
Abstract
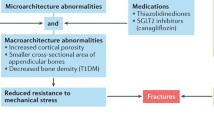
Diabetes increases the likelihood of suffering a fracture, and in the case of type 2 diabetes mellitus (T2D), low bone mass does not explain this loss in fracture resistance. Insulin contributes to the accrual of bone mass. As such, the elevated fracture risk among those with type 1 diabetes (T1D) could be due to a deficit in bone structure, especially if the diabetes is poorly controlled. Clinical studies involving computed tomography scans do suggest that low moment of inertia and low cross-sectional area of cortical bone accompany T1D. However, low bone mass does not typically accompany T2D, and fracture resistance arises from all the hierarchical levels comprising the organization of bone’s constituents. One consequence of diabetes, hyperglycemia, causes an increase in non-enzymatic collagen crosslinks, also known as advanced glycation end products (AGEs). Increases in AGEs within the bone matrix are strongly correlated with the age-related decrease in the ability of bone to dissipate energy during failure (toughness and fracture toughness). While elevated AGEs are associated with higher incidence of fracture, the mechanism by which non-enzymatic crosslinking lowers fracture resistance is not fully delineated. The general concept is that increases in AGEs within the matrix cause the collagen phase to become brittle, reducing the capacity of the tissue to resist the formation and propagation of microdamage. While certainly more research into the diabetic effects on fracture resistance is necessary before clear therapeutic options are identified, diabetes should be considered a problem of bone brittleness, not just one of low bone strength.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Both type 1 diabetes (T1D) and type 2 diabetes (T2D) increase the risk of experiencing a bone fracture [1–5]. In general, bone fractures are a widespread and costly problem [6, 7]. The expectation is that these costs will increase as the aging population grows [8] and the prevalence of diabetes increases [9], especially since diabetes impairs bone regeneration [10] and is associated with costly complications (e.g., non-unions). In addition to medical costs, fractures deleteriously impact the quality of life. For example, a third of Veterans treated for a hip fracture did not live beyond 1 year: an adjusted mortality rate of 29.8% [11, 12]. The cause for the disproportionate increase in fracture risk among diabetics is not well understood, but herein is a summary of what is currently known about the origins of fracture resistance and how diabetes potentially lowers fracture resistance.
Measured by the primary assessment tool of osteoporosis, dual energy X-ray absorptiometry (DXA) at the hip, areal bone mineral density (aBMD) was found to be lower in subjects with T1D than in age-matched control subjects [13], and in statistical models accounting for gender, race, menopausal status, and disease duration, low aBMD was associated with poor glycemic control [14]. Since aBMD is directly proportional to bone strength as determined from whole bone tests of cadaveric tissue (hip [15], radius [16], and spine [17]), this low aBMD could explain the higher risk of fracture among individuals with diabetes. On the other hand, T2D patients do not necessarily have low aBMD [18, 19], and in some reports, aBMD was actually higher in diabetics [20–22]. One possible cause then for the elevated fracture risk is the greater propensity for diabetics to fall compared to non-diabetics [23, 24]. Yet, in the clinical studies that accounted for history of falls and related risk factors (e.g., impaired vision), the association between T2D and increased fracture risk persisted [25–27]. Moreover, a meta-analysis of clinical studies concluded that people with T2D are at a higher-than-expected risk of a hip fracture than those who did not have the disease [4].
With regard to non-diabetics, the probability of suffering a fracture increases with age, and this increase is independent of the age-related decrease in aBMD [28, 29]. Why this occurs is presently unclear, though the limitations of DXA (e.g., projection method and insensitive to collagen) certainly play a role. To improve the ability of aBMD to predict fracture risk, an online risk assessment tool called FRAX was developed by the World Heath Organization. Based on certain risk factors and aBMD, it calculates the 10-year probability of suffering a major fracture and the 10-year probability of a hip fracture. Despite some improvement in predicting osteoporotic fractures with FRAX [30], T2D patients still have a higher fracture risk than individuals without diabetes for a given FRAX score [31]. In other words, diabetes likely increases fracture risk independent of the risk factors such as aBMD and age, which contribute to the FRAX scores [32]. Taken together, these observations point to the strong likelihood that diabetes affects bone in ways beyond changes in bone mass or density.
Hierarchical Organization of Bone
Bone of course has many interesting features that facilitate its function of sustaining loads. The primary constituents—type 1 collagen, mineral, and water—are organized in a hierarchical fashion with the resistance to fracture arising from each of the length scales (Fig. 1 and described by others [33–35]). At the level of the whole bone (macro-structure), resistance to bending by the femoral neck is dictated by the distribution of tissue about the centroid of the neck region. The quantification of this distribution is called the moment of inertia (aka second moment of area) in which periosteal perimeter has a greater contribution than endosteal perimeter to stiffness and strength [36]. Moving to a smaller length scale (microstructure), intra-cortical porosity or apparent bone density primarily determines the material strength of bone [37]. Material strength is independent of bone size or structure and is typically measured by destructive tests on machined, uniform specimens of bone (40 mm × 4 mm × 2 mm). In the case of the apparent strength of trabecular bone (typically 8 mm diameter cores), the volume of bone within the given total volume under loading (bone volume fraction or BV/TV) is the primary determinant with contributions from the architecture and morphology of the trabeculae [38, 39]. In particular, more plate-like trabeculae aligned with the axis of loading confer greater strength than more rod-like trabeculae aligned orthogonal to loading [40, 41].

