
Abstract
Background
Cerebral hypoxia is a frequent cause of secondary brain damage in patients with acute brain injury. Although hypercapnia can increase intracranial pressure, it may have beneficial effects on tissue oxygenation. We aimed to assess the effects of hypercapnia on brain tissue oxygenation (PbtO2).
Methods
This single-center retrospective study (November 2014 to June 2022) included all patients admitted to the intensive care unit after acute brain injury who required multimodal monitoring, including PbtO2 monitoring, and who underwent induced moderate hypoventilation and hypercapnia according to the decision of the treating physician. Patients with imminent brain death were excluded. Responders to hypercapnia were defined as those with an increase of at least 20% in PbtO2 values when compared to their baseline levels.
Results
On a total of 163 eligible patients, we identified 23 (14%) patients who underwent moderate hypoventilation (arterial partial pressure of carbon dioxide [PaCO2] from 44 [42–45] to 50 [49–53] mm Hg; p < 0.001) during the study period at a median of 6 (4–10) days following intensive care unit admission; six patients had traumatic brain injury, and 17 had subarachnoid hemorrhage. A significant overall increase in median PbtO2 values from baseline (21 [19–26] to 24 [22–26] mm Hg; p = 0.02) was observed. Eight (35%) patients were considered as responders, with a median increase of 7 (from 4 to 11) mm Hg of PbtO2, whereas nonresponders showed no changes (from − 1 to 2 mm Hg of PbtO2). Because of the small sample size, no variable independently associated with PbtO2 response was identified. No correlation between changes in PaCO2 and in PbtO2 was observed.
Conclusions
In this study, a heterogeneous response of PbtO2 to induced hypercapnia was observed but without any deleterious elevations of intracranial pressure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute brain injury poses a significant challenge to global health; traumatic brain injury (TBI) is the primary cause of mortality and disability in individuals under 45 years of age, whereas subarachnoid hemorrhage (SAH) accounts for more than 27% of life-years lost before 65 years of age, incurring substantial health care expenses and resulting in extensive long-term morbidity as well as high mortality rates [1, 2]. Consequently, contemporary management of acute brain injury patients predominantly focuses on the prevention and treatment of secondary brain injuries given that the primary injury is already present on hospital arrival and that no efficacious pharmacological agents have been discovered to enhance neurological recovery thus far [3,4,5].
Cerebral hypoxia is indeed a primary and prevalent contributor to secondary brain injury [6,7,8]; brain tissue oxygen pressure (PbtO2) monitoring serves as an invasive yet effective method for evaluating cerebral hypoxia in patients with brain injuries. Low PbtO2 values have been correlated with cerebral anaerobic metabolism and an elevated risk of mortality and poor functional outcomes in this context [9,10,11,12,13,14]. Low PbtO2 may be observed in various circumstances, including diminished cerebral blood flow (CBF) and/or cerebral perfusion pressure (CPP), intracranial hypertension, hypoxemia due to pulmonary disease, anemia, altered microcirculation, or excessive cellular metabolism and mitochondrial dysfunction [15]. Consequently, multiple therapeutic approaches can be employed to optimize PbtO2, encompassing the use of vasopressors (e.g., to increase CPP), intracranial hypertension treatments (e.g., osmotic therapy), red blood cell transfusions, ventilatory strategies to ameliorate hypoxia, temperature control, and/or sedation (e.g., to decrease cerebral oxygen consumption) [16,17,18]. Several studies have indeed indicated a favorable impact of PbtO2-based management strategies on mortality and functional outcomes in patients with acute brain injury, particularly in the case of TBI [3, 19].
Because PbtO2 reflects the oxygen content within the cerebral interstitial space, its value relies on the intricate balance between oxygen delivery (dependent on arterial oxygen content, CBF, and tissue diffusion) and oxygen consumption [17, 18]. Many of these interventions aim to augment CBF; a potentially effective intervention in this context involves the manipulation of carbon dioxide (CO2), a principal regulator of cerebrovascular tone. This phenomenon, known as “cerebral CO2 reactivity,” refers to the capacity of resistance arterioles to dilate or constrict in response to alterations in arterial partial pressure of carbon dioxide [PaCO2]. Notably, this mechanism appears to be preserved even when pressure autoregulation of CBF is disrupted, as demonstrated following aneurysmal SAH [20]. Specifically, it is the fluctuation of perivascular pH, secondary to broad PaCO2 ranges, that regulates vascular smooth muscle tone, fostering vasorelaxation with the elevation of PaCO2 and the subsequent decrease in pH, through various mediators (e.g., nitric oxide or prostaglandins) [21].
