
Abstract
Follicular helper T (Tfh) cells are increasingly recognized as participants in various autoimmune diseases, including Graves’ disease. Although many transcription factors and cytokines are known to regulate Tfh cells, the role of noncoding RNA in Tfh cells development and function is poorly understood. Twenty-three patients with GD, eleven patients with remitting GD, and twenty-four healthy controls were enrolled in the current study. The interaction of miRNA and target gene was predicted through software analysis and then validated by luciferase assay and Western blot. The levels of miR-346 in circulating CD4+ T cells and plasma were measured by qRT-PCR. The correlation of miR-346 levels with the percentages of CD4+CXCR5+T cells and autoantibody levels were also analyzed. Up-regulation of Bcl-6 and down-regulation of miR-346 in GD patients were observed, and miR-346 could inhibit Bcl-6 at both transcriptional and translational levels. Overexpression of miR-346 led to attenuating CD4+CXCR5+ T cells. The abnormal expression of miR-346 restored in GD patients after treatment. A negative correlation between levels of miR-346 and percentages of CD4+CXCR5+ T cells was confirmed in GD patients. Additionally, negative correlations between the levels of miR-346 in circulating CD4+ T cells and serum concentrations of TR-Ab, TG-Ab, and TPO-Ab were also revealed in GD patients. MiR-346 regulates CD4+CXCR5+ T cells by targeting Bcl-6, a positive regulator of Tfh cells, and might play an important role in the pathogenesis of Graves’ disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Upon antigen stimulation, naive CD4+ helper T cells differentiate into effector T cells and then participate in the adaptive immune response. Follicular helper T (Tfh) cells have been identified as a new subset of effector helper T cells that are essential in regulating the development of antigen-specific B cell immunity [1–3]. Tfh cells differentiation is critically regulated by specific transcription factor Bcl-6, which is characteristically expressed in Tfh cells but not other effector Th cells [4, 5]. Bcl-6 controls the Tfh cells development and causes a generalized inhibition of other CD4+ T cell differentiation pathways [6, 7]. Although an increasing number of molecular regulators in Tfh cells differentiation have been identified, possible factors that link the generation of Tfh cells to pathological conditions such as autoimmunity are largely unknown.
As a prototype of organ-specific autoimmune disease, Graves’ disease (GD) is manifested by diffuse goiter and positive anti-thyrotropin receptor antibody (TR-Ab). The disease is clinically diagnosed with elevated free triiodothyronine (FT3), free thyroxine (FT4), and thyroid-stimulating hormone (TSH) and is characterized by elevated production of autoantibodies, such as anti-thyroglobulin antibody (TG-Ab) and anti-thyroperoxidase antibody (TPO-Ab) [8–10]. Recently, it is reported that IL-21 in GD is correlated with these antibodies, which indicates it role in the pathogenesis in GD [11].
CD4+ Th1, Th2, and Th17 cells, characterized by the production of IFN-γ, IL-4, and IL-17, respectively, have been considered as effector helper T cells that mediate the pathogenesis of autoimmune thyroid disease [12–15]. Our recent studies have indicated that Tfh cells are involved in the pathogenesis of this disease [16]. Tfh cells exhibit a unique phenotype, expressing high level of CXCR5 [17–19]. CD4+ T cells that express CXCR5 can migrate to the follicles in response to CXCL13. Tfh cells in the germinal center participate in regulating both effector and memory B cell responses as well as antibody production [20]. The percentages of Tfh cells in peripheral blood from GD patients were increased and showed a close correlation with FT3 or FT4. Furthermore, a positive correlation between the percentages of Tfh cells and the concentration of autoantibody was found in GD patients [16]. Despite the lines of evidence described above, the underlying molecules regulating the Tfh cells development and function are still elusive.