The hierarchical arrangement of bone. Each length scale from mineralized collagen fibrils to the structure of the femoral neck confers resistance to fracture
The origins of strength also exist at the tissue level (nano-structure to ultra-structure). With respect to the mineral phase, increases in the degree of mineralization through secondary mineralization are thought to be one-way bisphosphonate therapy, which lowers remodeling activity, increases fracture resistance [42]. Direct correlations between crystallinity and material strength of human bone have been reported [43], but mineral is not necessarily the sole determinant of whether bone can sustain high stress (>125 MPa). Collagen also can influence strength of cortical bone. For example, collagen fibrils oriented in the longitudinal direction tend to confer greater axial strength than those oriented in the transverse direction [44]. Also, disruption of enzymatic collagen crosslinking can lower bone strength independent of any effects on mineral density [45].
An Engineering Perspective of Bone’s Resistance to Fracture
With multiple length scales contributing to fracture resistance, diabetes conceivably increases fracture risk in numerous ways from possibly disrupting cortical microstructure to altering the organic matrix. Complicating matters is that a decrease in bone strength is likely not the only effect of diabetes on fracture resistance. From an engineering perspective, material failure during service often involves fatigue-generated microcracks and the propagation of such cracks or flaws to a critical size upon which fracture occurs. As such, in addition to monotonic load-to-failure tests that measure modulus (linear slope of stress vs. strain curve), strength (peak stress), and toughness (area under the stress vs. strain curve) [36], there are other mechanical tests to characterize the ability of a material or bone to resist fracture such as fatigue (e.g., number of loading cycles to failure) and fracture toughness testing (e.g., resistance to crack propagation). Daily loads on the skeleton generate microdamage in bone with a stress fracture being the clinical manifestation of a fatigue-induced fracture [46, 47]. These fatigue fractures are one possible cause for the bone destruction that occurs with Charcot arthropathy, a common problem among diabetics [48, 49].
Unlike engineering materials, bone in vivo does have a repair mechanism for fatigue microdamage since bone remodeling can be activated to remove microcracks [50] and resorption sites are preferentially associated with microcracks [51]. Nonetheless, microdamage in bone tends to increase with age [52–54] and is inversely related to bone toughness [55]. Thus, how well the bone matrix can resist crack propagation is important to whether bone breaks, and moreover, this resistance is a function of osteonal density, porosity, compositional heterogeneity, collagen integrity, and other factors existing at each hierarchical level of bone’s organization [56–59]. At present, there is dearth of information on how diabetes affects many of these factors that influence microdamage initiation and resistance and ultimately the fracture resistance of bone.
Traditionally, osteoporosis is viewed as a problem of low bone mass causing reduced bone strength, and to some extent, elevated fracture risk with diabetes has been interpreted as an osteoporosis problem [60, 61]. Nonetheless, studies involving the mechanical testing of cadaveric bone have consistently found that aging affects the ability of bone to dissipate energy during failure (bone toughness) to a greater extent than it does the material strength of bone (Fig. 2) [62–65]. Basically, an older bone does not stretch (strain measured in change of length per original length) as far as a younger bone (Fig. 3), and this is related to an inability of the tissue to handle damage that forms during fracture. There are several engineering methods for quantifying the ability of bone to resist crack propagation (damage); and whether measuring the critical stress intensity factor [66] or the strain energy release rate [56], bone’s resistance to crack propagation (i.e., its fracture toughness) decreases with an increase in age. Moreover, the ability of bone tissue to demand greater energy to propagate a crack as the crack grows in length (i.e., R-curve behavior) is lost or reduced with aging [67, 68].

Mechanical properties as function of donor age. Whether measured using tensile tests (a) [61] or bending tests, the toughness of cortical bone decreases with aging to a greater extent the material strength (mean ± SD)

Aging differences in bone deformation. Aging reduces by half the degree of failure strain (left) and permanent strain (right) that bone can endure (mean ± SD)
Fatigue testing involves subjecting the specimen to repeated or cyclic loads that eventually cause failure, despite the service loads being much lower than the yield force of the material. Such loading of human tissue samples has found that aging affects the fatigue life of cortical bone in bending (compression and tension modes) [69, 70] as well as the fatigue strength in the shear mode [71]. The early studies on the fatigue properties of bone examined variables such as frequency of the applied cyclical load, specimen geometry, loading mode, and species [72], but certain determinants were identified. For example, Haversian porosity was inversely related to fatigue life [73]. The expectation is that diabetes like aging would affect the fatigue life of human cortical bone, but this is currently an untested hypothesis.
Possible Effect of Diabetes on Mechanical Properties of Bone
Since access to bone of sufficient size from humans with diabetes is extremely limited, little is still known about the full effect of diabetes on many of the mechanical properties of human bone. In two studies investigating bone from humans with diabetes, mechanical properties of bone were not different between middle-aged diabetics and elderly non-diabetics [74, 75]. Specifically, the modulus and strength of the metatarsals—acquired from foot amputations or an allograft bank—were not different between non-diabetic (72.3 ± 10 years old) and diabetic (51.3 ± 8 years old) donors as assessed by three-point bending [75]. In the subsequent study, Fleischli and co-workers tested machined, beam specimens from the tibia with the diabetic donors being between 46 and 61 years old and the non-diabetic donors being between 67 and 85 years old. There were no differences in material strength and fracture toughness suggesting that the effect of diabetes on the fracture resistance of human is akin to accelerated aging, but without age-match controls, this is only a supposition. In one other study investigating the mechanical behavior of cadaveric cortical bone from donors of varying age including one male, 74 years old donor with diabetes, the crack growth toughness was 14% less and crack initiation toughness was 40% less for the diabetic bone than for healthy young bone samples [68]. These percent differences were slightly greater (i.e., worse) than the percent differences between young and aged healthy bone. Given that an age-related decline in collagen integrity is associated with decreasing fracture resistance, diabetes could be a problem of bone brittleness, not just a loss of bone strength.