Although elevated PaCO2 has been integrated into PbtO2-guided therapy protocols, such as in the brain tissue oxygen monitoring and management in severe traumatic brain injury (BOOST-II) trial [22], and has been recommended in recent guidelines [23], increased PaCO2 may exert adverse effects on intracranial pressure (ICP) by increasing CBF, and its impact on PbtO2 warrants further understanding [24,25,26]. Consequently, the objective of this study was to evaluate the effects of induced hypercapnia on PbtO2 in patients with acute brain injuries and to determine the proportion of responders to this therapeutic approach.
Methods
Study Design
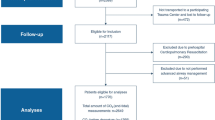
This retrospective analysis of prospectively collected data encompasses patients with acute brain injury secondary to SAH or TBI who were treated between November 2014 and June 2022 in the Department of Intensive Care at Hôpital Universitaire de Bruxelles, Belgium. All adult patients (> 18 years) were eligible if they required ICP and PbtO2 monitoring and underwent moderate hypoventilation and induced hypercapnia in accordance with local protocols [23, 27, 28]. Inclusion criteria were the following: (1) controlled mechanical ventilation; (2) treating physician’s decision to induce hypercapnia for at least 60 min (PaCO2 > 45 mm Hg, achieved via an absolute reduction in minute ventilation), with a PaCO2 change of at least 5 mm Hg compared with the baseline value; and (3) documented gas analyses at baseline and within 1 h of induced hypercapnia. The sole exclusion criterion was imminent death, resulting in early limitation of life-sustaining therapies. This study was approved by the Erasme Ethics Committee (P2022/449), which waived the requirement for informed consent.
Data Collection and Definitions
We collected demographic data, including age, sex, and comorbidities. Clinical severity scores on admission, such as the Acute Physiology and Chronic Health Evaluation II [29] score, were calculated for all patients, and neurological assessment was performed using the Glasgow Coma Scale [30]. ICP was continuously measured with intraparenchymal and interventricular probes (Neurovent-P, Raumedic, Helmbrechts, Germany, and IM3.ST_EU, Integra LifeSciences Corporation, Plainsboro, NJ, respectively). Intraparenchymal PbtO2 probes (Licox Integra LifeSciences Corporation, Plainsboro, NJ) were placed, whenever possible, into the hemisphere at the highest risk for secondary brain injury (e.g., near the injured/contused area in patients with TBI or in the area at risk for ischemia in patients with SAH) [28]. Probe location was confirmed via cerebral CT scan after placement. The probe proper functioning was tested with a 100% inspired fraction of oxygen test for a maximum of 5 min.
For each patient, we recorded PbtO2, pH, PaCO2, ICP, CPP, and minute ventilation at baseline and after induced hypercapnia. Baseline values were considered before minute ventilation was modified; average PbtO2, ICP, and CPP values over the 20 min preceding this time point were used. Patients underwent moderate hypoventilation for at least 60 min. To assess the effects of induced hypercapnia, the following arterial blood gas analysis (ABG) was performed within 45 and 60 min thereafter to exclude additional confounders on PbtO2 values; average PbtO2, ICP, and CPP values over the 20 min following this time point were used. No other therapeutic interventions were permitted during this period. Responders to induced hypercapnia were patients who exhibited a PbtO2 value increase of at least 20% compared with baseline values [31, 32]. Intracranial hypertension was defined as an ICP > 20 mm Hg for at least 5 min. Neurological outcomes were assessed using the Glasgow Outcome Scale at 6 months, and mortality was evaluated at intensive care unit (ICU) and hospital discharge.
Study Outcomes
The primary outcome was the absolute change in PbtO2 after induced hypercapnia compared with baseline. Secondary outcomes included the proportion of PbtO2 responders in the study cohort, the correlation between PaCO2 and PbtO2 changes, and different responses according to the type of brain injury and outcome.