MicroRNAs (miRNAs) represent a large family of endogenous noncoding RNAs that negatively regulate gene expression post-transcriptionally [21, 22]. It has been recognized that deregulated miRNAs are critically involved in multiple human diseases and may potentially serve as diagnostic markers [23, 24], prognostic markers, or therapeutic targets [25]. miRNAs also participate in the development of autoimmunity. For instance, up-regulated miR-146a expression has been found in peripheral blood mononuclear cells (PBMCs) from patients with rheumatoid arthritis [26]. Moreover, increased miR-21 expression is closely correlated with human lupus disease activity [27]. However, it is currently unclear whether and how miRNAs are involved in pathogenesis of Graves’ disease.
Here, we report that miR-346 plays a critical role in regulating CD4+CXCR5+T cells and hence contributes to the pathogenesis of GD. We found that miR-346 regulated CD4+CXCR5+ T cells through translational inhibition of Bcl-6 (NM_001706), a master regulator of Tfh cells development. In GD patients, miR-346 expression was down-regulated, which correlated with increased percentages of CD4+CXCR5+ T cells and disease severity. Collectively, our results suggest that miR-346 is a CD4+CXCR5+ T cells-associated miRNA which involved in the pathogenesis of Graves’ disease.
Materials and methods
Subjects and samples
Subjects were patients from the outpatient clinic of the Affiliated People’s Hospital of Jiangsu University with clinically defined GD or euthyroid-state GD. GD patients were diagnosed by clinically and biochemically verified hyperthyroidism and positive TR-Ab. The clinical evaluation included the patient’s history, physical examination, and thyroid ultrasound. The laboratory examination included serum levels of FT3, FT4, TR-Ab, thyroperoxidase antibody, and thyroglobulin antibody. Main clinical data of patients are shown in Table 1. 23 GD patients and 11 euthyroid-state GD were analyzed. All of the GD patients have a lower level of TSH. Serum concentrations of TR-Ab, TG-Ab, and TPO-Ab were measured by chemiluminescent immunoassay (MAGLUMI 2000 PLUS) according to the manufacturer’s protocol. Patients on methimazole therapy received 20–30 mg/day for the first phase, and the dose was reduced to 5–15 mg when patients achieved a remission. Patients treated with propylthiouracil took 300–500 mg/day for the first phase and 25–100 mg for maintaining remission. Twenty-four age- and sex-matched healthy subjects were included as controls, including 17 females and 7 males, ranging from 28 to 50 years old. All of the control subjects were healthy and free of a history of thyroid or autoimmune diseases. Blood samples were obtained from subjects after informed consent. All samples were taken in accordance with the regulations and approval of the Affiliated People’s Hospital of Jiangsu University.
Cell isolation and purification
PBMCs were purified from density-gradient centrifugation over Ficoll–Hypaque solution. CD4+ T cells were isolated from PBMC by biotin-conjugated anti-human CD4 monoclonal Ab (mAb) and anti-biotin microbeads (Miltenyi Biotec GmbH) according to the manufacturer’s instructions. Briefly, PBMCs were stained with biotin-conjugated anti-human CD4 mAb for 10 min, and then cells were washed with PBS and stained with anti-biotin microbeads for 15 min. Then, cells were washed, resuspended in PBS, and loaded onto the magnetic separation to isolate the CD4+ cells.
Flow cytometry analysis
Cells were collected and stained with relevant mAbs including phycoerythrin-cy5-conjugated anti-CD3, phycoerythrin (PE)-conjugated anti-CD4 (eBioscience, San Diego, CA), and Alexa Fluor 488-conjugated anti-CXCR5 (Becton Dickinson, San Jose, CA) mAb. Flow cytometry was performed using FACSCalibur Flow Cytometer (Becton Dickinson, Sparks, MD), and data were analyzed using WinMDI 2.8 software.
