
Abstract
Glioblastoma is the most common primary malignant brain tumor and one of the most aggressive tumors across all cancer types with remarkable resistance to any treatment. While immunotherapy has shown a robust clinical benefit in systemic cancers, its benefit is still under investigation in brain cancers. The broader use of immunotherapy in clinical trials for glioblastoma has highlighted the challenges of traditional methods of monitoring progression via imaging. Development of new guidelines, advanced imaging techniques, and immune profiling have emerged to counter premature diagnoses of progressive disease. However, these approaches do not provide a timely diagnosis and are costly and time consuming. Surgery is currently the standard of care for diagnosis of pseudoprogression in cases where MRI is equivocal. However, it is invasive, risky, and disruptive to patient’s lives and their oncological treatment. With its increased vascularity, glioblastoma is continually shedding tumor components into the vasculature including tumor cells, genetic material, and extracellular vesicles. These elements can be isolated from routine blood draws and provide a real-time non-invasive indicator of tumor progression. Liquid biopsy therefore presents as an attractive alternative to current methods to guide treatment. While the initial evaluation of liquid biopsy for brain tumors via identification of mutations in the plasma was disappointing, novel technologies and use of alternatives to plasma cell-free DNA analytes provide promise for an effective liquid biopsy approach in brain tumors. This review aims to summarize developments in the use of liquid biopsy to monitor glioblastoma, especially in the context of immunotherapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glioblastoma is the most common primary malignant brain tumor, comprising up to 50% of glioma incidence in all age groups (Hanif et al., 2017). Patients have a median survival of 14–15 months, despite advancements in current standard of care (Hanif et al., 2017). Current treatment consists of maximal safe surgical resection followed by radiotherapy and concurrent temozolomide (Davis, 2016; Delgado-López & Corrales-García, 2016). Given glioblastoma’s high propensity for relapse, there has been considerable interest in developing novel treatments that can prolong survival and quality of life (Sherriff et al., 2013).
Immunotherapy for Brain Tumors
Recently, immunotherapy has emerged as a viable therapeutic option against difficult-to-treat solid tumors including melanoma, non-small cell lung carcinoma, and renal cell carcinoma (Coppin et al., 2004; Farina et al., 2017; Luke et al., 2017; Vansteenkiste et al., 2013). The arsenal of immunotherapeutic options against such tumors ranges from the use of vaccines and oncolytic viruses to immune checkpoint inhibition (Desjardins et al., 2018; Sampson et al., 2010; Schumacher et al., 2014). Vaccine therapies aim to use epitopes that occur only on glioma tumor cells, such as EGFRvIII or mutant IDH1, to prime the immune system to recognize and lyse these cells. Oncolytic virus therapy seeks to both directly cause tumor cell oncolysis via infection in combination with the release of intratumoral antigens from lysed cells for recognition by the immune system, leading to an immune response. Through the inhibition of tolerogenic markers like programmed cell death protein 1 (PD-1), cytotoxic t-lymphocyte-associated protein 4 (CTLA-4), T-cell immunoglobulin and mucin domain-3 (TIM-3), lymphocyte-activation gene 3 (LAG-3), glucorticoid-induced tumor necrosis factor receptor (GITR), and T cell immunoreceptor with Ig and ITIM domains (TIGIT), there are drastic changes to the tumor microenvironment including increased infiltration of lymphocytes, anti-tumor polarization of tumor-associated macrophages, and depletion of regulatory T cells (Gubin et al., 2018; Jackson et al., 2019; Luksik et al., 2017; Markovic et al., 2018; Nicholas et al., 2013; Selby et al., 2013; Simpson et al., 2013). Traditional views of the brain as a distinct immunologically privileged state with limited immune activity have been replaced in favor of a more nuanced paradigm depicting the brain as privy to ongoing immune surveillance and potent immune responses (Lim et al., 2018). This paradigm shift paved the way for trialing immune checkpoint inhibitors (ICI) therapy in glioblastoma.
However, despite the initial success of ICIs seen in a variety of solid tumors, glioblastoma did not exhibit the same results. Checkmate 143 was the first randomized phase III clinical trial to trial nivolumab, an anti-PD-1 ICI, against bevacizumab, an anti-vascular endothelial growth factor therapy, in recurrent glioblastoma. Their recently published study shows that there was no improved survival benefit in patients treated with nivolumab monotherapy as compared to bevacizumab (Reardon et al., 2020). However, despite these initial results, there is still promising work being done with ICIs in the setting of glioblastoma. New approaches aim to sensitize the tumor microenvironment to ICI therapy or to harness the ability of ICIs to create a pro-immunogenic milieu and synergize with other forms of therapy (Medikonda et al., 2020; Preusser et al., 2015). Additionally, neoadjuvant anti-PD-1 treatment with subsequent adjuvant treatment after surgical resection has shown survival benefit in patients with glioblastoma (Cloughesy et al., 2019). Given this increased interest in the role of ICIs with regard to glioblastoma, it is imperative that there are clear and consistent guidelines on monitoring treatment response and identifying patient responders. Prematurely stopping a treatment may prevent a patient from benefitting from a promising therapy, while failing to stop a treatment when there is a lack of response may lead to unwarranted toxicities and may prevent/exclude patients from other trials with agents that could prove more effective.