Clinical Observations of Diabetic Effects on Bone Structure
There is evidence that individuals lacking the ability to properly generate insulin may develop smaller bones. Using peripheral quantitative computed tomography (pQCT), Saha et al. [76] found that the radius and tibia of T1D adolescents, especially boys, had a smaller cross-sectional area (CSA) than did these bones of appropriately matched non-diabetic adolescents. A smaller CSA translates to weaker bones assuming no difference in inherent tissue quality. Another pQCT study by Bechtold et al. [77] indicated that as T1D adolescents reached 14 and 15 years of age, their cortical CSA normalized, becoming equivalent to the cortical CSA of non-diabetics at the same age.
Structural deficits in bone are not necessarily apparent in individuals with T2D. In one study, pQCT-derived cortical thickness (Ct.Th) and CSA were similar between T2D subjects and non-diabetic controls [78], but in another study with a larger cohort, cortical CSA was smaller in the T2D subjects, albeit this was offset by a higher volumetric BMD, than for the controls [79]. Assessing trabecular bone of the spine, a QCT study reported that differences in volumetric BMD between T2D and non-diabetic women and men were not independent of differences in body mass index [80]. As for postmenopausal women, those with T2D did not develop bones with a markedly different structure compared to non-diabetics other than a smaller cortical area at the distal tibia as measured by high-resolution (HR) pQCT [81]. For another relatively small cohort of elderly women in which bones were assessed by HR-pQCT, Burghardt et al. [82] observed greater cortical porosity in the distal radius of T2D subjects compared to bones of otherwise healthy women. In all likelihood, poorly controlled diabetes can increase fracture risk of T1D patients through structural changes and affect T2D patients through reductions in the ratio of structural strength to body weight, which is typically elevated with diabetes.
Potential Effects of Diabetes on Bone Tissue
At the nano-length scale, non-enzymatic collagen crosslinks or advanced glycation end products (AGEs) have been implicated in the diabetes-related decrease in fracture resistance. As animals age, AGEs accumulate in a variety of connective tissues [83, 84]—including bone [85–87]—even though bone undergoes turnover throughout life. The formation of these crosslinks involves the Maillard reaction with sugars resulting in less soluble collagen [88]. As such, hyperglycemia promotes glycation-mediated crosslinking in the tissues of individuals with poorly controlled diabetes. Such excessive crosslinking of the organic matrix could potentially increase fracture risk [89]. In a prospective study of 432 elderly Japanese women, urinary pentosidine (a biomarker for AGEs) was a significant predictor of vertebral fracture in addition to traditional risk factors such as aging and areal BMD [90]. In a French cohort of 396 postmenopausal women, fracture risk was higher for women with high urinary pentosidine, but this biomarker was not independent of the other osteoporosis risk factors [91]. Moreover, two different research groups report that a high level of pentosidine in urine or serum was associated with greater fracture risk in patients with T2D [92, 93]. Lastly, bone taken from hip fracture patients was found to have higher concentration of pentosidine in the tissue than bone of age-matched, postmortem controls [94].
Given these clinical observations, the question arises of whether AGEs mechanistically reduce bone’s resistance to fracture. Mechanical tests of cadaveric tissue consistently find that the age-related decrease in both bone toughness and bone fracture toughness correlate with an increase in pentosidine [65, 85, 95]. Moreover, inducing non-enzymatic crosslinking by incubating cortical or trabecular bone in high concentrations of sugar in vitro reduced fracture properties related to post-yield energy dissipation mechanisms [87, 96], but not strength [97]. This glycation process increases the stiffness of the collagen phase (i.e., demineralized bone) [98] and affects the ability of the collagen to dissipate energy [87]. In vivo, the non-enzymatic crosslinking could occur while mineralization is still an active process, and unlike enzymatic crosslinks that facilitate fibrillation and mineralization [65], these AGEs could potentially disrupt proper collagen–mineral interactions. The nature of such disruptions is presently unknown.
Hypothetically, AGEs cause the collagen to behave in a brittle fashion (i.e., fail at low strain, though resistant to stress) reducing energy dissipation by collagen fibril stretching and sliding within the mineral phase. With the loss of energy dissipation at the local tissue level, microcracks more readily propagate through bone tissue with higher concentration of AGEs. Several studies provide supporting evidence of this hypothesis: collagen fibrils undergo more deformation than mineral crystals at the same apparent tissue strain before and after the yield point of bone (onset of damage) [99, 100], this collagen fibril strain is lower in aged bone with higher AGEs than in young cortical bone [101], and the local tissue strain immediately surrounding a propagating crack is lower in aged bone than young cortical bone such that the extent of crack deflection at lamellar interfaces is reduced with aging [102]. Since crosslinking is likely affecting the contribution of collagen to energy dissipation mechanisms, diabetes likely affects fracture resistance through time-dependent changes to the organic matrix.
Diabetic Effects on Rodent Bone
As with many preclinical studies of disease, rodents have been widely used to study the effects of diabetes on bone (recent findings summarized in Table 1). Starting in 1952, diabetes (induced by alloxan) was found to affect bone structure in rats [103]. Consistently since then, the loss of insulin production (more commonly induced by streptozotocin or STZ) causes a reduction in the structural strength of rodent long bones (mice [104] and rats [105–109]). That is, bones from T1D rodents are narrower with a thinner cortex (Fig. 4), and therefore, break at a lower force or torque than bones from control rodents. There is also a loss of trabecular bone with diabetes [104, 110]. Inconsistently, STZ-induced diabetes decreased the degree of mineralization of cortical bone in male mice [104] and female Wistar rats [111] but did not affect the mineralization density of the cortical tissue (Ct.TMD) in male Fischer 344 and Sprague–Dawley rats [109]. In addition, T1D has not always affected material strength or toughness across studies (Table 1), and the loss in fatigue life was related to the diabetic effect on bone structure, not tissue quality [109]. Variations in the duration of T1D among studies may explain these observed differences in the diabetic effects on the biomechanical properties of bone. We recently found that with increasing duration of T1D, the strength vs. structure relationship (i.e., peak force vs. moment of inertia) changes from that of normal mice, and bones exhibit less post-yield work-to-failure, a measure of brittleness (Fig. 5) [104].