RNA isolation and real-time PCR
Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Random primer and M-MLV Reverse Transcriptase (Toyobo, Osaka, Japan) were used for Bcl-6 reverse transcription. Gene transcripts were quantified by real-time quantitative PCR performed with the Bio-Rad SYBR green super mix (Bio-Rad, Hercules, CA). Primer sequences were as follows: Bcl-6, sense, 5′-CACACTCGAATTCACTCTG-3′; and antisense, 5′-TATTGCACCTTGGTGTTGG-3′. The gene was normalized to β-actin with the following primers: sense, 5′-CACGAAACTACCTTCAACTCC-3′; and antisense, 5′-CATACTCCTGCTTGCTGATC-3′. microRNA in plasma was isolated with NucleoSpin® miRNA Plasma (Macherey–Nagel GmbH & Co. KG). miRNA qRT-PCR Primer Set (Ribo Life Science Co. Ltd., China) and M-MLV Reverse Transcriptase (Takara Biotechnology, China) were used for miR-346 and U6 reverse transcription. Gene transcripts were quantified by real-time quantitative PCR performed with the Bio-Rad SYBR green super mix (Bio-Rad). Data were analyzed by Bio-Rad CFX Manager software.
Transfection
Jurkat cells were transfected with the miR-346 mimics or negative control oligos at different doses (100, 150, and 200 nM) using the Entranster-R (Engreen Biosystem, China) according to the manufacturers’ instructions.
Western blot analysis
Proteins extracted from cells were prepared as described previously [28]. Proteins were separated by sodium-dodecyl-sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto immobilon PVDF membranes (Bio-Rad), and probed with Bcl-6 antibody (eBioscience) and β-actin antibody (Abcam, UK), followed by chemiluminescent detection (Champion Chemical).
Luciferase reporter assay
HEK293T cells were maintained in DMEM (Gibco) in 24-well plates and transfected with Lipofectamine 2000 reagent (Invitrogen). A Dual-Luciferase reporter gene construct (200 ng per well) and microRNA mimics or negative control (50nM) were co-transfected. Cell extracts were prepared 48 h after transfection, and luciferase activity was measured with the Dual-Luciferase Reporter Assay system (Promega Corporation, Beijing, China). Wild-type or mutated 3′ UTR sequences of Bcl-6 were cloned into the psiCHECK-2 vector (Promega) as described [29].
Statistical analysis
A two-tailed Student’s t test was applied for statistical comparison of two groups or, where appropriated, a Mann–Whitney test for nonparametric data (serum miR-346). A P value of 0.05 or less was considered significant. Correlations between variables were determined by Pearson’s correlation coefficient. Data were analyzed with GraphPad Prism 5 software (GraphPad Software).
Results
Up-regulation of Bcl-6 in circulating CD4+ T cells from GD patients
Recent studies showed that the transcriptional factor Bcl-6 is critically involved in regulating Tfh cells differentiation. Here, we found that the levels of Bcl-6 mRNA in circulating CD4+ T cells were significantly increased in GD patients (Fig. 1a). This result was consistent with our previous findings that increased percentages of Tfh cells were found in peripheral blood from GD patients [16].

An inverse correlation between the expression of miR-346 and Bcl-6 in GD patients. Quantitative RT-PCR was used to analyze the expression of Bcl-6 (a), miR-346 (b), miR-181d (c), and miR-30b (d) in circulating CD4+ T cells from GD patients and healthy controls (n = 10). Each data point represents an individual subject; horizontal lines show the mean. *P < 0.05; ns no significant differences
miR-346 functionally targets to Bcl-6 molecule
We applied the prediction programs PicTar and miRanda to identify which miRNAs were involved in regulating Bcl-6 expression. Through base-pairing interaction between miRNA seed sequence and the 3′UTR of target genes, we found that a sequence motif of the 3′UTR of Bcl-6 matches with miR-346, miR-181d, and miR-30b. Next, we examined the levels of miRNAs expression in circulating CD4+ T cells from GD patients by quantitative RT-PCR analysis. miR-346 was found to be significantly down-regulated, while miR-181d was up-regulated in GD patients (Fig. 1b, c). However, no significant difference in miR-30b expression was detected between GD patients and control subjects (Fig. 1d). As miRNAs are known to negatively regulate their target genes, miR-346 was selected for further studies.
