to the Neurocritical Care Unit at Addenbrooke’s Hospital, Cambridge, in whom an increase in ventilation (20% increase in respiratory minute volume) was performed over 50 min as part of a standard clinical CO2 reactivity test.
ABP was monitored invasively using a pressure monitoring kit (Baxter Healthcare CA, USA; Sidcup, UK) at the radial artery, zeroed at the level of the heart, whereas an intraparenchymal probe (Codman & Shurtleff, MA, USA or Camino Laboratories, CA, USA) was used to monitor ICP. FV was measured from the middle cerebral artery with a 2-MHz probe and monitored with the Doppler Box (DWL Compumedics, Germany) or Neuroguard (Medasonics, Fremont, CA, USA). All the monitoring was performed as part of the standard protocol for the management of traumatic brain injury [1]. Patient monitoring was approved by the local Ethics Committee (REC97/291).
The raw data signals were digitised at a sampling frequency of 50 Hz using an analogue–digital converter (DT9801 and DT9803, Data Translation, Marlboro, MA, USA), and recorded using ICM +® software (Cambridge Enterprise, ltd, Cambridge, UK, http://icmplus.neurosurg.cam.ac.uk). All the analyses on the recorded raw waveforms were performed over 10-s long-sliding windows using the same software ICM+.
Critical closing pressure (CrCP) was calculated using the Windkessel model of cerebral arterial flow [2] according to the following formula:
where R a is the composite cerebrovascular resistance estimated as:
C a is the lumped cerebral arterial compliance estimated as:
AMP here denotes the amplitude of the first, Fourier, harmonic of the pulse component of ABP, and CaBV respectively.
CaBV is the cerebral arterial blood volume, which is derived from FV by means of time integration:
and finally, HR is the heart rate.
Both R a and C a formulae contain unknown factor S a (directly, or indirectly inside the CaBV formula, as above) used to convert blood flow velocity measurements into blood flow, which cancels out when the product of the two (denoted as time constant τ in the results) is used in the CrCP formula.
Subsequently, vascular wall tension (WT) was estimated as (Dewey et al.): WT = CrCP − ICP, which when using the impedance model becomes:
In addition, the compliance of the cerebrospinal space C i was also calculated as:
The exact derivation of all the above formulae with detailed explanations can be found elsewhere [2, 3].
For statistical analysis of the effects of hypocapnia, mean values of the studied parameters at baseline and during the hypotension period were compared using univariate tests (paired t test).
Results
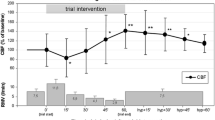
A representative example of the behaviour of the studied parameters during the 20% increase in the respiratory rate is shown in Fig. 1 and the statistical comparisons of baseline versus hypocapnic period values are given in Table 1. PacO2, ICP, FV and CrCP decreased significantly, whereas ABP, CPP, C i , R a , and WT increased significantly. The other parameters, HR and C a , did not change. For completeness measures of cerebral blood flow autoregulation, the mean index, Mx, and the pressure reactivity index, PRx, were also added, showing in both cases intact reactivity at both levels of CO2, with no significant difference between the two levels. Also, the diastolic closing margin (DCM), defined as the difference between the diastolic arterial blood pressure and CrCP, was calculated at both levels, and the difference was found to be non-significant.

Typical example of time trends of the haemodynamic parameters studied during one episode of a 20% increase in ventilation-induced mild hypocapnia
Discussion
The increase in ventilation led to a state of mild hypocapnia in all cases. As expected, this produced a decrease in both ICP and TCD blood FV, evidence of induced vasoconstriction. Not surprisingly, the compliance of the CSF space (C i ) increased significantly because of the decrease in the cerebral blood volume. Cerebrovascular time constant τ also increased, despite the opposing behaviour of R a and C a . The former increased as a direct effect of the vasoconstriction whereas the latter decreased in individual cases, but as a group statistically not significantly, and in all cases, the change in R a prevailed over the change in C a . Similarly, WT and ICP changed in the opposite direction, with WT increasing significantly during the hypocapnia period. In this case, however, those opposite effects were largely balanced out; thus, the resulting critical closing pressure, being the sum of WT and ICP, decreased only a little, though statistically significantly.
What is even more important is that the diastolic closing margin (DCM), which provides a “safety” window for arterial blood pressure before the first ischaemic threshold for the cerebral blood flow is reached during the diastolic phase of the heart cycle, not only did not decrease but even showed a tendency to increase.
It is also worth noting that cerebral autoregulation was not significantly affected by this low degree of hypocapnia. The pressure reactivity index (Prx) showed a general tendency towards a slight improvement, but the mean flow index (Mx) did not change at all.
Of course, these global measures still do not necessarily reflect what happens in local, vulnerable areas of the injured brain. However, the finding that the increase in ventilation induced mild hypocapnia caused a reduction in ICP. There was no evidence of any other accompanying significant haemodynamic consequences, though, which seems to suggest that the use of the mild form of hypocapnia might indeed be a safe option. This is because it is unlikely to produce the adverse effects observed under conditions of pronounced hypocapnia.
Last, the authors would like to accentuate the usefulness of the presented methodology for a more thorough interpretation of the TCD measurements. This sort of analysis providing clinically important additional insights into the cerebral haemodynamics would have not been possible without the support of dedicated real-time analysis software such as ICM+.
Conclusions
During hypocapnia in TBI patients, ICP decreases and WT increases. CrCP increases slightly as the rise in wall tension outweighs the decrease in ICP. Closing margins remained unchanged, suggesting that the risk of hypocapnia-induced ischaemia might be low.
References
Menon DK. Cerebral protection in severe brain injury: physiological determinants of outcome and their optimisation. Br Med Bull. 1999;55(1):226–58.
Varsos GV, et al. Critical closing pressure determined with a model of cerebrovascular impedance. J Cereb Blood Flow Metab. 2013;33(2):235–43.
Kim DJ, Kasprowicz M, Carrera E, et al. The monitoring of relative changes in compartmental compliances of brain. Physiol Meas. 2009;30(7):647–59.
Acknowledgements
The authors wish to express gratitude to the staff of Neurocritical Care Unit, Addenbrooke’s Hospital, Cambridge, UK, for their help and support with the ICM+ brain monitoring project.
Disclosure: ICM+® is software licensed by Cambridge Enterprise Ltd., UK; P.S. and M.C. have a financial interest in a part of the licensing fee.
Conflicts of interest statement
We declare that we have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this paper

Cite this paper
Smielewski, P., Steiner, L., Puppo, C., Budohoski, K., Varsos, G.V., Czosnyka, M. (2018). Effect of Mild Hypocapnia on Critical Closing Pressure and Other Mechanoelastic Parameters of the Cerebrospinal System. In: Heldt, T. (eds) Intracranial Pressure & Neuromonitoring XVI. Acta Neurochirurgica Supplement, vol 126. Springer, Cham. https://doi.org/10.1007/978-3-319-65798-1_29
Download citation
DOI: https://doi.org/10.1007/978-3-319-65798-1_29
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-65797-4
Online ISBN: 978-3-319-65798-1
eBook Packages: MedicineMedicine (R0)