Statistical Analysis
Descriptive statistics were computed for all variables. Numeric variables were described either as median and interquartile intervals (25–75%) or mean and standard deviation. Categorical variables were described as proportions. Normally distributed continuous variables were compared using Student’s t-test, and asymmetrically distributed variables were compared using the Mann–Whitney U-test. For comparing paired measurements, normally distributed continuous variables were compared using paired t-tests, and asymmetrically distributed variables were compared using the Wilcoxon rank-sum test. For comparing and evaluating the differences in variation before and after an intervention in different subgroups, we used a generalized mixed model for repeated measures; generalized mixed linear models describe the relationship between a dependent variable (responders) and independent variables (e.g., PbtO2 and other physiological variables) using coefficients that can vary with respect to one or more time points (e.g., baseline and after moderate hypoventilation) and using nonnormal data distribution. To investigate possible baseline variables associated with PbtO2 responders, we performed a univariable logistic regression analysis according to Firth’s method because of the small sample size. The Firth method is a penalized log-likelihood function that mitigates bias introduced by rare events on a data set or small sample size [33]. The chosen baseline variables in the univariate logistic regression were selected according to clinical and physiological relevance. Odds ratios and 95% confidence intervals (CIs) were computed for each variable; a p value < 0.05 was considered statistically significant. Similarly, we performed a univariable linear regression model to investigate the association of baseline line physiological and clinical variables with absolute delta PbtO2 (difference between PbtO2 after hyperventilation and PbtO2 at baseline). For the statistical analysis, we used Prism 9.5.0 by GraphPad Software, LLC.
Results
Study Population
In a cohort of 163 patients with acute brain injury with TBI or SAH who were monitored with ICP and PbtO2, 23 (14%) underwent induced hypercapnia during the study period and were included in the analysis; among these, six had TBI and 17 had SAH. The median age was 52 years (interquartile range: 40–62 years), with arterial hypertension and alcohol consumption being the most common comorbidities. The median Glasgow Coma Scale and Acute Physiology and Chronic Health Evaluation II scores on admission were 7 (3–11) and 16 (14–21), respectively. Intracranial hypertension at any point during the ICU stay was observed in 11 of 23 (48%) patients. The primary characteristics of the study population are detailed in Table 1.
Induced Hypercapnia and PbtO2
Induced hypercapnia was initiated at a median of 6 (4–10) days following ICU admission. Table 2 presents the alterations in primary collected variables before and after induced hypercapnia. As anticipated, PaCO2 significantly rose from baseline (44 [42–45] to 50 [49–53] mm Hg; p < 0.001) following the reduction in minute ventilation, resulting in a significant decrease in pH (from 7.40 [7.36–7.45] to 7.35 [7.31–7.39]; p < 0.001).
A significant overall increase in median PbtO2 values from baseline (21 [19–26] to 24 [22–26] mm Hg; p = 0.02) was observed after reducing minute ventilation (Fig. 1 and Table 2). However, there was no linear correlation between changes in PaCO2 and changes in PbtO2 in the overall population, as shown in Supplemental Fig. S1 (r = − 0.028 [95% CI − 0.45 to 0.40]). In a univariable linear regression, baseline CPP was associated with changes in PbtO2 after induced hypercapnia (β 0.138 [95% CI 0.012 to 0.263], p = 0.03). No multivariable linear regression was possible because of the limited sample size (Supplemental Table S1).

Changes in brain oxygenation (PbtO2) before and after induced hypercapnia, according to PbtO2 response. Differences between groups were assessed using a generalized mixed model. Data are presented as median and interquartile range
We identified eight (35%) responders to induced hypercapnia, with a median PbtO2 increase of 7 (5–11) mm Hg (from 19 [18–21] to 25 [24–30] mm Hg; p = 0.014), in comparison with nonresponders (n = 15; changes in PbtO2 from − 1 to 2 mm Hg and absolute PbtO2 values from 24 [21–27] to 23 [21–27] mm Hg; p = 0.64; Fig. 1).
Responders more frequently had preexisting heart disease and liver cirrhosis than others (Table 1); no other significant differences in baseline physiological values were observed between groups, as shown in Supplemental Table S2. In the univariable analysis, no baseline physiological variables were associated with PbtO2 responders (Supplemental Table S3). No multivariable analysis was performed because of the limited sample size. Also, no correlation between the change in PaCO2 and the change in PbtO2 was observed (Fig. 2).

Spearman correlation of changes in PaCO2 (delta PaCO2) due to a decrease in minute ventilation and changes in brain oxygenation (delta PbtO2) in responders (in green) and nonresponders (in light blue). CI confidence interval, PaCO2 arterial partial pressure of carbon dioxide, PbtO2 brain tissue partial pressure of oxygen
Supplemental Table S4 displays alterations in PaCO2 and PbtO2 response according to various subgroups. We observed a significant PbtO2 increase at hypercapnia in patients with SAH and in those with unfavorable neurological outcome when compared with others. The changes in PaCO2, ICP, and CPP before and after induced hypercapnia, according to PbtO2 response, are reported in Fig. 3. In all nonresponders, ICP remained below 20 mm Hg, whereas among PbtO2 responders, three patients had an ICP > 20 mm Hg at hypercapnia (the maximum level of ICP was 30 mm Hg). However, this did not result in a CPP < 60 mm Hg or a PbtO2 < 20 mm Hg.