Micro-CT-derived renderings of the femur mid-shaft. The bones of the diabetic rodent are smaller with thinner cortices than those of the control rodents

Diabetic effects on bone brittleness. With increasing duration of diabetes, bone from male mice became brittle relative to control, as measured by post-yield work-to-fracture (mean ± SEM)
There are a variety of rat models of T2D including the Zucker Diabetic Fatty (ZDF) [112, 113], the Zucker Diabetic Sprague–Dawley (ZDSD) [113], Goto-Kakizaki [114], and the WBN/Kob [115], and each exhibits a difference in various bone parameters compared to non-diabetic controls (Table 1). The long bones of the ZDF male rats, which become hyperglycemic by 13 weeks of age, have similarities to those of STZ-treated rats in that they are shorter and narrower with a lower structural strength than controls [109, 112, 113]. This is also the case for male ZDSD rats, though the impairment in growth was reported to be less [113]. The cortical and trabecular bone tissue also have lower mineralization density (mgHA/cm3, as measured by peripheral or pQCT) in these diabetic rats relative to controls. Interestingly, when accounting for structural differences using the moment of inertia (also measured by pQCT), the estimated material strength of long bones was not different between ZDF or ZDSD rats and respective controls [112, 113]. The cause of this dissociation between BMD and material strength is not known, but likely involves heretofore unmeasured changes in the collagen phase of bone interacting with changes in the tissue mineral density. A study involving obese and non-obese T2D rats by Reinwald et al. [113] reported that the post-yield energy to failure, as determined by a three-point bending test of the femur, was markedly lower for the ZDF rats at 33 weeks of age than for the age-matched controls. Increases in AGEs within the bone tissue have been documented for STZ-treated rats [109] and the WBN/KoB rats [115], and it is likely that a similar increase occurs in the ZDF and other T2D rats and mice.
Clinical Directions
While diabetes increases the risk of suffering a fracture, the therapeutic options for preventing a fracture are limited. There is no definitive evidence that bisphosphonates are an effective treatment for individuals with diabetes. Nonetheless, a retrospective review of fracture cases in Denmark investigated the use of bisphosphonates in the context of diabetes and found that T1D or T2D did not appear to affect the ability of bisphosphonates to lower fracture risk [116]. With regard to other diabetic complications involving bone, anti-resorptive therapy has had some success in alleviating the symptoms of Charcot neuroarthropathy [117, 118], although recent studies found no beneficial effects of bisphosphonates on time to resolution or total immobilization time [119, 120]. Since the higher-than-expected fracture incidence among those with T2D is not associated with low BMD, there is no clear rationale to treat T2D patients with a bisphosphonate if the patient’s BMD values are not low. There is however a clear rationale for investigating how diabetes lowers bone’s resistance to fracture. Doing so could lead to effective therapeutic options for lowering the fracture risk of diabetics.
References
Nicodemus KK, Folsom AR. Type 1 and type 2 diabetes and incident hip fractures in postmenopausal women. Diabetes Care. 2001;24(7):1192–7.
Bonds DE, Larson JC, Schwartz AV, Strotmeyer ES, Robbins J, Rodriguez BL, et al. Risk of fracture in women with type 2 diabetes: the Women’s Health Initiative Observational Study. J Clin Endocrinol Metab. 2006;91(9):3404–10.
Schwartz AV, Sellmeyer DE. Diabetes, fracture, and bone fragility. Curr Osteoporos Rep. 2007;5(3):105–11.
Janghorbani M, Van Dam RM, Willett WC, Hu FB. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am J Epidemiol. 2007;166(5):495–505.
Vestergaard P, Rejnmark L, Mosekilde L. Diabetes and its complications and their relationship with risk of fractures in type 1 and 2 diabetes. Calcif Tissue Int. 2009;84(1):45–55.
Lewiecki EM. Management of osteoporosis. Clin Mol Allergy. 2004;2(1):9.
Atik OS, Gunal I, Korkusuz F. Burden of osteoporosis. Clin Orthop Relat Res. 2006;443:19–24.
Gullberg B, Johnell O, Kanis JA. World-wide projections for hip fracture. Osteoporos Int. 1997;7(5):407–13.
Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27(5):1047–53.
Thrailkill KM, Liu L, Wahl EC, Bunn RC, Perrien DS, Cockrell GE, et al. Bone formation is impaired in a model of type 1 diabetes. Diabetes. 2005;54(10):2875–81.
Lapcevic WA, French DD, Campbell RR. All-cause mortality rates of hip fractures treated in the VHA: do they differ from medicare facilities? J Am Med Dir Assoc. 2010;11(2):116–9.
Bass E, French DD, Bradham DD, Rubenstein LZ. Risk-adjusted mortality rates of elderly veterans with hip fractures. Ann Epidemiol. 2007;17(7):514–9.
Tuominen JT, Impivaara O, Puukka P, Ronnemaa T. Bone mineral density in patients with type 1 and type 2 diabetes. Diabetes Care. 1999;22(7):1196–200.
Danielson KK, Elliott ME, LeCaire T, Binkley N, Palta M. Poor glycemic control is associated with low BMD detected in premenopausal women with type 1 diabetes. Osteoporos Int. 2009;20(6):923–33.
Boehm HF, Eckstein F, Wunderer C, Kuhn V, Lochmueller EM, Schreiber K, et al. Improved performance of hip DXA using a novel region of interest in the upper part of the femoral neck: in vitro study using bone strength as a standard of reference. J Clin Densitom. 2005;8(4):488–94.
Muller ME, Webber CE, Bouxsein ML. Predicting the failure load of the distal radius. Osteoporos Int. 2003;14(4):345–52.
