
Abstract
Objectives
Accumulating evidence have suggested microRNAs (miRNAs) play important roles in the pathogenesis of systemic lupus erythematosus (SLE). Here we aimed to explore aberrant expression of miRNAs in CD4+ T cells from SLE patients and their potential function in SLE pathogenesis.
Methods
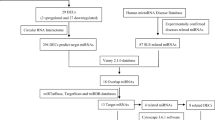
First, next-generation sequencing was performed on CD4+ T cells from four SLE patients and three healthy controls (HCs). Candidate miRNAs were then validated in CD4+ T cells from 97 patients with SLE, 16 patients with rheumatoid arthritis, and 12 HCs using qRT-PCR. Then the relationship between the candidate miRNA and clinical characteristics was analyzed. Bioinformatics analysis and validation of the target genes of the candidate miRNA were performed.
Results
A total of 66 upregulated miRNAs and 70 downregulated miRNAs were found between SLE and normal CD4+ T cells samples. miR-199a-3p was identified significant upregulation in the CD4+ T cells of lupus patients. High expression of miR-199a-3p was correlated with several clinical characteristics including low C3 level, positive anti-dsDNA antibody, high ESR level, active lupus nephritis, and active disease activity. When distinguishing active LN from non-LN or active lupus from stable lupus, the AUCs of miR-199a-3p were 0.68 and 0.70, respectively. And the expression of miR-199a-3p, involved in JAK-STAT signaling pathway, was negatively correlated with the STAM expression in CD4+ T cells of SLE.
Conclusion
Our study suggested a novel and promising role of miR-199a-3p in CD4+ T cells for SLE. Further studies are needed to precisely determine the function of miR-199a-3p in this disease.
Key Points • Aberrant expression of miRNAs in CD4+ T cells and their potential function in SLE pathogenesis remained unclear. • miR-199a-3p in CD4+ T cells plays a novel role in the pathogenesis of SLE and serves as a potential target for SLE. |
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Systemic lupus erythematosus (SLE) is a chronic and complex autoimmune disease, mediated by pathogenic autoantibodies, with severe organ damages such as nephritis [1]. Various factors such as genetics, environment, and immunoregulatory factors contributed to the development of SLE. As the etiology of SLE remains unclear, SLE is still associated with high morbidity and a 5-year mortality rate of more than 20% [2]. Recently, there are a growing number of studies with a focus on identification of novel biomarkers to prevent disease progress and improve clinical outcomes of SLE. Therefore, better understanding of diagnostic targets associated with SLE can play a key role in the disease diagnosis and prognostication.
Although the disease pathogenesis is still unclear, multiple studies have suggested that cytokines and immune cells including dendritic cells, neutrophils, and lymphocytes are recognized as key contributors to the pathogenesis of SLE [1, 3]. Moreover, there is an increasing validation of the essential role of CD4+ T cells in pathogenic mechanism of SLE [4,5,6]. A loss of the healthy balance of activity between effector and regulatory CD4+ T cells correlates with the development of SLE [7]. A better understanding of the molecular defects including abnormal cytokine production, aberrant cell signaling events and gene regulation, and aberrant epigenetic reprogramming underlying the dysfunctional CD4+ T cells in SLE is critical to contribute to the autoimmune disease development, better diagnosis, therapy, and perhaps prevention of SLE [8,9,10].
microRNAs (miRNAs) are a class of endogenous noncoding RNAs that regulate various inflammatory, immunologic, and oncogenic pathways by mediating protein translation [11]. Recently, it has been demonstrated that miRNAs were early promise as important molecules in the SLE pathogenesis. miRNAs are involved in lupus susceptibility, diagnoses and assessment, disease activity, and major organ damages by affecting a wide range of post-transcriptional genes and facilitating the development of lupus [12, 13]. A recent study has identified the clinical value and promising biomarkers of miR-124-3p and miR-377-3p in plasma for the diagnosis of SLE [14]. Another study found that levels of miR-21, miR-25, and miR-186 were positively correlated with SLEDAI scores and miR-146a was negatively correlated lupus disease activity in SLE patients [15], suggesting that miRNAs have translational potential and serve as novel therapeutic targets in patients with SLE. These data show a dynamic interplay between disease pathogenesis and miRNA expression.
In this study, we evaluated miRNA expression profiles in CD4+ T cells from SLE patients and HCs using next-generation sequencing (NGS). Secondly, we selected the candidate upregulated miR-199a-3p and evaluated the association of this miRNA with the clinical and laboratory characteristics and disease activity of SLE patients. Lastly, we identified the potential targets that are regulated by miRNAs.