Changes in arterial partial pressure of carbon dioxide (PaCO2), intracranial pressure (ICP) and cerebral perfusion pressure (CCP) before and after induced hypercapnia, according to brain oxygen pressure (PbtO2) response. Differences between groups were assessed using a generalized mixed model. Data are presented as median and interquartile range
Discussion
In this single-center retrospective physiological study, the response to therapeutic hypoventilation was heterogeneous, leading to a significant increase in PbtO2 in one third of patients. Overall, ICP remained within the normal ranges but increased in responders, accompanied by a decrease in CPP, although the difference before and after hypoventilation was not statistically significant. There was no correlation between the change in PaCO2 and the change in PbtO2. Because of the small cohort, we were unable to identify baseline characteristics associated with a significant PbtO2 response to hypercapnia. However, baseline CPP was directly associated with changes in PbtO2 after induced hypercapnia.
In patients with TBI, Okonkwo et al. demonstrated that the use of PbtO2-guided therapy, employing a specific and intricate protocol, reduced the incidence of brain hypoxia compared with patients who underwent ICP-guided therapy alone [22]. Furthermore, two meta-analyses reported that combined ICP/PbtO2-guided therapy was associated with improved neurological outcomes when compared to standard ICP-guided therapy in this setting [34, 35]. In SAH, low PbtO2 levels have been linked to various pathological pathways, such as reduced CBF, lung injury with hypoxemia, and/or anemia [28]; consequently, strategies aimed at increasing brain perfusion can elevate PbtO2 levels in some of these patients [17].
One possible strategy to increase PbtO2 is to promote vasodilation by inducing hypercapnia [25, 26]. Increased PaCO2 results in vasodilation of cerebral arterioles, leading to increased CBF. This response, known as cerebrovascular reactivity, is a crucial mechanism to maintain appropriate oxygen delivery to brain tissue in response to metabolic demands. The underlying mechanism for CO2-induced vasodilation involves changes in the pH of the cerebrospinal fluid and the release of various vasodilatory mediators, such as nitric oxide (NO), adenosine, and prostaglandins [21, 36]. Interestingly, this phenomenon is heterogeneous and varies in different areas of the brain and in different individuals [37,38,39]. By increasing CBF, mild hypercapnia could improve cerebral oxygenation, as shown in animal models of acute brain injury [40]. In a study including patients with SAH, Stetter et al. showed that temporary mild hypercapnia can increase CBF and brain tissue oxygen saturation with no relevant adverse effects, especially on ICP [25]. In our study, responders to hypercapnia concomitantly showed an increase in ICP, probably because of an increased cerebral blood volume [24, 41, 42]. Hypercapnia also influences blood flow in other organs; in pulmonary circulation, it induces vasoconstriction, which is known as the “hypoxic pulmonary vasoconstriction response” [43]. This response serves to divert blood flow away from poorly ventilated lung areas with high CO2 levels, thus optimizing oxygenation; no significant drop in PaO2 values was observed in our study. In systemic circulation, hypercapnia typically causes vasodilation, enhancing blood flow to tissues [44], with some notable exception regarding kidneys and to some extension the skeletal muscle [45, 46]; nonetheless, no changes in CPP were observed in our study. As such, our findings suggest that hypercapnia may affect cerebral perfusion and oxygenation in patients with brain injury without detrimental effects or adverse consequences for other organs.
Notably, a significant PbtO2 response was observed in only one third of the patients. One possible explanation for these findings is the loss of cerebrovascular reactivity following an acute brain injury [47, 48]. Several mechanisms contribute to the loss of cerebrovascular reactivity, such as endothelial dysfunction (resulting from an imbalance in the production of vasodilatory and vasoconstrictive mediators), smooth muscle cell dysfunction (stemming from structural and functional changes in these cells, impairing their ability to respond to vasodilatory stimuli), neural dysregulation (arising from alterations in perivascular nerves that modulate cerebral vascular tone through the release of vasoactive neurotransmitters), or altered metabolic coupling (pertaining to the relationship between neuronal activity and CBF) [49,50,51,52]. Unfortunately, our study did not specifically assess which of these mechanisms might be present in nonresponder patients. Although we observed a more substantial PbtO2 response in patients with SAH in an exploratory descriptive analysis, it remains unknown whether the underlying brain injury may have different effects on cerebrovascular reactivity. Moreover, a larger PbtO2 response was observed in patients with unfavorable neurological outcome, who might have also exhibited a higher probability of neural dysfunction or altered metabolic coupling. This observation may contradict our hypothesis that the loss of cerebrovascular reactivity is the primary factor behind the varying PbtO2 responses, although it remains an exploratory analysis. Further research is needed to explore the potential mechanisms and their impact on the response of PbtO2 to induced hypercapnia in this setting.