There were two sites of the 3′UTR of Bcl-6, namely UTR1 and UTR2 (Fig. 2a). To directly prove that Bcl-6 is the functional target of miR-346, four recombined reporters were transfected into human 293T cells with miR-346 mimics, respectively (Fig. 2b). We found that miR-346 inhibited the luciferase activity of reporters containing the wild-type UTR1 or UTR2, but not that of the reporter with a mutated 3′UTR unable to bind miR-346 (Fig. 2c). As Bcl-6 is expressed in Jurkat cells (Fig. 2d), miR-346 suppressed the expression of Bcl-6 mRNA in a dose-dependent manner (Fig. 2e). Manipulation of miR-346 in Jurkat cells also regulated the amount of endogenous Bcl-6 protein level (Fig. 2f). Together, these results indicate that miR-346 functions at both transcriptional and translational levels.

Bcl-6 is a functional target of miR-346. a Mutations in the Bcl-6 3′ UTR matched the miR-346. b Double-enzyme digestion production of four recombinant plasmids. M1 λ-EcoT14I digest; 1–4 double-enzyme digestion production of recombinant plasmids containing wild-type (UTR1 + 2), mutated UTR1, mutated UTR2, and mutated UTR(1 + 2) fragments, respectively; M2: DL2000 DNA Marker. c luciferase activity of reporter carrying the mutated (Mut) or wild-type Bcl-6 3′ UTR co-transfected into HEK293T cells with miR-346 mimics or negative control (NC). d RT-PCR products of Bcl-6 in Jurkat cells and PBMCs. e Expression of miR-346 (left) and Bcl-6 mRNA (right) in transfected Jurkat cells. f Immunoblot analysis of Bcl-6 protein in transfected Jurkat cells. Results are representative of three independent experiments. **P < 0.01
miR-346 attenuated CD4+CXCR5+ T cells in vitro
Circulating CD4+ cells from healthy donor were purified by magnetic beads separation. Next, we examined the percentage of CD4+CXCR5+ T cells in groups which transfected with miR-346 mimics or control. Manipulation of miR-346 in CD4+ cells regulated the amount of miR-346 and endogenous Bcl-6 mRNA (Fig. 3a, b). We found decreased percentage of CD4+CXCR5+ T cells in miR-346-transfected group than that in the control group (Fig. 3c).

Regulatory effect of miR-346 on CD4+CXCR5+ T cells phenotype. miR-346 mimics or negative control were transfected into CD4+ cells, and the levels of miR-346 (a) and Bcl-6 (b) were detected. c Proportions of CD4+CXCR5+ T cells from different groups were analyzed by flow cytometry. Results are representative of three independent experiments
Down-regulated miR-346 in circulating CD4+T cells from GD patients
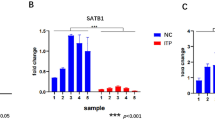
From clinical samples (Table 1), we found that miR-346 expression was significantly lower in circulating CD4+T cells of GD patients than in those of age-matched controls or patients after treatment (Fig. 4a). Consistently, a negative correlation was found between levels of Bcl-6 and miR-346 transcripts in circulating CD4+T cells of GD patients (Fig. 4b). There was also a negative correlation between the levels of miR-346 and the percentages of CD4+CXCR5+ T cells from GD patients (Fig. 4c). Moreover, miR-346 expression was significantly lower in plasma of patients with GD than in those of age-matched controls (Fig. 4D). There was a negative but modest correlation between miR-346 levels and CD4+CXCR5+ T cells from patients with GD (Fig. 4E). These results demonstrate that decreased expression of miR-346 correlates with increased circulating CD4+CXCR5+ T cells.