Carter DR, Bouxsein ML, Marcus R. New approaches for interpreting projected bone densitometry data. J Bone Miner Res. 1992;7(2):137–45.
Buysschaert M, Cauwe F, Jamart J, Brichant C, De Coster P, Magnan A, et al. Proximal femur density in type 1 and 2 diabetic patients. Diabete Metab. 1992;18(1):32–7.
Hampson G, Evans C, Petitt RJ, Evans WD, Woodhead SJ, Peters JR, et al. Bone mineral density, collagen type 1 alpha 1 genotypes and bone turnover in premenopausal women with diabetes mellitus. Diabetologia. 1998;41(11):1314–20.
Schwartz AV. Diabetes mellitus: Does it affect bone? Calcif Tissue Int. 2003;73(6):515–9.
de L II, van der Klift M, de Laet CE, van Daele PL, Hofman A, Pols HA. Bone mineral density and fracture risk in type-2 diabetes mellitus: the Rotterdam Study. Osteoporos Int. 2005;16(12):1713–20.
Vestergaard P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes–a meta-analysis. Osteoporos Int. 2007;18(4):427–44.
Wallace C, Reiber GE, LeMaster J, Smith DG, Sullivan K, Hayes S, et al. Incidence of falls, risk factors for falls, and fall-related fractures in individuals with diabetes and a prior foot ulcer. Diabetes Care. 2002;25(11):1983–6.
Schwartz AV, Hillier TA, Sellmeyer DE, Resnick HE, Gregg E, Ensrud KE, et al. Older women with diabetes have a higher risk of falls: a prospective study. Diabetes Care. 2002;25(10):1749–54.
Forsen L, Meyer HE, Midthjell K, Edna TH. Diabetes mellitus and the incidence of hip fracture: results from the Nord-Trondelag Health Survey. Diabetologia. 1999;42(8):920–5.
Schwartz AV, Sellmeyer DE, Ensrud KE, Cauley JA, Tabor HK, Schreiner PJ, et al. Older women with diabetes have an increased risk of fracture: a Prospective Study. J Clin Endocrinol Metab. 2001;86(1):32–8.
Ottenbacher KJ, Ostir GV, Peek MK, Goodwin JS, Markides KS. Diabetes mellitus as a risk factor for hip fracture in Mexican American older adults. J Gerontol A Biol Sci Med Sci. 2002;57(10):M648–53.
De Laet CE, Van Hout BA, Burger H, Weel AE, Hofman A, Pols HA. Hip fracture prediction in elderly men and women: validation in the Rotterdam study. J Bone Miner Res. 1998;13(10):1587–93.
Kanis JA, Johnell O, Oden A, Dawson A, De Laet C, Jonsson B. Ten year probabilities of osteoporotic fractures according to BMD and diagnostic thresholds. Osteoporos Int. 2001;12(12):989–95.
Pressman AR, Lo JC, Chandra M, Ettinger B. Methods for assessing fracture risk prediction models: experience with FRAX in a large integrated health care delivery system. J Clin Densitom. 2011;14(4):407–15. doi:10.1016/j.jocd.2011.06.006.
Schwartz AV, Vittinghoff E, Bauer DC, Hillier TA, Strotmeyer ES, Ensrud KE, et al. Association of BMD and FRAX score with risk of fracture in older adults with type 2 diabetes. Jama. 2011;305(21):2184–92. doi:10.1001/jama.2011.715.
Giangregorio L, Leslie W, Lix L, Johansson H, Oden A, McCloskey E, et al. FRAX underestimates fracture risk in patients with diabetes. J Bone Miner Res. 2011;. doi:10.1002/jbmr.556.
Rho JY, Kuhn-Spearing L, Zioupos P. Mechanical properties and the hierarchical structure of bone. Med Eng Phys. 1998;20(2):92–102.
Gupta HS, Zioupos P. Fracture of bone tissue: the ‘hows’ and the ‘whys’. Med Eng Phys. 2008;30(10):1209–26.
Ritchie RO. How does human bone resist fracture? Ann N Y Acad Sci. 2010;1192:72–80. doi:10.1111/j.1749-6632.2009.05232.x.
Turner CH, Burr DB. Basic biomechanical measurements of bone: a tutorial. Bone. 1993;14(4):595–608.
Martin RB. Determinants of the mechanical properties of bones. J Biomech. 1991;24(Suppl 1):79–88.
Teo JC, Si-Hoe KM, Keh JE, Teoh SH. Relationship between CT intensity, micro-architecture and mechanical properties of porcine vertebral cancellous bone. Clin Biomech (Bristol, Avon). 2006;21(3):235–44. doi:10.1016/j.clinbiomech.2005.11.001.
Zysset PK. A review of morphology-elasticity relationships in human trabecular bone: theories and experiments. J Biomech. 2003;36(10):1469–85.
Garrison JG, Slaboch CL, Niebur GL. Density and architecture have greater effects on the toughness of trabecular bone than damage. Bone. 2009;44(5):924–9. doi:10.1016/j.bone.2008.12.030.
Shi X, Liu XS, Wang X, Guo XE, Niebur GL. Type and orientation of yielded trabeculae during overloading of trabecular bone along orthogonal directions. J Biomech. 2010;43(13):2460–6. doi:10.1016/j.jbiomech.2010.05.032.
Boivin GY, Chavassieux PM, Santora AC, Yates J, Meunier PJ. Alendronate increases bone strength by increasing the mean degree of mineralization of bone tissue in osteoporotic women. Bone. 2000;27(5):687–94.
Yerramshetty JS, Akkus O. The associations between mineral crystallinity and the mechanical properties of human cortical bone. Bone. 2008;42(3):476–82.
Martin RB, Lau ST, Mathews PV, Gibson VA, Stover SM. Collagen fiber organization is related to mechanical properties and remodeling in equine bone. A comparison of two methods. J Biomech. 1996;29(12):1515–21.