This study has inherent limitations. First, it is important to acknowledge that our study relied on the accuracy of the medical records, and it is possible that relevant data may not have been consistently reported because of its retrospective design. Second, as a single-center study, the generalizability of our results to other centers may be limited. Furthermore, the decision to induce hypercapnia was determined by the treating physician and not protocolized, which could impact the generalizability of our findings. However, it is worth noting that this decision was typically made in cases in which ICP was within normal limits and PbtO2 was low. Third, we had a small patient cohort, which limited our analysis and statistical inference; therefore, our results should be interpreted with caution. The small sample size of this study limits the generalizability of its findings and impacts their applicability in the clinical domain. Studies with a limited number of participants may not adequately represent the broader population or patient groups, leading to potential biases and reduced external validity. Clinicians need robust evidence from additional studies with larger and diverse cohorts to confidently implement the routine use of induced hypercapnia as a valid tool to increase PbtO2. Moreover, additional research is required to understand the heterogeneous effect of such therapy on tissue oxygenation after an acute brain injury. Fourth, we did not evaluate the probe position, which may have impacted our results, as cerebrovascular reactivity is not uniform throughout the brain. Fifth, the threshold used to define “PbtO2 responders” is somewhat arbitrary, although it has been used in previous studies. The decision to use a relative increase in PbtO2, rather than an absolute increase, was based on the understanding that areas with reduced CBF and impending ischemia may exhibit a lower increase in PbtO2 in response to specific challenges (e.g., an increase in mean arterial pressure, inspired fraction of oxygen, red blood cells transfusions, or PaCO2) as compared to normally perfused areas [53]. Therefore, assessing the relative increase in PbtO2 would provide a more accurate evaluation of the response to therapies. Lastly, it is important to consider that the effect of PaCO2 on PbtO2 may not follow a linear relationship, which could have influenced our results. Additionally, because we did not have continuous measures of CO2, we were unable to assess whether there is a threshold effect associated with the PbtO2 response to hypercapnia. Furthermore, it is worth noting that practices might have changed during the study period, and these changes could have influenced our subgroup analyses.
Conclusions
In this study, patients exhibited a heterogeneous PbtO2 response to moderate hypoventilation and induced hypercapnia. Future studies should focus on identifying which patients may benefit from this approach and the optimal strategy for implementing induced mild hypercapnia.
Data Availability
Because of ethical restrictions, the data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request. All data generated after the analysis during this study are included in this published article and in its supplementary material.
References
Lantigua H, Ortega-Gutierrez S, Schmidt JM, Lee K, Badjatia N, Agarwal S, et al. Subarachnoid hemorrhage: who dies, and why? Crit Care. 2015;19:309. https://doi.org/10.1186/s13054-015-1036-0.
Popescu C, Anghelescu A, Daia C, Onose G. Actual data on epidemiological evolution and prevention endeavours regarding traumatic brain injury. J Med Life. 2015;8:272–7.
Hays LMC, Udy A, Adamides AA, Anstey JR, Bailey M, Bellapart J, et al. Effects of brain tissue oxygen (PbtO2) guided management on patient outcomes following severe traumatic brain injury: a systematic review and meta-analysis. J Clin Neurosci. 2022;99:349–58. https://doi.org/10.1016/j.jocn.2022.03.017.
Taccone FS, De Oliveira Manoel AL, Robba C, Vincent JL. Use a “GHOST-CAP” in acute brain injury. Crit Care. 2020;24:89. https://doi.org/10.1186/s13054-020-2825-7.
Wartenberg KE, Schmidt JM, Claassen J, Temes RE, Frontera JA, Ostapkovich N, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med. 2006;34:617–23. https://doi.org/10.1097/01.ccm.0000201903.46435.35.
Lazaridis C, Robertson CS. The role of multimodal invasive monitoring in acute traumatic brain injury. Neurosurg Clin N Am. 2016;27:509–17. https://doi.org/10.1016/j.nec.2016.05.010.
O’Leary RA, Nichol AD. Pathophysiology of severe traumatic brain injury. J Neurosurg Sci. 2018;62:542–8. https://doi.org/10.23736/S0390-5616.18.04501-0.
Lazaridis C, Rusin CG, Robertson CS. Secondary brain injury: predicting and preventing insults. Neuropharmacology. 2019;145:145–52. https://doi.org/10.1016/j.neuropharm.2018.06.005.