Down-regulation of miR-346 in patients with Graves’ disease. a Quantitative PCR analysis of miR-346 expression in circulating CD4+ cells from normal controls (n = 23) and patients with GD (n = 23) or remitting GD (n = 11). b The correlation between the levels of miR-346 and Bcl-6 in circulating CD4+ cells. c correlation of miR-346 in circulating CD4+ cells with percentages of CD4+CXCR5+ T cells was analyzed. d Quantitative PCR was applied to analyze miR-346 expression in plasma from normal controls and patients with GD or remitting GD. e the correlation between the levels of miR-346 in plasma and percentages of CD4+CXCR5+ T cells was analyzed. Each data point represents an individual subject; horizontal lines show the mean. ***P < 0.001; *P < 0.05; ns no significant differences
Decreased miR-346 expression correlates with high levels of autoantibodies in GD patients
In GD patients, TR-Ab is a typical autoantibody which is known to simulate the TSH binding to TSHR. TG-Ab and TPO-Ab are also critical antibodies because about 80 % of GD patients are positive regarding these parameters [9, 30]. Our data showed that miR-346 in circulating CD4+ T cells was negatively correlated with the concentrations of TR-Ab, TG-Ab, and TPO-Ab in GD patients (Fig. 5).

The correlation between miR-346 and relevant autoantibodies. The correlation of miR-346 in circulating CD4+ T cells with the TR-Ab (a), TG-Ab (b), and TPO-Ab (c) in plasma was analyzed (n = 23)
Discussion
Accumulating evidence suggests that Tfh cells are critically involved in the pathogenesis of autoimmune diseases, including Graves’ disease [16]. Regulators of Tfh cells differentiation are considered to be potentially essential for clinical diagnosis and treatment of various autoimmune disorders. Here, we report that miR-346 is involved in the pathogenesis of Graves’ disease possibly by targeting Bcl-6 in CD4+CXCR5+T cells. We have shown that miR-346 expression in circulating CD4+T cells or plasma correlated with Graves’ disease activity. The level of miR-346 was substantially down-regulated in GD patients and restored in post-treatment patients. A negative correlation was found between the levels of miR-346 in circulating CD4+T cells and serum autoantibody levels.
Our data suggest that miR-346 is a CD4+CXCR5+ T cells-associated miRNA. Firstly, miR-346 expression inversely correlated with the levels of Bcl-6 mRNA and the percentages of CD4+CXCR5+ T cells in patients with Graves’ disease. Secondly, overexpression of miR-346 in circulating CD4+ T cells from healthy donor caused down-regulation of Bcl-6 mRNA and reduced the percentage of CD4+CXCR5+ T cells in vitro. Our findings may facilitate the identification of a new target for manipulating the generation of CD4+CXCR5+ T cells in related diseases. Further exploration of the function of miR-346, especially in other cell types of the innate immune system, will be of great importance for understanding the cell differentiation network of the immune system.
Generally, miRNAs are believed to function in vivo by targeting multiple functionally related proteins or a key protein target [31–33]. In the immune system, miR-155 controls T cells proliferation by down-regulating CTLA-4 in patients with atopic dermatitis [34] or targeting GSK3β in cardiac allograft rejection [35]. There is strong evidence that miR-9 controls B cell survival and differentiation by regulating a key target, PRDM-1 [36, 37]. Our data suggest that miR-346 regulates CD4+CXCR5+ T cells mainly by targeting Bcl-6. As a functional miR-346 target, Bcl-6 represents a distinct regulator of Tfh cells differentiation. Accumulated data have shown that Bcl-6 effectively promotes Tfh cells differentiation by inhibiting other CD4+T cell differentiation pathways [4, 5]. Our results have shown that Bcl-6 expression, both at mRNA and protein levels, was substantially down-regulated by miR-346 in Jurkat cells. Moreover, miR-346 inhibited the luciferase activity of a reporter containing the wild-type Bcl-6 3′ UTR, but not that of a reporter with a mutated 3′ UTR unable to bind miR-346. This indicates that miR-346 directly targets to Bcl-6 molecule.