Methods
Subjects and study design
This study included 132 participants, and their clinical data are listed in Supplementary Table 1. Four SLE patients and three healthy subjects were used in the primary small RNA profiling study, and the validation cohort consisted of 97 patients with SLE, 16 patients with rheumatoid arthritis (RA), and 12 HCs. All patients fulfilled the classification criteria for SLE [16] and RA [17], respectively, and the enrollment was completed at the Department of Rheumatology, Renji Hospital South Campus, between June 2017 and December of 2018. The SLE disease activity index (SLEDAI) [18] and the Systemic Lupus International Collaborating Clinics (SLICC) [19] were assessed, and all patients with > 500mg/24h proteinuria were defined as experiencing active lupus nephritis at the time of enrollment. SLE patients with SLEDAI score > 6 were defined as active disease activity, while SLE patients with SLEDAI score ≤ 6 were defined as stable disease activity. Among 97 enrolled SLE patients, 33 SLE patients were experiencing active disease, other patients with stable disease activity used stable doses of glucocorticoids and immunosuppressive drugs for the past 1 month. The mean stable dose of glucocorticoids patients received was 15.15 ± 13.14 mg/day, and 46 SLE patients were taking immunosuppressants at the time of the study (Supplementary Table 1). Based on the glucocorticoid dose patients receiving, SLE patients were divided into three groups (< 20 mg/day / ≥ 20 mg/day and < 60 mg/day / ≥ 60 mg/day). Among the 45 patients with nephritis, 27 (76.47%) were confirmed by renal biopsy, of 20 with class III/IV and 7 with class II/V. Any subjects with acute or chronic infections were excluded, and the study protocol was approved by the institutional ethics committee, and written informed consents were obtained from all participants.
CD4 + T cell and RNA isolation
Total RNA containing miRNA was isolated from the CD4+ T cells as previously described [10]. CD4+ T cells were separated from HCs and patients’ blood samples using Ficoll gradient centrifugation and human CD4+ T cell isolation kit (BD Biosciences) according to manufacturer’s protocols. Samples from the validation cohort were used for qRT-PCR.
NGS profiling
Total RNA was extracted from the CD4+ T cells using the TRIzol Reagent (Invitrogen), and the quality of the RNA samples was evaluated by using a Nanodrop 2000c Spectrophotometer (Thermo Fisher Scientific Inc.) and then size-fractionated on a 15% polyacrylamide gel electrophoresis (PAGE) gel to collect the 18–60nt fraction. The 5′ and 3′ RNA adapters were ligated to the RNA pool, and then this was subjected to RT-PCR to generate the sequencing libraries. PCR products were purified and then sequenced using a HiSeq 2000 sequencing system (BGI Tech). Differential expression of miRNAs was considered significant at a |fold change| > 2 and P-value < 0.05.
GO and KEGG pathway analysis
The functions of all differentially expressed miRNAs and target mRNAs were investigated using Gene Ontology (GO) annotations and Kyoto Encyclopedia of Genes and Genome (KEGG) pathway analysis. Hierarchical clustering of the differentially expressed genes (DEGs) according to the biological process (BP), cellular component (CC), and molecular function (MF) categories was performed by GO analysis to elucidate genetic regulatory networks (http://www.geneontology.org). Pathway analysis using graphical diagrams was performed to explore DEG pathways using the KEGG database (http://www.genome.jp/kegg/). Significance was determined by P-value and FDR.
Target gene prediction and bioinformatics analysis of miRNAs
Potential target genes for differentially expressed miRNAs and the candidate miRNA were predicted using the miRanda (https://www.genewiz.com.cn/), TargetScan (http://www.targetscan.org/), PicTar (http://pictar.mdc-berlin.de) software. Subsequently, the functions of potential miRNAs were investigated by GO and KEGG analysis. A pathway network was constructed for the candidate miRNA and target genes using Cytoscape software. Detailed pathway was analyzed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) database (https://david.ncifcrf.gov). The prediction of miRNA targets in 3′UTRs and coding sequence using RNAhybrid tool (http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/) [20].
qRT-PCR validation of identified miRNAs and miRNA target gene
As our previous study described [21], isolated total miRNA (1.0μg) was polyadenylated at 37°C for 20 min with poly(A) polymerase (NEB), and mRNA reverse transcription was performed using the Superscript II Reverse Transcriptase kit (Takara) as previously described [10]. All samples and experiments were performed in biological triplicate, and the data were analyzed using the comparative cycle threshold (Ct) method. U6 or GAPDH was used as an internal control, and the relative quantification was calculated using the following equation: Amount of gene = 2−ΔΔCt, where ΔCt = Ctgenes − Ct U6/GAPDH. Specific primers for genes are listed in Supplementary Table 2.