Monitoring of Immune Response in Brain Tumors via Imaging Modalities
Traditional methods of monitoring treatment response of glioblastoma to a variety of therapies including radiation, chemotherapy, and surgical resection involved the use of imaging via computed tomography (CT) or magnetic resonance imaging (MRI). Response evaluation criteria in solid tumors (RECIST) guidelines published in 2000 and revised in 2009 characterized a tumoricidal response as shrinkage on imaging, while disease progression was operationalized as enlargement or development of additional foci (Kasten et al., 2019; Wolchok et al., 2009). However, such guidelines failed to account for inflammatory infiltration and response that can present as apparent tumor enlargement when using standard CT/MRI techniques. This presumed progression is then ultimately followed by tumor regression and corresponding clinical improvement. The series of events describing tumor response following an initial trajectory of tumor progression is deemed ‘pseudoprogression’ (Frelaut et al., 2020; Kasten et al., 2019). Although pseudoprogression can be seen as a result of radiation or chemotherapy, immunotherapy and ICIs are particularly susceptible to pseudoprogression given their enhancement of immune infiltration and morphogenic changes to tumor makeup (Chamberlain, 2008; Gubin et al., 2018; Markovic et al., 2018; Mieghem et al., 2013).
To address the shortcomings of traditional imaging criteria like the RECIST guidelines with regard to pseudoprogression, the response assessment in neuro-oncology (RANO) guidelines were published in 2010 (Wen et al., 2010). The RANO criteria aimed to account for pseudoprogression through the use of advanced imaging techniques, like fluid-attenuated inversion recovery (T2/FLAIR) MRI, and a revised definition of disease progression that allowed time to distinguish between pseudoprogression and true progression. Increasing enhancement or additional foci specifically in the treatment area in the first 12 weeks of therapy would not meet the criteria for disease progression. Given that immunotherapy in glioblastoma has garnered interest largely in the last decade, the 2010 RANO criteria were focused primarily on distinguishing pseudoprogression in response to radiation or chemotherapy (Wen et al., 2010). In 2015, the RANO group published specific criteria for immunotherapy called the iRANO guidelines (Okada et al., 2015). This set of recommendations employed a similar use of a more flexible timeline to account for pseudoprogression in that new foci of lesions within the initial 6 months of immunotherapy do not constitute progressive disease.
In addition to updated imaging criteria, the technology for discerning pseudoprogression via imaging has advanced considerably. Radiomics, a new field that uses imaging features from various modalities such as MRI, CT, and PET and algorithmically combines these features to identify subtle differences in these images by clinical condition, not possible through standard imaging analysis, holds considerable enthusiasm (Kumar et al., 2012). This methodology is currently under investigation in many non-CNS cancers with promising results. Sun et al. conducted a retrospective multicohort study in which they examined patients with advanced solid tumors who were treated with immunotherapy (Sun et al., 2018). They developed and validated a radiomic biomarker predictive of immunotherapy response using RNA sequencing data and pretreatment CT scans from 135 patients enrolled in the MOSCATO trial to evaluate cytotoxic T cell tumor infiltration (Sun et al., 2018). This biomarker was validated in an independent patient cohort from The Cancer Genome Atlas (AUC = 0.67) and subsequently validated in two other cohorts (Sun et al., 2018). This study demonstrated the connection between imaging data, tumor phenotype, and clinical responses to immunotherapy. Subsequently, Korpics et al. validated the radiomics biomarker developed by the Sun group (Korpics et al., 2019) in a different clinical setting. They used this algorithm to examine the impact of anti-PD-1 and stereotactic radiosurgery (SRS) in 68 patients with 139 metastatic solid tumors on progression-free survival (PFS) and overall survival (OS) (Korpics et al., 2019). Patients with a low radiomic signature score were significantly less likely to respond to SRS and had worse PFS and worse OS (Korpics et al., 2019). Furthermore, Trebeschi et al. analyzed 1055 primary and metastatic lesions from 203 patients with non-small cell lung cancer (NSCLC) and advanced melanoma treated with anti-PD1 in order to create a radiomics biomarker to predict treatment response at the lesion level (Trebeschi et al., 2019). On combining lesion-specific predictions at the patient level, they achieved an AUC 0.76 (p < 0.001) (Trebeschi et al., 2019).