Oxlund H, Barckman M, Ortoft G, Andreassen TT. Reduced concentrations of collagen cross-links are associated with reduced strength of bone. Bone. 1995;17(4 Suppl):365S–71S.
Muir P, Johnson KA, Ruaux-Mason CP. In vivo matrix microdamage in a naturally occurring canine fatigue fracture. Bone. 1999;25(5):571–6.
Kaeding CC, Najarian RG. Stress fractures: classification and management. Phys Sportsmed. 2010;38(3):45–54. doi:10.3810/psm.2010.10.1807.
Jones BH, Harris JM, Vinh TN, Rubin C. Exercise-induced stress fractures and stress reactions of bone: epidemiology, etiology, and classification. Exerc Sport Sci Rev. 1989;17:379–422.
Chantelau E, Richter A, Schmidt-Grigoriadis P, Scherbaum WA. The diabetic Charcot foot: MRI discloses bone stress injury as trigger mechanism of neuroarthropathy. Exp Clin Endocrinol Diabetes. 2006;114(3):118–23.
Mori S, Burr DB. Increased intracortical remodeling following fatigue damage. Bone. 1993;14(2):103–9.
Burr DB, Martin RB, Schaffler MB, Radin EL. Bone remodeling in response to in vivo fatigue microdamage. J Biomech. 1985;18:189–200.
Schaffler MB, Choi K, Milgrom C. Aging and matrix microdamage accumulation in human compact bone. Bone. 1995;17(6):521–5.
Norman TL, Wang Z. Microdamage of human cortical bone: incidence and morphology in long bones. Bone. 1997;20(4):375–9.
Zioupos P. Accumulation of in vivo fatigue microdamage and its relation to biomechanical properties in ageing human cortical bone. J Microsc. 2001;201(Pt 2):270–8.
Zioupos P. Accumulation of in vivo fatigue microdamage and its relation to biomechanical properties in ageing human cortical bone. J Microsc. 2001;201(2):270–8.
Yeni YN, Brown CU, Wang Z, Norman TL. The influence of bone morphology on fracture toughness of the human femur and tibia. Bone. 1997;21(5):453–9.
Yeni YN, Brown CU, Norman TL. Influence of bone composition and apparent density on fracture toughness of the human femur and tibia. Bone. 1998;22(1):79–84.
Zioupos P, Currey JD, Hamer AJ. The role of collagen in the declining mechanical properties of aging human cortical bone. J Biomed Mater Res. 1999;45(2):108–16.
Wang XD, Masilamani NS, Mabrey JD, Alder ME, Agrawal CM. Changes in the fracture toughness of bone may not be reflected in its mineral density, porosity, and tensile properties. Bone. 1998;23(1):67–72.
Issa C, Zantout MS, Azar ST. Osteoporosis in men with diabetes mellitus. J Osteoporos. 2011;2011:651867. doi:10.4061/2011/651867.
Wongdee K, Charoenphandhu N. Osteoporosis in diabetes mellitus: possible cellular and molecular mechanisms. World J Diabetes. 2011;2(3):41–8. doi:10.4239/wjd.v2.i3.41.
Burstein AH, Reilly DT, Martens M. Aging of bone tissue: mechanical properties. J Bone Joint Surg Am. 1976;58(1):82–6.
McCalden RW, McGeough JA, Barker MB, Court-Brown CM. Age-related changes in the tensile properties of cortical bone. The relative importance of changes in porosity, mineralization, and microstructure. J Bone Joint Surg Am. 1993;75(8):1193–205.
Zioupos P, Currey JD, Hamer AJ. The role of collagen in the declining mechanical properties of aging human cortical bone. J Biomed Mater Res. 1999;45(2):108–16.
Nyman JS, Roy A, Tyler JH, Acuna RL, Gayle HJ, Wang X. Age-related factors affecting the postyield energy dissipation of human cortical bone. J Orthop Res. 2007;25(5):646–55.
Zioupos P, Currey JD. Changes in the stiffness, strength, and toughness of human cortical bone with age. Bone. 1998;22(1):57–66.
Nalla RK, Kruzic JJ, Kinney JH, Ritchie RO. Effect of aging on the toughness of human cortical bone: evaluation by R-curves. Bone. 2004;35(6):1240–6.
Koester KJ, Barth HD, Ritchie RO. Effect of aging on the transverse toughness of human cortical bone: evaluation by R-curves. J Mech Behav Biomed Mater. 2011;4(7):1504–13. doi:10.1016/j.jmbbm.2011.05.020.
Zioupos P, Wang XT, Currey JD. The accumulation of fatigue microdamage in human cortical bone of two different ages in vitro. Clin Biomech (Bristol, Avon). 1996;11(7):365–75.
Diab T, Sit S, Kim D, Rho J, Vashishth D. Age-dependent fatigue behaviour of human cortical bone. Eur J Morphol. 2005;42(1–2):53–9.
Zioupos P, Gresle M, Winwood K. Fatigue strength of human cortical bone: Age, physical, and material heterogeneity effects. J Biomed Mater Res A. 2007;86(3):627–36.
Kruzic JJ, Ritchie RO. Fatigue of mineralized tissues: cortical bone and dentin. J Mech Behav Biomed Mater. 2008;1(1):3–17.
Carter DR, Hayes WC, Schurman DJ. Fatigue life of compact bone–II. Effects of microstructure and density. J Biomech. 1976;9(4):211–8.
Fleischli JG, Laughlin TJ, Athanasiou K, Lanctot DR, Lavery L, Wang X, et al. Effect of diabetes mellitus on the material properties of the distal tibia. J Am Podiatr Med Assoc. 2006;96(2):91–5.
Fleischli JG, Laughlin TJ, Lavery LA, Shah B, Lanctot D, Agrawal CM, et al. The effects of diabetes mellitus on the material properties of human metatarsal bones. J Foot Ankle Surg. 1998;37(3):195–8.