The antithyroid drugs now approved for the clinical treatment of Graves’ disease work mainly by suppressing B cells from secreting antibody and inhibiting synthesis of thyroid hormones. Drugs that target T cells, however, have not yet been applied to Graves’ disease therapy. Notably, our data show that miR-346 is up-regulated in a group of patients after antithyroid drugs treatment, which indicates that miR-346 expression correlates with disease severity. Elucidation of the specific signals that regulate miR-346 expression in disease will enhance the understanding of how this miRNA-mediated mechanism contributes to the pathogenesis of Graves’ disease and help to identify new therapeutic approaches by targeting miR-346 for Graves’ disease treatment.
In summary, we found that miR-346 could regulate CD4+CXCR5+ T cells by targeting Bcl-6. In GD patients, the expression of miR-346 was down-regulated, which correlated with increased proportion of CD4+CXCR5+ T cells and disease severity. Collectively, our results may facilitate the validation of miR-346 as a new therapeutic target for the treatment of patients with Graves’ disease.
References
S. Crotty, Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29, 621–663 (2011)
J. Rolf, S.E. Bell, D. Kovesdi, M.L. Janas, D.R. Soond, L.M. Webb, S. Santinelli, T. Saunders, B. Hebeis, N. Killeen, K. Okkenhaug, M. Turner, Phosphoinositide 3-kinase activity in T cells regulates the magnitude of the germinal center reaction. J. Immunol. 185(7), 4042–4052 (2010)
J. Ma, C. Zhu, B. Ma, J. Tian, S.E. Baidoo, C. Mao, W. Wu, J. Chen, J. Tong, M. Yang, Z. Jiao, H. Xu, L. Lu, S. Wang, Increased frequency of circulating follicular helper T cells in patients with rheumatoid arthritis. Clin. Dev. Immunol. 2012, 827480 (2012)
S. Crotty, R.J. Johnston, S.P. Schoenberger, Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat. Immunol. 11(2), 114–120 (2010)
R.J. Johnston, A.C. Poholek, D. DiToro, I. Yusuf, D. Eto, B. Barnett, A.L. Dent, J. Craft, S. Crotty, Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325(5943), 1006–1010 (2009)
R.I. Nurieva, Y. Chung, G.J. Martinez, X.O. Yang, S. Tanaka, T.D. Matskevitch, Y.H. Wang, C. Dong, Bcl6 mediates the development of T follicular helper cells. Science 325(5943), 1001–1005 (2009)
D. Yu, S. Rao, L.M. Tsai, S.K. Lee, Y. He, E.L. Sutcliffe, M. Srivastava, M. Linterman, L. Zheng, N. Simpson, J.I. Ellyard, I.A. Parish, C.S. Ma, Q.J. Li, C.R. Parish, C.R. Mackay, C.G. Vinuesa, The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 31(3), 457–468 (2009)
M. Rotondi, L. Chiovato, Vitamin D deficiency in patients with Graves’ disease: probably something more than a casual association. Endocrine 43(1), 3–5 (2013)
A.P. Weetman, Autoimmune thyroid disease. Autoimmunity 37(4), 337–340 (2004)
E. Szczepanek-Parulska, M. Ruchala, A. Hernik, Unexpected conversion from hypothyroidism to an euthyroid state due to Graves’ disease in a patient with an ectopic thyroid. Endocrine 46(3), 684–685 (2014)
J. Zhang, H. Zeng, M. Ren, H. Yan, M. Xu, Z. Feng, W. Liang, C. Yang, H. Cheng, H. Ding, L. Yan, Interleukin-21 is associated with disease activity in patients with Graves’ disease. Endocrine 46(3), 539–548 (2014)
T. Kocjan, B. Wraber, U. Repnik, S. Hojker, Changes in Th1/Th2 cytokine balance in Graves’ disease. Pflugers Arch. 440(5 Suppl), R94–95 (2000)