Data statistics
Statistical analysis was performed using SPSS 22.0 software. Data are presented as mean ± SD. Statistical significance between groups was determined by Student’s t test or Mann-Whitney U test. P-value < 0.05 was considered to represent statistically significant difference. Expression graphs were analyzed using GraphPad Prism version 6.0. Receiver operating characteristic (ROC) curve analysis was performed for CD4+ T cell miRNAs, and the areas under curve (AUC) were calculated using R software. The liner correlation analysis of miR-199a-3p expression with the target genes levels of patients was done.
Results
Overview of RNA-seq data in CD4 + T cells for SLE patients and HCs
At the exploratory cohort, CD4+ T cells from 4 newly diagnosed SLE patients and 3 HCs were collected for small sequencing. Overall, 1264 differentially expressed miRNAs were detected in the CD4+ T cells of SLE patients compared with HCs, including 710 upregulated miRNAs and 554 downregulated ones. Principal component analysis (PCA) results of miRNA profiles indicated that the gene expression patterns of SLE patients were different from those of HC group (Supplementary Figure 1). And the distribution of clinical SLE samples indicated the diversity and abundance of miRNAs varied. A total of 136 differentially expressed miRNAs were identified, with 70 downregulated miRNAs and 66 upregulated miRNAs in SLE patients compared with HCs (fold change > |2.0|, P < 0.05) (Fig. 1A, B and Supplementary Table 1). And upregulation of miR-199a-3p and miR-199b-3p was shared in SLE patients (Fig. 1A, B).

Differential expression of CD4+ T cell miRNAs between patients with systemic lupus erythematosus (SLE) and healthy controls (HCs) in the exploratory cohort. A Heatmap of differentially expressed miRNAs of CD4+ T cell between SLE patients (n=4) and HCs (n=3). The red indicates upregulated miRNAs, and the green indicates downregulated miRNAs. B The Volcano plot of differentially expressed miRNAs of CD4+ T cell between SLE patients and HCs. The red points indicate upregulated miRNAs (n = 66), and the blue points indicate downregulated miRNAs (n = 70). C Gene ontology (GO) analysis of differentially expressed miRNAs. The red indicates biological process (BP), the blue indicates cellular component (CC), and the blue indicates molecular function (MF). D KEGG pathway analysis of differentially expressed miRNAs. The size of the spots represents the number of target genes of differentially expressed miRNAs, and the color of the spots represents the P-value
Bioinformatics analysis of differentially expressed miRNAs in CD4 + T cells of SLE patients and HCs
To further explore the significant pathways of differentially expressed miRNA in CD4+ T cells of SLE patients, GO analysis and KEGG Pathway analysis were performed. GO analysis showed that the target genes of differentially expressed miRNA were involved in some essential biological functions. In the BP category, enriched terms included regulation of transcription, transport, and multicellular organismal development process. In the CC category, enrich terms included cytosol, nucleoplasm, and nucleolus. In the MF category, enriched terms included metal ion binding, DNA binding, and nucleotide binding (Fig. 1C). KEGG pathway analysis showed that the target genes of the differentially expressed miRNAs participated in the Janus kinase/signal transduction and activator of transcription (JAK-STAT) signaling pathway signaling pathway, MAPK signaling pathways, and hypoxia inducible factor-1 (HIF-1) signaling pathway (Fig. 1D), which are well-known important signaling pathways in the pathogenesis of SLE. The pathway analysis manifested that the most enriched pathways for these miRNA were related to inflammatory regulation pathways, especially autoimmune diseases, suggesting that aberrant miRNAs in SLE patients might be substantially involved in the pathogenesis of SLE.
Differential expression of miR-199a-3p in CD4 + T cells of SLE
Consider that miR-199a-3p and miR-199b-3p were also identified upregulation in the CD4+ T cells of lupus patients, which is consistent with upregulation of miR-199-3p in PBMCs of SLE patients in previous study [22, 23]. Recently, it has been reported that miR-199a-3p modulated the function of dendritic cells and might be a potential therapeutic strategy for patients with autoimmune diseases [24]. This feature suggested that miR-199a-3p and miR-199b-3p play a key role in autoimmune disease. Therefore, we selected miR-199a-3p for the validation in the CD4+ T cell samples by qRT-PCR. We found that the expression of miR-199a-3p in the validation cohort showed significantly upregulated in CD4+ T cells of SLE in comparison with that in HCs and RA patients (Fig. 2A), showing the same change on the miRNA sequencing.