Saha MT, Sievanen H, Salo MK, Tulokas S, Saha HH. Bone mass and structure in adolescents with type 1 diabetes compared to healthy peers. Osteoporos Int. 2009;20(8):1401–6.
Bechtold S, Putzker S, Bonfig W, Fuchs O, Dirlenbach I, Schwarz HP. Bone size normalizes with age in children and adolescents with type 1 diabetes. Diabetes Care. 2007;30(8):2046–50.
Melton LJ 3rd, Riggs BL, Leibson CL, Achenbach SJ, Camp JJ, Bouxsein ML, et al. A bone structural basis for fracture risk in diabetes. J Clin Endocrinol Metab. 2008;93(12):4804–9.
Petit MA, Paudel ML, Taylor BC, Hughes JM, Strotmeyer ES, Schwartz AV et al. Bone mass and strength in older men with type 2 diabetes: The Osteoporotic Fractures in Men Study. J Bone Miner Res. 2010;25(2):285–91.
Register TC, Lenchik L, Hsu FC, Lohman KK, Freedman BI, Bowden DW, et al. Type 2 diabetes is not independently associated with spinal trabecular volumetric bone mineral density measured by QCT in the Diabetes Heart Study. Bone. 2006;39(3):628–33. doi:10.1016/j.bone.2006.03.003.
Shu A, Yin MT, Stein E, Cremers S, Dworakowski E, Ives R, et al. Bone structure and turnover in type 2 diabetes mellitus. Osteoporos Int. 2011;. doi:10.1007/s00198-011-1595-0.
Burghardt AJ, Issever AS, Schwartz AV, Davis KA, Masharani U, Majumdar S, et al. High-resolution peripheral quantitative computed tomographic imaging of cortical and trabecular bone microarchitecture in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2010;95(11):5045–55. doi:10.1210/jc.2010-0226.
Sell DR, Monnier VM. Structure elucidation of a senescence cross-link from human extracellular matrix. Implication of pentoses in the aging process. J Biol Chem. 1989;264(36):21597–602.
Zieman S, Kass D. Advanced glycation end product cross-linking: pathophysiologic role and therapeutic target in cardiovascular disease. Congest Heart Fail. 2004;10(3):144–9. quiz 50-1.
Wang X, Shen X, Li X, Agrawal CM. Age-related changes in the collagen network and toughness of bone. Bone. 2002;31(1):1–7.
Tomasek JJ, Meyers SW, Basinger JB, Green DT, Shew RL. Diabetic and age-related enhancement of collagen-linked fluorescence in cortical bones of rats. Life Sci. 1994;55(11):855–61.
Tang SY, Zeenath U, Vashishth D. Effects of non-enzymatic glycation on cancellous bone fragility. Bone. 2007;40(4):1144–51.
Monnier VM, Mustata GT, Biemel KL, Reihl O, Lederer MO, Zhenyu D, et al. Cross-linking of the extracellular matrix by the maillard reaction in aging and diabetes: an update on “a puzzle nearing resolution”. Ann N Y Acad Sci. 2005;1043:533–44.
Saito M, Marumo K. Collagen cross-links as a determinant of bone quality: a possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteoporos Int. 2010;21(2):195–214.
Shiraki M, Kuroda T, Tanaka S, Saito M, Fukunaga M, Nakamura T. Nonenzymatic collagen cross-links induced by glycoxidation (pentosidine) predicts vertebral fractures. J Bone Miner Metab. 2008;26(1):93–100.
Gineyts E, Munoz F, Bertholon C, Sornay-Rendu E, Chapurlat R. Urinary levels of pentosidine and the risk of fracture in postmenopausal women: the OFELY study. Osteoporos Int. 2010;21(2):243–50.
Schwartz AV, Garnero P, Hillier TA, Sellmeyer DE, Strotmeyer ES, Feingold KR, et al. Pentosidine and increased fracture risk in older adults with type 2 diabetes. J Clin Endocrinol Metab. 2009;94(7):2380–6.
Yamamoto M, Yamaguchi T, Yamauchi M, Yano S, Sugimoto T. Serum pentosidine levels are positively associated with the presence of vertebral fractures in postmenopausal women with type 2 diabetes. J Clin Endocrinol Metab. 2008;93(3):1013–9.
Saito M, Fujii K, Soshi S, Tanaka T. Reductions in degree of mineralization and enzymatic collagen cross-links and increases in glycation-induced pentosidine in the femoral neck cortex in cases of femoral neck fracture. Osteoporos Int. 2006;17(7):986–95.
Nyman JS, Ni Q, Nicolella DP, Wang X. Measurements of mobile and bound water by nuclear magnetic resonance correlate with mechanical properties of bone. Bone. 2008;42(1):193–9.
Vashishth D, Gibson GJ, Khoury JI, Schaffler MB, Kimura J, Fyhrie DP. Influence of nonenzymatic glycation on biomechanical properties of cortical bone. Bone. 2001;28(2):195–201.
Viguet-Carrin S, Farlay D, Bala Y, Munoz F, Bouxsein ML, Delmas PD. An in vitro model to test the contribution of advanced glycation end products to bone biomechanical properties. Bone. 2008;42(1):139–49.
Reddy GK. Glucose-mediated in vitro glycation modulates biomechanical integrity of the soft tissues but not hard tissues. J Orthop Res. 2003;21(4):738–43.
Gupta HS, Seto J, Wagermaier W, Zaslansky P, Boesecke P, Fratzl P. Cooperative deformation of mineral and collagen in bone at the nanoscale. Proc Natl Acad Sci USA. 2006;103(47):17741–6.
Dong XN, Almer JD, Wang X. Post-yield nanomechanics of human cortical bone in compression using synchrotron X-ray scattering techniques. J Biomech. 2011;44(4):676–82. doi:10.1016/j.jbiomech.2010.11.003.