Vath A, Kunze E, Roosen K, Meixensberger J. Therapeutic aspects of brain tissue pO2 monitoring after subarachnoid hemorrhage. Acta Neurochir Suppl. 2002;81:307–9. https://doi.org/10.1007/978-3-7091-6738-0_78.
Chen HI, Stiefel MF, Oddo M, Milby AH, Maloney-Wilensky E, Frangos S, et al. Detection of cerebral compromise with multimodality monitoring in patients with subarachnoid hemorrhage. Neurosurgery. 2011;69:53–63. https://doi.org/10.1227/NEU.0b013e3182191451.
Kett-White R, Hutchinson PJ, Al-Rawi PG, Gupta AK, Pickard JD, Kirkpatrick PJ. Adverse cerebral events detected after subarachnoid hemorrhage using brain oxygen and microdialysis probes. Neurosurgery. 2002;50:1213–21. https://doi.org/10.1097/00006123-200206000-00008.
Maloney-Wilensky E, Gracias V, Itkin A, Hoffman K, Bloom S, Yang W, et al. Brain tissue oxygen and outcome after severe traumatic brain injury: a systematic review. Crit Care Med. 2009;37:2057–63. https://doi.org/10.1097/CCM.0b013e3181a009f8.
Oddo M, Levine JM, Mackenzie L, Frangos S, Feihl F, Kasner SE, et al. Brain hypoxia is associated with short-term outcome after severe traumatic brain injury independently of intracranial hypertension and low cerebral perfusion pressure. Neurosurgery. 2011;69:1037–45. https://doi.org/10.1227/NEU.0b013e3182287ca7.
van den Brink WA, van Santbrink H, Steyerberg EW, Avezaat CJ, Suazo JA, Hogesteeger C, et al. Brain oxygen tension in severe head injury. Neurosurgery. 2000;46:868–76. https://doi.org/10.1097/00006123-200004000-00018.
Haitsma IK, Maas AI. Advanced monitoring in the intensive care unit: brain tissue oxygen tension. Curr Opin Crit Care. 2002;8:115–20. https://doi.org/10.1097/00075198-200204000-00005.
Bohman LE, Heuer GG, Macyszyn L, Maloney-Wilensky E, Frangos S, Le Roux PD, et al. Medical management of compromised brain oxygen in patients with severe traumatic brain injury. Neurocrit Care. 2011;14:361–9. https://doi.org/10.1007/s12028-011-9526-7.
Gouvea Bogossian E, Diaferia D, Ndieugnou Djangang N, Menozzi M, Vincent JL, Talamonti M, et al. Brain tissue oxygenation guided therapy and outcome in non-traumatic subarachnoid hemorrhage. Sci Rep. 2021;11:16235. https://doi.org/10.1038/s41598-021-95602-6.
Kovacs M, Peluso L, Njimi H, De Witte O, Gouvêa Bogossian E, Quispe-Cornejo A, et al. Optimal cerebral perfusion pressure guided by brain oxygen pressure measurement. Front Neurol. 2021;12:732830. https://doi.org/10.3389/fneur.2021.732830.
Gouvea Bogossian E, Diosdado A, Barrit S, Al Barajraji M, Annoni F, Schuind S, et al. The impact of invasive brain oxygen pressure guided therapy on the outcome of patients with traumatic brain injury: a systematic review and meta-analysis. Neurocrit Care. 2022;37:779–89. https://doi.org/10.1007/s12028-022-01613-0.
Schmieder K, Jarus-Dziedzic K, Wronski J, Harders A. CO2 reactivity in patients after subarachnoid haemorrhage. Acta Neurochir (Wien). 1997;139:1038–41. https://doi.org/10.1007/BF01411557.
Brian JE Jr. Carbon dioxide and the cerebral circulation. Anesthesiology. 1998;88:1365–86. https://doi.org/10.1097/00000542-199805000-00029.
Okonkwo DO, Shutter LA, Moore C, Temkin NR, Puccio AM, Madden CJ, et al. Brain oxygen optimization in severe traumatic brain injury phase-II: a phase II randomized trial. Crit Care Med. 2017;45:1907–14. https://doi.org/10.1097/CCM.0000000000002619.
Chesnut R, Aguilera S, Buki A, Bulger E, Citerio G, Cooper DJ, et al. A management algorithm for adult patients with both brain oxygen and intracranial pressure monitoring: the Seattle International Severe Traumatic Brain Injury Consensus Conference (SIBICC). Intensive Care Med. 2020;46:919–29. https://doi.org/10.1007/s00134-019-05900-x.
Godoy DA, Rovegno M, Lazaridis C, Badenes R. The effects of arterial CO2 on the injured brain: two faces of the same coin. J Crit Care. 2021;61:207–15. https://doi.org/10.1016/j.jcrc.2020.10.028.