Comparison of relative expression levels of miR-199a-3p in CD4+ T cells of patients with systemic lupus erythematosus (SLE). A Expression of miR-199a-3p in CD4+ T cells of patients with SLE (n=97), patients with rheumatoid arthritis (RA) (n=16), and HCs (n=12). Data are presented as 2−ΔΔCt relative to U6 expression.*, P<0.05; **, P<0.01; ****, P<0.0001. B Expression of miR-199a-3p in CD4+ T cells between SLE patients with negative and positive anti-dsDNA antibody. C Expression of miR-199a-3p between SLE patients with active lupus nephritis (LN) or non-LN. D Expression of miR-199a-3p between stable SLE patients and active SLE patients
The correlation between miR-199a-3p expression in CD4 + T cells and SLE clinical characteristics
Furthermore, we investigated the correlation between the expression of miR-199a-3p and laboratory and clinical characteristics of 97 SLE patients in the validation cohort (Table 1). Interestingly, the results demonstrated that upregulated miR-199a-3p expression was significantly correlated in SLE patients with positive dsDNA (P = 0.001, Fig. 2B), high TNF-α level (P < 0.0001), high ESR level (P = 0.008), low hemoglobin level (P < 0.05), high triglyceride level (P < 0.05), and positive urine leucocyte (P = 0.04) (Table 1). Moreover, we found that miR-199a-3p expression was higher in patients with active lupus nephritis than those non-LN patients (P < 0.05, Fig. 2C). However, no statistically significant differences were detected in miR-199a-3p expression levels and renal function (serum creatinine, albumin, and 24-h urine protein), class of renal biopsy, or other clinical manifestations (Table 1 and Supplementary Table 4). To further investigate different levels of disease severity, SLE patients were divided into the stable group (SLEDAI score ≤ 6) and active group (SLEDAI score > 6). The results showed that compared with stable SLE patients, the active SLE patients had significantly higher expression level of miR-199a-3p based on both the SLEDAI score (P<0.01, Fig. 2D).
Furthermore, we investigated the relationship between treatments (doses of glucocorticoids and immunosuppressives) and the levels of miR-199a-3p. We found that the expression level of miR-199a-3p showed no significant difference among different doses of glucocorticoids groups and types of immunosuppressives treatments (Supplementary Figure 2). These results further suggest that miR-199a-3p has a strong association with SLE.
The potential clinical value of miR-199a-3p expression in CD4 + T cells in SLE
ROC curve analysis was used to further detect the potential utility of miR-199a-3p as a diagnostic biomarker of SLE. When distinguishing SLE patients from healthy persons and RA patients, AUCs for miR-199a-3p were 97.96% and 89.86% in the validation cohort, respectively (Fig. 3A and B). When distinguishing SLE patients with non-LN from SLE patients with LN, AUCs for miR-199a-3p were 67.60% (Fig. 3C). When distinguishing stable SLE patients from active SLE patients, AUCs for miR-199a-3p were 70.22% (Fig. 3D). These results indicated that miR-199a-3p might have good clinical value for SLE and active SLE.

The potential diagnostic value of miR-199a-3p expression in CD4+ T cells of SLE. A, B Receiver-operating characteristic (ROC) curves of miR-199a-3p, showing the ability to distinguish SLE patients from healthy controls (A) and RA patients (B). C ROC curves of miR-199a-3p, showing the ability to distinguish active SLE patients from stable SLE patients. D ROC curves of miR-199a-3p, showing the ability to distinguish LN from the non-LN group
Predicted functions and pathways and validation of miR-199a-3p in CD4 + T cells
In order to investigate the potential function of miR-199a-3p in CD4+ T cells in SLE, we predicted the target genes of miR-199a-3p and putative function using miRanda software, GO analysis, and KEGG pathway analysis (Supplementary Table 5). GO analysis showed that the target genes of miR-199a-3p were enriched in regulation of transcription, positive regulation of MAPK cascade, positive regulation of NF-kB signaling (BP category), nucleus, nucleoplasm, cytoplasm (CC category) and metal ion binding, DNA binding, and protein binding (MF category) (Fig. 4A), which is highly consistent with those of the target genes of differentially expressed miRNAs. Pathway analysis showed that the target genes of miR-199a-3p were involved in JAK-STAT signaling pathway and glyoxylate and dicarboxylate metabolism (Fig. 4B). Intriguingly, both the target genes of miR-199a-3p and the target genes of differentially expressed miRNAs participate in JAK-STAT signaling pathway, which is an essential signaling pathway in the pathogenesis of SLE [25, 26]. As shown in Fig. 4C, the JAK-STAT signaling was visualized using DAVID database. After applying the criteria of binding energy < −20 kcal/mol, strict matching of score >168 of the miRNA region with the 3′UTR region of the target gene, a co-expression network of miR-199a-3p and target genes was built (Fig. 4D). More importantly, we found that signal transducing adapter molecule (STAM), the target gene of miR-199a-3p, is a key gene in the JAK-STAT signaling. Furthermore, the STAM expression level is negatively correlated with the level of miR-199a-3p (Fig. 5A). We also found one perfect match seed between miR-199a-3p and STAM (Fig. 5B). The binding energy of miR-199a-3p with STAM mRNA was −25.0 kcal/mol. These suggest that the upregulated miR-199a-3p might target in the SLE to regulate inflammation.