Zimmermann EA, Schaible E, Bale H, Barth HD, Tang SY, Reichert P, et al. Age-related changes in the plasticity and toughness of human cortical bone at multiple length scales. Proc Natl Acad Sci USA. 2011;108(35):14416–21. doi:10.1073/pnas.1107966108.
Chan KS, Chan CK, Nicolella DP. Relating crack-tip deformation to mineralization and fracture resistance in human femur cortical bone. Bone. 2009;45(3):427–34. doi:10.1016/j.bone.2009.01.468.
Hernberg CA. The bone structure in alloxan-induced diabetes mellitus in rats. Acta Med Scand. 1952;142(4):274–83.
Nyman JS, Even JL, Jo CH, Herbert EG, Murry MR, Cockrell GE, et al. Increasing duration of type 1 diabetes perturbs the strength-structure relationship and increases brittleness of bone. Bone. 2011;48(4):733–40. doi:10.1016/j.bone.2010.12.016.
Dixit PK, Ekstrom RA. Decreased breaking strength of diabetic rat bone and its improvement by insulin treatment. Calcif Tissue Int. 1980;32(3):195–9.
Einhorn TA, Boskey AL, Gundberg CM, Vigorita VJ, Devlin VJ, Beyer MM. The mineral and mechanical properties of bone in chronic experimental diabetes. J Orthop Res. 1988;6(3):317–23.
Reddy GK, Stehno-Bittel L, Hamade S, Enwemeka CS. The biomechanical integrity of bone in experimental diabetes. Diabetes Res Clin Pract. 2001;54(1):1–8.
Zhang SQ, Chen GH, Lu WL, Zhang Q. Effects on the bones of vanadyl acetylacetonate by oral administration: a comparison study in diabetic rats. J Bone Miner Metab. 2007;25(5):293–301.
Silva MJ, Brodt MD, Lynch MA, McKenzie JA, Tanouye KM, Nyman JS, et al. Type 1 diabetes in young rats leads to progressive trabecular bone loss, cessation of cortical bone growth, and diminished whole bone strength and fatigue life. J Bone Miner Res. 2009;24(9):1618–27.
Silva MJ, Brodt MD, Lynch MA, McKenzie JA, Tanouye KM, Nyman JS, et al. Type 1 diabetes in young rats leads to progressive trabecular bone loss, cessation of cortical bone growth, and diminished whole bone strength and fatigue life. J Bone Miner Res. 2009;24(9):1618–27. doi:10.1359/jbmr.090316.
Facchini DM, Yuen VG, Battell ML, McNeill JH, Grynpas MD. The effects of vanadium treatment on bone in diabetic and non-diabetic rats. Bone. 2006;38(3):368–77.
Prisby RD, Swift JM, Bloomfield SA, Hogan HA, Delp MD. Altered bone mass, geometry and mechanical properties during the development and progression of type 2 diabetes in the Zucker diabetic fatty rat. J Endocrinol. 2008;199(3):379–88.
Reinwald S, Peterson RG, Allen MR, Burr DB. Skeletal changes associated with the onset of type 2 diabetes in the ZDF and ZDSD rodent models. Am J Physiol Endocrinol Metab. 2009;296(4):E765–74.
Ahmad T, Ohlsson C, Saaf M, Ostenson CG, Kreicbergs A. Skeletal changes in type-2 diabetic Goto-Kakizaki rats. J Endocrinol. 2003;178(1):111–6.
Saito M, Fujii K, Mori Y, Marumo K. Role of collagen enzymatic and glycation induced cross-links as a determinant of bone quality in spontaneously diabetic WBN/Kob rats. Osteoporos Int. 2006;17(10):1514–23.
Vestergaard P, Rejnmark L, Mosekilde L. Are antiresorptive drugs effective against fractures in patients with diabetes? Calcif Tissue Int. 2011;88(3):209–14. doi:10.1007/s00223-010-9450-4.
Jude EB, Selby PL, Burgess J, Lilleystone P, Mawer EB, Page SR, et al. Bisphosphonates in the treatment of Charcot neuroarthropathy: a double-blind randomised controlled trial. Diabetologia. 2001;44(11):2032–7.
Selby PL, Young MJ, Boulton AJ. Bisphosphonates: a new treatment for diabetic Charcot neuroarthropathy? Diabet Med. 1994;11(1):28–31.
Pakarinen TK, Laine HJ, Maenpaa H, Mattila P, Lahtela J. The effect of zoledronic acid on the clinical resolution of Charcot neuroarthropathy: a pilot randomized controlled trial. Diabetes Care. 2011;34(7):1514–6. doi:10.2337/dc11-0396.
Game FL, Catlow R, Jones GR, Edmonds ME, Jude EB, Rayman G, et al. Audit of acute Charcot’s disease in the UK: the CDUK Study. Diabetologia. 2011;. doi:10.1007/s00125-011-2354-7.
Zhang L, Liu Y, Wang D, Zhao X, Qiu Z, Ji H, et al. Bone biomechanical and histomorphometrical investment in type 2 diabetic Goto-Kakizaki rats. Acta Diabetol. 2009;46(2):119–26.
Kawashima Y, Fritton JC, Yakar S, Epstein S, Schaffler MB, Jepsen KJ, et al. Type 2 diabetic mice demonstrate slender long bones with increased fragility secondary to increased osteoclastogenesis. Bone. 2009;44(4):648–55.
Acknowledgments
The author thanks Dr. John Fowlkes for insightful comments on earlier versions of the present paper. Also, he thanks Dr. Colm Morrissey for providing the histological image of trabecular bone.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nyman, J.S. Effect of Diabetes on the Fracture Resistance of Bone. Clinic Rev Bone Miner Metab 11, 38–48 (2013). https://doi.org/10.1007/s12018-012-9124-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12018-012-9124-z