Stetter C, Weidner F, Lilla N, Weiland J, Kunze E, Ernestus RI, et al. Therapeutic hypercapnia for prevention of secondary ischemia after severe subarachnoid hemorrhage: physiological responses to continuous hypercapnia. Sci Rep. 2021;11:11715. https://doi.org/10.1038/s41598-021-91007-7.
Westermaier T, Stetter C, Kunze E, Willner N, Holzmeier J, Weiland J, et al. Controlled hypercapnia enhances cerebral blood flow and brain tissue oxygenation after aneurysmal subarachnoid hemorrhage: results of a phase 1 study. Neurocrit Care. 2016;25:205–14. https://doi.org/10.1007/s12028-016-0246-x.
Meyfroidt G, Bouzat P, Casaer MP, Chesnut R, Hamada SR, Helbok R, et al. Management of moderate to severe traumatic brain injury: an update for the intensivist. Intensive Care Med. 2022;48:649–66. https://doi.org/10.1007/s00134-022-06702-4. (Erratum in: Intensive Care Med. 2022;48(7):989–91).
Gouvea Bogossian E, Battaglini D, Fratino S, Minini A, Gianni G, Fiore M, et al. The role of brain tissue oxygenation monitoring in the management of subarachnoid hemorrhage: a scoping review. Neurocrit Care. 2023. https://doi.org/10.1007/s12028-023-01680-x.
Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med. 1985;13:818–29.
Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 2017;2:81–4.
Coppalini G, Duvigneaud E, Diosdado A, Migliorino E, Schuind S, Creteur J, et al. Effect of inotropic agents on oxygenation and cerebral perfusion in acute brain injury. Front Neurol. 2022;13:963562. https://doi.org/10.3389/fneur.2022.963562.
Gouvea-Bogossian E, Rass V, Lindner A, Iaquaniello C, Miroz JP, Cavalcante-Dos-Santos E, et al. Factors associated with brain tissue oxygenation changes after RBC transfusion in acute brain injury patients. Crit Care Med. 2022;50:e539–47. https://doi.org/10.1097/CCM.0000000000005460.
Firth D. Bias reduction of maximum likelihood estimates. Biometrika. 1993;80:27–38. https://doi.org/10.1093/biomet/80.1.27.
Nangunoori R, Maloney-Wilensky E, Stiefel M, Park S, Andrew Kofke W, Levine JM, et al. Brain tissue oxygen-based therapy and outcome after severe traumatic brain injury: a systematic literature review. Neurocrit Care. 2012;17:131–8. https://doi.org/10.1007/s12028-011-9621-9.
Xie Q, Wu HB, Yan YF, Liu M, Wang ES. Mortality and outcome comparison between brain tissue oxygen combined with intracranial pressure/cerebral perfusion pressure-guided therapy and intracranial pressure/cerebral perfusion pressure-guided therapy in traumatic brain injury: a meta-analysis. World Neurosurg. 2017;100:118–27. https://doi.org/10.1016/j.wneu.2016.12.097.
Harper AM, Glass HI. Effect of alterations in the arterial carbon dioxide tension on the blood flow through the cerebral cortex at normal and low arterial blood pressures. J Neurol Neurosurg Psychiatry. 1965;28:449–52. https://doi.org/10.1136/jnnp.28.5.449.
Ito H, Kanno I, Ibaraki M, Suhara T, Miura S. Relationship between baseline cerebral blood flow and vascular responses to changes in PaCO2 measured by positron emission tomography in humans: implication of inter-individual variations of cerebral vascular tone. Acta Physiol (Oxf). 2008;193:325–30. https://doi.org/10.1111/j.1748-1716.2008.01847.x.
Ito H, Yokoyama I, Iida H, Kinoshita T, Hatazawa J, Shimosegawa E, et al. Regional differences in cerebral vascular response to PaCO2 changes in humans measured by positron emission tomography. J Cereb Blood Flow Metab. 2000;20:1264–70. https://doi.org/10.1097/00004647-200008000-00011.
Ito H, Ibaraki M, Kanno I, Fukuda H, Miura S. Changes in the arterial fraction of human cerebral blood volume during hypercapnia and hypocapnia measured by positron emission tomography. J Cereb Blood Flow Metab. 2005;25:852–7. https://doi.org/10.1038/sj.jcbfm.9600076.
Hawryluk GW, Phan N, Ferguson AR, Morabito D, Derugin N, Stewart CL, et al. Brain tissue oxygen tension and its response to physiological manipulations: influence of distance from injury site in a swine model of traumatic brain injury. J Neurosurg. 2016;125:1217–28. https://doi.org/10.3171/2015.7.JNS15809.