The pathway analysis of the target genes of miR-199a-3p. A Gene ontology (GO) analysis of the target genes of miR-199a-3p. B KEGG pathway analysis of the target genes of miR-199a-3p. C The JAK-STAT signaling pathway analysis by the database for annotation, visualization, and integrated discovery (DAVID) tool. D The network map for miR-199a-3p-target gene network prediction. The blue nodes around the red node were the predicted target genes that interacted with miR-199a-3p. The yellow nodes mean the key predicted target genes

miR-199a-3p targets STAM. A The relationship between target gene STAM expression and miR-199a-3p expression. B Predicted target sites in the STAM 3′UTR based on the miR-199a-3p seed region
Discussion
In the present study, we performed the microRNA expression profiling in CD4+ T cells of SLE patients compared with healthy individuals using NSG. We have identified 136 miRNAs with significantly differential expression and validated miR-199a-3p expression with significant upregulation in CD4+ T cells of SLE patients compared with HC group. miR-199a-3p was related to SLE disease activity and could be a potential diagnostic biomarker. Moreover, miR-199a-3p expression displayed a tendency of negative correlation with STAM expression, involved in the JAK-STAT signaling pathway in CD4+ T cells of SLE patients.
Over the past years, a great number of studies have addressed the significance of the dysregulated T-cell response as a trigger in SLE [26]. CD4+ T cells are a subset of T lymphocytes involving in the immune and inflammation. However, they are well-known for their roles in the pathogenesis of autoimmune disorders like SLE [6]. Currently, miRNAs have been demonstrated to contribute to the pathogenesis of lupus by affecting immune responses and the production of proinflammatory cytokines [27]. Further, miRNAs expression alteration in CD4+ T cells during SLE leads to a list of transcriptional dysregulations in several genes [9]. High-expressed miR-152-3p in CD4+ T cells of SLE patients was associated with facial erythema, joint pain, dsDNA, and IgG antibody and promoted inflammatory response by regulating the DNMT1/MyD88 signaling pathway [28]. miRNA-26a in CD4+ T cells has been found to decrease with a glucose limitation by targeting enhancer of a DNA methylated regulator and correlate with lupus disease activity [29]. And we found the differentially expressed miRNAs participated in several key immune responses and SLE-related processes, including JAK-STAT signaling pathway, MAPK signaling pathways, and HIF-1 signaling pathway. Specially, the JAK-STAT signaling pathway is implicated in the pathogenesis of inflammation and various autoimmune disorders including SLE, RA, psoriasis, and dermatomyositis [30, 31]. Also, we found that upregulation expression of miR-199a-3p in lupus CD4+ T cells may relate to JAK-STAT signaling pathway in SLE.
Latest study found that levels of miR21, miR25, miR186, and miR146a in PBMCs of SLE patients correlated with SLEDAI in SLE patients [14]. It also has demonstrated that the combination of plasma miRNA-124-3p and miRNA-377-3p displayed high clinical application for diagnosis [15]. Different from recent studies that compared patients with SLE with healthy control subjects in plasma or PBMCs [14, 15], we studied a specific T-cell subset, CD4+ T cells, from three groups including patients with different autoimmune disease. In our study, miR-199a-3p expression in CD4+ T cell validation in a larger population demonstrates that miR-199a-3p may play an essential part in SLE. The increase in miR-199a-3p was related to positive dsDNA, high ESR level, and high TNF-α level, which revealed that miR-199a-3p was associated with immune responses and inflammatory processes in SLE. Likewise, high miR-199a-3p expressions were observed to correlate with renal damage and SLE disease activity in a significant manner. And high miR-199a-3p was relevant to high triglyceride level, indicating that miR-199a-3p may participate in lipid metabolism in SLE. It is well documented that dysregulated lipid metabolism in T cells impacts the pathogenesis of autoimmune diseases [32]. Additionally, ROC curve results demonstrated that miR-199a-3p may have good clinical value for distinguishing SLE patients from healthy individuals and RA patients. The diagnostic efficiency for miR-199a-3p in CD4+ T cells was comparable with miRNA-124-3p and miRNA-377-3p in plasma samples of SLE [14]. Furthermore, miR-199a-3p also has clinical value for active SLE and LN patients. And the relative expression levels of miR-124-3p and miR-377-3p in both plasma and PBMCs had no difference in treated and untreated SLE patients [14]. We also did not find the strong relationship of the miR-199a-3p with the treatments that patients received, which implied that miR-199a-3p may constitute readily detectable biomarkers in SLE patients. Similarly, Su X et al. reported upregulation of miR-199-3p in PBMCs of SLE patients and confirmed the correlation between miR-199-3p and IL-10 production [22]. These findings suggested the potential role of miR-199a-3p as biomarker and may be involved in the SLE pathology.