Y. Nagayama, K. Watanabe, M. Niwa, S.M. McLachlan, B. Rapoport, Schistosoma mansoni and alpha-galactosylceramide: prophylactic effect of Th1 Immune suppression in a mouse model of Graves’ hyperthyroidism. J Immunol 173(3), 2167–2173 (2004)
T. Nanba, M. Watanabe, N. Inoue, Y. Iwatani, Increases of the Th1/Th2 cell ratio in severe Hashimoto’s disease and in the proportion of Th17 cells in intractable Graves’ disease. Thyroid 19(5), 495–501 (2009)
S. Wang, S.E. Baidoo, Y. Liu, C. Zhu, J. Tian, J. Ma, J. Tong, J. Chen, X. Tang, H. Xu, L. Lu, T cell-derived leptin contributes to increased frequency of T helper type 17 cells in female patients with Hashimoto’s thyroiditis. Clin. Exp. Immunol. 171(1), 63–68 (2013)
C. Zhu, J. Ma, Y. Liu, J. Tong, J. Tian, J. Chen, X. Tang, H. Xu, L. Lu, S. Wang, Increased frequency of follicular helper T cells in patients with autoimmune thyroid disease. J. Clin. Endocrinol. Metab. 97(3), 943–950 (2012)
D. Breitfeld, L. Ohl, E. Kremmer, J. Ellwart, F. Sallusto, M. Lipp, R. Forster, Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J. Exp. Med. 192(11), 1545–1552 (2000)
C.H. Kim, L.S. Rott, I. Clark-Lewis, D.J. Campbell, L. Wu, E.C. Butcher, Subspecialization of CXCR5+ T cells: B helper activity is focused in a germinal center-localized subset of CXCR5+ T cells. J. Exp. Med. 193(12), 1373–1381 (2001)
P. Schaerli, K. Willimann, A.B. Lang, M. Lipp, P. Loetscher, B. Moser, CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J. Exp. Med. 192(11), 1553–1562 (2000)
N. Fazilleau, L. Mark, L.J. McHeyzer-Williams, M.G. McHeyzer-Williams, Follicular helper T cells: lineage and location. Immunity 30(3), 324–335 (2009)
D.P. Bartel, MicroRNAs: target recognition and regulatory functions. Cell 136(2), 215–233 (2009)
J. Tian, K. Rui, S. Wang, Roles of miRNAs in regulating the differentiation and maturation of myeloid-derived suppressor cells. Med. Hypotheses 83(2), 151–153 (2014)
J. Lu, G. Getz, E.A. Miska, E. Alvarez-Saavedra, J. Lamb, D. Peck, A. Sweet-Cordero, B.L. Ebert, R.H. Mak, A.A. Ferrando, J.R. Downing, T. Jacks, H.R. Horvitz, T.R. Golub, MicroRNA expression profiles classify human cancers. Nature 435(7043), 834–838 (2005)
X. Tang, X. Tian, Y. Zhang, W. Wu, J. Tian, K. Rui, J. Tong, L. Lu, H. Xu, S. Wang, Correlation between the frequency of Th17 cell and the expression of MicroRNA-206 in patients with dermatomyositis. Clin. Dev. Immunol. 2013, 7 (2013)
T. Thum, C. Gross, J. Fiedler, T. Fischer, S. Kissler, M. Bussen, P. Galuppo, S. Just, W. Rottbauer, S. Frantz, M. Castoldi, J. Soutschek, V. Koteliansky, A. Rosenwald, M.A. Basson, J.D. Licht, J.T. Pena, S.H. Rouhanifard, M.U. Muckenthaler, T. Tuschl, G.R. Martin, J. Bauersachs, S. Engelhardt, MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 456(7224), 980–984 (2008)
K.M. Pauley, M. Satoh, A.L. Chan, M.R. Bubb, W.H. Reeves, E.K. Chan, Upregulated miR-146a expression in peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthr. Res. Ther. 10(4), R101 (2008)
E. Stagakis, G. Bertsias, P. Verginis, M. Nakou, M. Hatziapostolou, H. Kritikos, D. Iliopoulos, D.T. Boumpas, Identification of novel microRNA signatures linked to human lupus disease activity and pathogenesis: miR-21 regulates aberrant T cell responses through regulation of PDCD4 expression. Ann. Rheum. Dis. 70(8), 1496–1506 (2011)