Zoerle T, Lombardo A, Colombo A, Longhi L, Zanier ER, Rampini P, et al. Intracranial pressure after subarachnoid hemorrhage. Crit Care Med. 2015;43:168–76. https://doi.org/10.1097/CCM.0000000000000670.
Godoy DA, Lubillo S, Rabinstein AA. Pathophysiology and management of intracranial hypertension and tissular brain hypoxia after severe traumatic brain injury: an integrative approach. Neurosurg Clin N Am. 2018;29:195–212. https://doi.org/10.1016/j.nec.2017.12.001.
Lumb AB, Slinger P. Hypoxic pulmonary vasoconstriction: physiology and anesthetic implications. Anesthesiology. 2015;122:932–46. https://doi.org/10.1097/ALN.0000000000000569.
Marhong J, Fan E. Carbon dioxide in the critically ill: too much or too little of a good thing? Respir Care. 2014;59:1597–605. https://doi.org/10.4187/respcare.03405.
Moriyama S, Ichinose M, Dobashi K, Matsutake R, Sakamoto M, Fujii N, et al. Hypercapnia elicits differential vascular and blood flow responses in the cerebral circulation and active skeletal muscles in exercising humans. Physiol Rep. 2022;10:e15274. https://doi.org/10.14814/phy2.15274.
Chapman CL, Schlader ZJ, Reed EL, Worley ML, Johnson BD. Renal and segmental artery hemodynamic response to acute, mild hypercapnia. Am J Physiol Regul Integr Comp Physiol. 2020;318:R822–7. https://doi.org/10.1152/ajpregu.00035.2020.
Zeiler FA, Ercole A, Czosnyka M, Smielewski P, Hawryluk G, Hutchinson PJA, et al. Continuous cerebrovascular reactivity monitoring in moderate/severe traumatic brain injury: a narrative review of advances in neurocritical care. Br J Anaesth. 2020;124:440–53. https://doi.org/10.1016/j.bja.2019.11.031.
Bøthun ML, Haaland ØA, Moen G, Logallo N, Svendsen F, Thomassen L, et al. Impaired cerebrovascular reactivity may predict delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. J Neurol Sci. 2019;407:116539. https://doi.org/10.1016/j.jns.2019.116539.
Phillips AA, Chan FH, Zheng MM, Krassioukov AV, Ainslie PN. Neurovascular coupling in humans: physiology, methodological advances and clinical implications. J Cereb Blood Flow Metab. 2016;36:647–64. https://doi.org/10.1177/0271678X15617954.
Lidington D, Kroetsch JT, Bolz SS. Cerebral artery myogenic reactivity: The next frontier in developing effective interventions for subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2018;38:17–37. https://doi.org/10.1177/0271678X17742548.
Lidington D, Wan H, Bolz SS. Cerebral autoregulation in subarachnoid hemorrhage. Front Neurol. 2021;12:688362. https://doi.org/10.3389/fneur.2021.688362.
Claassen J, Thijssen DHJ, Panerai RB, Faraci FM. Regulation of cerebral blood flow in humans: physiology and clinical implications of autoregulation. Physiol Rev. 2021;101:1487–559. https://doi.org/10.1152/physrev.00022.2020.
Hosmann A, Schnackenburg P, Rauscher S, Hopf A, Bohl I, Engel A, et al. Brain tissue oxygen response as indicator for cerebral lactate levels in aneurysmal subarachnoid hemorrhage patients. J Neurosurg Anesthesiol. 2022;34:193–200. https://doi.org/10.1097/ANA.0000000000000713.
Funding
None.
Author information
Authors and Affiliations
Contributions
MA, EGB, and FST contributed to conception and design of the study. MA, MS, LP, and FA collected data. EGB and MA performed data curation. EGB, MA, and FST performed the statistical analysis. MA, EGB, and FST wrote the first draft of the manuscript. KD and SS revised the manuscript for intellectual content and English editing. All authors contributed to manuscript revision and read and approved the submitted version.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no competing interests regarding this article.
Ethical Approval/Informed Consent
The study followed ethical guidelines, and the Erasme Hospital Ethics Committee approved this study (P2022/449). Written informed consent for study participation was waived by the ethics committee.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Anderloni, M., Schuind, S., Salvagno, M. et al. Brain Oxygenation Response to Hypercapnia in Patients with Acute Brain Injury. Neurocrit Care 40, 750–758 (2024). https://doi.org/10.1007/s12028-023-01833-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-023-01833-y