In this study, we found that miR-199a-3p in lupus CD4+ T cells was negatively related to STAM, involved in the JAK-STAT signaling pathway. STAM acts downstream of JAK, once induced by IL-2 or other cytokines, in human hematopoietic cells [33]. Many important biological effects are mediated by JAK-STAT signaling in which alternative second effectors have been assumed [34], such as STAM. Actually, the role of certain miRNAs in the regulation of immune responses and development of immune cells is becoming more evident [10]. Various studies on miR-199a show that this miRNA is involved in multiple different autoimmune diseases and cancers. Elevation of miR-199a expression has been shown in PBMCs of SLE and LN [23, 35]. Inhibition of miR-199a expression was found to significantly downregulate the secretion of TNF-α and IL-1β in LN [23], and upregulated miR-199-3p was found to promote IL-10 production [22]. Ghadiri et al. found upregulated miR-199a in CD4+ T cells from multiple sclerosis patients compared with HCs [36]. The miR-199 family is located on chromosomes 19 and 1 [37]. miR-199a/214 cluster genes are encoded by the Dynamin 3 opposite strand (DNM3os). Our results suggest a novel role and potential therapeutic targets of miR-199a-3p for SLE.
There are still several limitations in the present study. First, this study with a relatively small sample size is not enough to support that miR-199a-3p is a promising biomarker. Second, this study has lack of function studies for whether STAM is a direct downstream target of miR-199a-3p in SLE. Third, it is unclear whether miR-199a-3p is specific for CD4+ T cells in SLE. We did not examine the miR-199a-3p expression in other immune cells, for example, B cells, CD8+ T cells, and monocytes. Another important limitation of the study is the absence of essential pro- and anti-inflammatory cytokines in stages of remission or activity of SLE.
Our study provided a CD4+ T cell miRNA profiling and a correlation between miRNAs and clinical characteristics in SLE. The significant correlation between the overexpressed miR-199a-3p and SLE disease activity further shows that miR-199a-3p plays an important role in the pathogenesis of SLE and could act as a potential target. miR-199a-3p might be related to the STAM and JAK-STAT signaling pathway in CD4+ T cells of SLE. Further studies are needed to investigate miR-199a-3p in a larger SLE cohort and the functional mechanisms of miR-199a-3p in SLE.
Data availability
All data generated or analyzed in this study are included in this article and its Supplementary materials.
References
Tsokos GC (2011) Systemic lupus erythematosus. N Engl J Med 365(22):2110–2121. https://doi.org/10.1056/NEJMra1100359
Doria A, Gatto M, Zen M, Iaccarino L, Punzi L (2014) Optimizing outcome in SLE: treating-to-target and definition of treatment goals. Autoimmun Rev 13(7):770–777. https://doi.org/10.1016/j.autrev.2014.01.055
Moulton VR, Suarez-Fueyo A, Meidan E, Li H, Mizui M, Tsokos GC (2017) Pathogenesis of human systemic lupus erythematosus: a cellular perspective. Trends Mol Med 23(7):615–635. https://doi.org/10.1016/j.molmed.2017.05.006
Morel L (2017) Immunometabolism in systemic lupus erythematosus. Nat Rev Rheumatol 13(5):280–290. https://doi.org/10.1038/nrrheum.2017.43
Teng X, Brown J, Choi SC, Li W, Morel L (2020) Metabolic determinants of lupus pathogenesis. Immunol Rev 295(1):167–186. https://doi.org/10.1111/imr.12847
Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, Sobel ES, Brusko TM, Morel L (2015) Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med 7(274):274ra218. https://doi.org/10.1126/scitranslmed.aaa0835
Berretta F, St-Pierre J, Piccirillo CA, Stevenson MM (2011) IL-2 contributes to maintaining a balance between CD4+Foxp3+ regulatory T cells and effector CD4+ T cells required for immune control of blood-stage malaria infection. J Immunol 186(8):4862–4871. https://doi.org/10.4049/jimmunol.1003777
Moulton VR, Tsokos GC (2015) T cell signaling abnormalities contribute to aberrant immune cell function and autoimmunity. J Clin Invest 125(6):2220–2227. https://doi.org/10.1172/jci78087