Radiomics in neuro-oncology is a newer field with a paucity of research studies available in the literature compared to other cancer types. Various studies have shown the promise of radiomics as a tool with significant potential for high-grade glioma diagnosis (Artzi et al., 2014; Chaddad et al., 2018; Gevaert et al., 2014; Jamshidi et al., 2014; Lee et al., 2015), but few studies have specifically addressed the issue of treatment response. Elshafeey et al. used dynamic susceptibility contrast (DSC) and dynamic contrast-enhanced perfusion (DCE) MRI images to construct a radiomics model to differentiate pseudoprogression from disease progression in a retrospective cohort of 98 patients with glioblastoma treated with immunotherapy (Elshafeey et al., 2019). This model yielded high accuracy with a sensitivity of 91.36%, at a specificity of 88.24%. However, these findings need to be validated in an independent cohort of glioblastoma patients to assure generalizability of the methodology. Nevertheless, this study provided a theoretical framework for future studies to further assess the value of machine learning and deep learning algorithms in neuroradiology.
Despite these promising studies, many of them face limitations and challenges. One major challenge is the small sample size explored in these studies that are often preliminary and exploratory in nature. It will be important to incorporate these new radiomic tools in large multi-center randomized clinical trials in order to better validate these algorithms. Furthermore, one of the biggest limitations of this field is data reproducibility. Given the novelty of this field as well as the massive amounts of data that it generates, there are so much variety and a lack of standardization in image acquisition, nomenclature, calculation of radiomics features, and statistical and machine learning methods (Limkin & Sun, 2020). Standardization of these various components would help limit technical and procedural heterogeneity and improve the generalizability of the data produced in these studies. Given the large amounts of data produced by these radiomic studies, data sharing becomes an issue, especially with the use of larger trials involving multiple hospital sites and health networks—the cultural, administrative, regulatory, and personal issues will be difficult to navigate and creative solutions will be required (Nelson, 2009). These limitations highlight the need for exploring easily implemented and less costly imaging-based methods for determining disease progression versus therapeutic response in immunotherapy trials.
Immune-Based Predictors of Immunotherapeutic Response for Brain Tumors
One natural extension of efforts of non-invasive disease monitoring during the course of immunotherapy is the assessment of the surface marker expression on immune cells or cytokine levels in the peripheral blood during the course of immunotherapy. We here highlight some of the efforts that have been used to track immune responses during the course of immunotherapy trials.
Crane et al. showed that ex vivo stimulation of T cells isolated from PBMCs of patients treated with a HSPPC96 (Heat shock protein–peptide complexes-96) vaccine, pre- and post-vaccination, produced a significant T cell proliferation in responders but failed to stimulate a similar proliferative effect in non-responders (Crane et al., 2013). The immune response was associated with an improvement in overall survival of these patients.
Another T cell surface marker that has shown promise in assessing immune response is ICOS (Inducible T-cell COStimulator) or CD278. ICOS is a molecule expressed in activated T cells. The number of CD4 + ICOS + cells in the peripheral blood was significantly correlated with treatment response and improvement of overall survival in a clinical trial of CTLA-4 administration of patients with melanoma (Carthon et al., 2010). This marker, however, has not been assessed in an immunotherapy glioblastoma clinical trial so far.
The use of cytokine expression and other surface markers of inflammation in the peripheral blood as a tool for disease monitoring during treatment has not shown consistent and clinically useful results so far and therefore we will not expand on the subject.
An exciting new avenue for monitoring of treatment response during immunotherapy has been recently provided by the field of immunogenomics. More specifically, the use of T cell receptor sequencing technology has provided a way to non-invasively assess the emergence of T cell receptor (TCR) repertoires in response to immunotherapy. Zhang et al. utilized the TCR sequencing technology for patients enrolled in a HSPPC-96 trial of primary glioblastoma (Zhang et al., 2020). They TCR-sequenced tumor infiltrating lymphocytes (TILs) of patients with great response (long-term survivors) and no response (short-term survivors) and found that the responders had a narrower TCR repertoire indicating a more coordinated immune response. These clones appeared to be shared among the responder group and not be present in the non-responder group. While this group used TILs obtained from tumor biopsy and not peripheral blood, their results highlight the use of this technology to assess response to treatment.