J. Tian, J. Ma, K. Ma, H. Guo, S.E. Baidoo, Y. Zhang, J. Yan, L. Lu, H. Xu, S. Wang, β-Glucan enhances antitumor immune responses by regulating differentiation and function of monocytic myeloid-derived suppressor cells. Eur. J. Immunol. 43(5), 1220–1230 (2013)
J.F. Chen, E.M. Mandel, J.M. Thomson, Q. Wu, T.E. Callis, S.M. Hammond, F.L. Conlon, D.Z. Wang, The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 38(2), 228–233 (2006)
A.P. Weetman, Graves’ disease. N. Engl. J. Med. 343(17), 1236–1248 (2000)
C. Du, C. Liu, J. Kang, G. Zhao, Z. Ye, S. Huang, Z. Li, Z. Wu, G. Pei, MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat. Immunol. 10(12), 1252–1259 (2009)
K.P. Hoefig, V. Heissmeyer, MicroRNAs grow up in the immune system. Curr. Opin. Immunol. 20(3), 281–287 (2008)
C. Xiao, K. Rajewsky, MicroRNA control in the immune system: basic principles. Cell 136(1), 26–36 (2009)
E. Sonkoly, P. Janson, M.L. Majuri, T. Savinko, N. Fyhrquist, L. Eidsmo, N. Xu, F. Meisgen, T. Wei, M. Bradley, J. Stenvang, S. Kauppinen, H. Alenius, A. Lauerma, B. Homey, O. Winqvist, M. Stahle, A. Pivarcsi, MiR-155 is overexpressed in patients with atopic dermatitis and modulates T-cell proliferative responses by targeting cytotoxic T lymphocyte-associated antigen 4. J Allergy Clin Immunol. 126(3), 581–589 (2010). e581–520
Z. Feng, Y. Xia, M. Zhang, J. Zheng, MicroRNA-155 regulates T cell proliferation through targeting GSK3beta in cardiac allograft rejection in a murine transplantation model. Cell. Immunol. 281(2), 141–149 (2013)
X. Huang, X. Zhou, Z. Wang, F. Li, F. Liu, L. Zhong, X. Li, X. Han, Z. Wu, S. Chen, T. Zhao, CD99 triggers upregulation of miR-9-modulated PRDM1/BLIMP1 in Hodgkin/Reed-Sternberg cells and induces redifferentiation. Int. J. Cancer 131(4), E382–394 (2012)
J. Lin, T. Lwin, J.J. Zhao, W. Tam, Y.S. Choi, L.C. Moscinski, W.S. Dalton, E.M. Sotomayor, K.L. Wright, J. Tao, Follicular dendritic cell-induced microRNA-mediated upregulation of PRDM1 and downregulation of BCL-6 in non-Hodgkin’s B-cell lymphomas. Leukemia 25(1), 145–152 (2011)
Acknowledgments
This work was supported by the Specialized Research Fund for the Doctoral Program of Higher Education (Grant No. 20133227110008), National Natural Science Foundation of China (Grant Nos. 31100648, 81072453), Specialized Project for Clinical Medicine of Jiangsu Province (Grant No. BL2014065), Health Department Foundation of Jiangsu Province (Grant No. Z201312), Science and technology support program (Social Development) of Zhenjiang (Grant Nos. SH2013040, SH2014039) Graduate Student Research and Innovation Program of Jiangsu Province (Grant Nos. CXZZ13_0700, KYLX_1074), Jiangsu Province “333” Project, and Priority Academic Program Development of Jiangsu Higher Education Institutions.
Conflict of interest
The authors have no financial conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Juan Chen, Jie Tian, and Xinyi Tang have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Chen, J., Tian, J., Tang, X. et al. MiR-346 regulates CD4+CXCR5+ T cells in the pathogenesis of Graves’ disease. Endocrine 49, 752–760 (2015). https://doi.org/10.1007/s12020-015-0546-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-015-0546-5