Wang Z, Chang C, Lu Q (2017) Epigenetics of CD4+ T cells in autoimmune diseases. Curr Opin Rheumatol 29(4):361–368. https://doi.org/10.1097/bor.0000000000000393
Geng G, Wang H, Xin W, Liu Z, Chen J, Danting Z, Han F, Ye S (2021) tRNA derived fragment (tRF)-3009 participates in modulation of IFN-α-induced CD4(+) T cell oxidative phosphorylation in lupus patients. J Transl Med 19(1):305. https://doi.org/10.1186/s12967-021-02967-3
Schetter AJ, Heegaard NH, Harris CC (2010) Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis 31(1):37–49. https://doi.org/10.1093/carcin/bgp272
Stypinska B, Paradowska-Gorycka A (2015) Cytokines and MicroRNAs as candidate biomarkers for systemic lupus erythematosus. Int J Mole Sci 16(10):24194–24218. https://doi.org/10.3390/ijms161024194
Carlsen AL, Schetter AJ, Nielsen CT, Lood C, Knudsen S, Voss A, Harris CC, Hellmark T, Segelmark M, Jacobsen S, Bengtsson AA, Heegaard NH (2013) Circulating microRNA expression profiles associated with systemic lupus erythematosus. Arthritis Rheum 65(5):1324–1334. https://doi.org/10.1002/art.37890
Yan L, Jiang L, Wang B, Hu Q, Deng S, Huang J, Sun X, Zhang Y, Feng L, Chen W (2022) Novel microRNA biomarkers of systemic lupus erythematosus in plasma: miR-124-3p and miR-377-3p. Clin Biochem 107:55–61. https://doi.org/10.1016/j.clinbiochem.2022.05.004
Singh RP, Hahn BH, Bischoff DS (2022) Identification and contribution of inflammation-induced novel MicroRNA in the pathogenesis of systemic lupus erythematosus. Front Immunol 13:848149. https://doi.org/10.3389/fimmu.2022.848149
Hochberg MC (1997) Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40(9):1725. https://doi.org/10.1002/art.1780400928
van der Heijde D, Aletaha D, Carmona L, Edwards CJ, Kvien TK, Kouloumas M, Machado P, Oliver S, de Wit M, Dougados M (2015) 2014 Update of the EULAR standardised operating procedures for EULAR-endorsed recommendations. Ann Rheum Dis 74(1):8–13. https://doi.org/10.1136/annrheumdis-2014-206350
Gladman DD, Ibañez D, Urowitz MB (2002) Systemic lupus erythematosus disease activity index 2000. J Rheumatol 29(2):288–291
Petri M, Orbai AM, Alarcón GS, Gordon C, Merrill JT, Fortin PR, Bruce IN, Isenberg D, Wallace DJ, Nived O, Sturfelt G, Ramsey-Goldman R, Bae SC, Hanly JG, Sánchez-Guerrero J, Clarke A, Aranow C, Manzi S, Urowitz M, Gladman D, Kalunian K, Costner M, Werth VP, Zoma A, Bernatsky S, Ruiz-Irastorza G, Khamashta MA, Jacobsen S, Buyon JP, Maddison P, Dooley MA, van Vollenhoven RF, Ginzler E, Stoll T, Peschken C, Jorizzo JL, Callen JP, Lim SS, Fessler BJ, Inanc M, Kamen DL, Rahman A, Steinsson K, Franks AG Jr, Sigler L, Hameed S, Fang H, Pham N, Brey R, Weisman MH, McGwin G Jr, Magder LS (2012) Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 64(8):2677–2686. https://doi.org/10.1002/art.34473
Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R (2004) Fast and effective prediction of microRNA/target duplexes. Rna 10(10):1507–1517. https://doi.org/10.1261/rna.5248604
Guo G, Wang H, Shi X, Ye L, Wu K, Lin K, Ye S, Li B, Zhang H, Lin Q, Ye S, Xue X, Chen C (2018) NovelmiRNA-25 inhibits AMPD2 in peripheral blood mononuclear cells of patients with systemic lupus erythematosus and represents a promising novel biomarker. J Transl Med 16(1):370. https://doi.org/10.1186/s12967-018-1739-5
Su X, Ye L, Chen X, Zhang H, Zhou Y, Ding X, Chen D, Lin Q, Chen C (2019) MiR-199-3p promotes ERK-mediated IL-10 production by targeting poly (ADP-ribose) Polymerase-1 in patients with systemic lupus erythematosus. Chem Biol Interact 306:110–116. https://doi.org/10.1016/j.cbi.2019.04.015
Ye H, Su B, Ni H, Li L, Chen X, You X, Zhang H (2018) microRNA-199a may be involved in the pathogenesis of lupus nephritis via modulating the activation of NF-κB by targeting Klotho. Mol Immunol 103:235–242. https://doi.org/10.1016/j.molimm.2018.10.003
Xiong A, Wang J, Mao XL, Jiang Y, Fan Y (2019) MiR-199a-3p modulates the function of dendritic cells involved in transplantation tolerance by targeting CD86. Hla 94(6):493–503. https://doi.org/10.1111/tan.13677
Alunno A, Padjen I, Fanouriakis A, Boumpas DT (2019) Pathogenic and therapeutic relevance of JAK/STAT signaling in systemic lupus erythematosus: integration of distinct inflammatory pathways and the prospect of their inhibition with an oral agent. Cells 8(8). https://doi.org/10.3390/cells8080898
Dang WZ, Li H, Jiang B, Nandakumar KS, Liu KF, Liu LX, Yu XC, Tan HJ, Zhou C (2019) Therapeutic effects of artesunate on lupus-prone MRL/lpr mice are dependent on T follicular helper cell differentiation and activation of JAK2-STAT3 signaling pathway. Phytomedicine 62:152965. https://doi.org/10.1016/j.phymed.2019.152965
Zan H, Tat C, Casali P (2014) MicroRNAs in lupus. Autoimmunity 47(4):272–285. https://doi.org/10.3109/08916934.2014.915955
Tao B, Xiang W, Li X, He C, Chen L, Xia X, Peng T, Peng L, Yang X, Zhong C (2021) Regulation of Toll-like receptor-mediated inflammatory response by microRNA-152-3p-mediated demethylation of MyD88 in systemic lupus erythematosus. Inflamm Res 70(3):285–296. https://doi.org/10.1007/s00011-020-01433-y
Karantanos T, Christofides A, Bardhan K, Li L, Boussiotis VA (2016) Corrigendum: regulation of T cell differentiation and function by EZH2. Front Immunol 7:346. https://doi.org/10.3389/fimmu.2016.00346
You H, Xu D, Zhao J, Li J, Wang Q, Tian X, Li M, Zeng X (2020) JAK inhibitors: prospects in connective tissue diseases. Clin Rev Allergy Immunol 59(3):334–351. https://doi.org/10.1007/s12016-020-08786-6
Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM (2017) JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs 77(5):521–546. https://doi.org/10.1007/s40265-017-0701-9
Ryu H, Kim J, Kim D, Lee JE, Chung Y (2019) Cellular and molecular links between autoimmunity and lipid metabolism. Mol Cells 42(11):747–754. https://doi.org/10.14348/molcells.2019.0196
Mesilaty-Gross S, Reich A, Motro B, Wides R (1999) The Drosophila STAM gene homolog is in a tight gene cluster, and its expression correlates to that of the adjacent gene ial. Gene 231(1–2):173–186. https://doi.org/10.1016/s0378-1119(99)00053-0
Watanabe S, Itoh T, Arai K (1996) JAK2 is essential for activation of c-fos and c-myc promoters and cell proliferation through the human granulocyte-macrophage colony-stimulating factor receptor in BA/F3 cells. J Biol Chem 271(21):12681–12686. https://doi.org/10.1074/jbc.271.21.12681
Dai Y, Huang YS, Tang M, Lv TY, Hu CX, Tan YH, Xu ZM, Yin YB (2007) Microarray analysis of microRNA expression in peripheral blood cells of systemic lupus erythematosus patients. Lupus 16(12):939–946. https://doi.org/10.1177/0961203307084158
Ghadiri N, Emamnia N, Ganjalikhani-Hakemi M, Ghaedi K, Etemadifar M, Salehi M, Shirzad H, Nasr-Esfahani MH (2018) Analysis of the expression of mir-34a, mir-199a, mir-30c and mir-19a in peripheral blood CD4+T lymphocytes of relapsing-remitting multiple sclerosis patients. Gene 659:109–117. https://doi.org/10.1016/j.gene.2018.03.035
Chen R, Alvero AB, Silasi DA, Kelly MG, Fest S, Visintin I, Leiser A, Schwartz PE, Rutherford T, Mor G (2008) Regulation of IKKbeta by miR-199a affects NF-kappaB activity in ovarian cancer cells. Oncogene 27(34):4712–4723. https://doi.org/10.1038/onc.2008.112
Acknowledgements
We thank all patients and healthy donors who participated in this study.
Funding
This research was supported by the Clinical Research Plan of SHDC (SHDC2020CR1015B) to YS and Cultivating Funds of Renji Hospital South Campus, Shanghai Jiaotong University School of Medicine (2016PWGZR05) to GG.
Author information
Authors and Affiliations
Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Ye had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. WH, YS.
Acquisition of data. WH, GG, ZD, HF.
Analysis and interpretation of data. WH, GG, YS.
Corresponding author
Ethics declarations
Disclosures
None.
Ethics approval and consent to participate
The research protocol was approved by the Renji Hospital of Shanghai Jiaotong University School of Medicine. All subjects who participated in this study provided written informed consent.
Consent for publication
Not applicable.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, H., Geng, G., Zhang, D. et al. Analysis of microRNA-199a-3p expression in CD4+ T cells of systemic lupus erythematosus. Clin Rheumatol 42, 1683–1694 (2023). https://doi.org/10.1007/s10067-023-06534-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-023-06534-7