Cloughesy et al. assessed the efficacy of neoadjuvant anti-PD-1 in the treatment of glioblastoma and unlike the use of adjuvant anti-PD-1, they found a significant number of patients with treatment response (Cloughesy et al., 2019). They assessed the TCR repertoire at several timepoints during the course of anti-PD-1 treatment and suggested that T cell expansion at the neo-adjuvant setting can increase tumor-specific T cell clones. While the study did not specifically address T cell clonality at timepoints of response and recurrence/disease progression of each patient, the fact that responses to neoadjuvant anti-PD-1 treatment were associated with an increase in tumor-specific T cell clones could prove useful in tracking immune responses during and after the course of immunotherapy and should be further explored in future clinical trials.
Overall, blood-based immune monitoring approaches appear to be of limited use in predicting treatment response or failure during the course of treatment. This is mainly due to the low sensitivity and specificity of the approaches, their lack of generalizability, the expensive and complicated protocols required for these assays, and the low throughput of the current technologies.
Liquid Biopsy for Disease Monitoring During Brain Tumor Treatment
A strategy that has garnered interest for monitoring tumor progression is using biofluid analytes such as circulating tumor cells, nucleic acids, or exosomes shed by the tumors in patient blood or cerebrospinal fluid (CSF) (Crowley et al., 2013; Pantel & Alix-Panabières, 2013, 2019; Zhang et al., 2017). These methods have been shown to correlate with tumor burden in patients and early detection for therapeutic response (Dawson et al., 2013; Diehl et al., 2008). Liquid biopsies also offer several advantages compared to traditional imaging or biopsy methods. Firstly, liquid biopsies tend to be easier for patients; in that, they are subjected to timely blood draws that can be done anywhere as opposed to scheduled, long imaging appointments. Secondly, they offer more information than a static image such as tumor genetics and specific biomarkers (Di Meo et al., 2017). Furthermore, non-invasive liquid biopsies have the advantage of posing no risk for iatrogenic injury during traditional biopsies, especially in eloquent brain areas. In the modern era of precision medicine, clinicians understand that genetic and epigenetic modifications contribute to unique tumor morphologies and phenotypes. Since liquid biopsies sample a wide array of the genetic material originating directly from the tumor and perhaps tumor-related immunological changes in the circulation, they provide valuable data that can be used to predict prognosis and therapeutic response (Mathai et al., 2019). This is especially advantageous in glioblastoma given its exceptional heterogeneity with respect to its tumor microenvironment (Soeda et al., 2015). Imaging methods and the traditional gold standard for solid tumors, biopsy, cannot accurately account for the dissimilar phenotypes in the intratumoral niche. Such advantages make liquid biopsy an attractive avenue of both diagnostic and prognostic means. Table 1 provides a summary of notable liquid biopsy studies discussed in-depth later during this review.
Circulating Tumor Cells
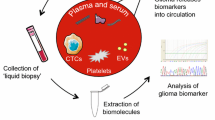
The first documented paper on circulating tumor cells (CTCs) in 1869 proposed that metastasis occurred when tumor cells were shed by the primary tumor into the circulation to seed new tumor sites (Wang et al., 2017). We now understand that CTCs are primary tumor cells that have undergone an epithelial-to-mesenchymal transition and have been released from the basement membrane via a variety of matrix metalloproteases (Chistiakov & Chekhonin, 2018). These released cells employ immune evasion and host mimicry, along with their increased mobility, to survive in the vasculature and search for other sites permissive of cell proliferation and invasion. With their enhanced capacity for survival, these cells not only contribute to metastasis but also can explain recurrence after treatment after the CTCs switch from a dormant, quiescent state to a proliferative one (Gao et al., 2017). This is especially pertinent to glioblastoma since glioblastoma has an extracranial metastasis rate of only 0.4–0.5% (Lun et al., 2011). Given that CTCs retain many of the genetic and phenotypic qualities of the original tumor, these cells can be harnessed to glean information about the characteristics of the original tumor (Fig. 1a).

Schematic representation of liquid biopsy approaches for gliomas. High-grade gliomas can shed circulating tumor cells, DNA, RNA as well as extracellular vesicles in the circulation or the CSF. Analysis of these analytes through a blood draw or a lumbar puncture could allow for real-time monitoring of tumor dynamics. Created with BioRender.com
One of the primary hurdles with CTC isolation in glioblastoma patients is that glioma cells did not express EpCAM, a surface glycoprotein marker present on many carcinomas and commonly used for CTC detection (Moldenhauer et al., 1987). Instead, researchers had to develop alternate methods to identify glioblastoma CTCs. In 2014, Muller et al. found that CTCs were present in 29 of 141 patients, or approximately 20% of glioblastoma patients, when isolated using the marker glial fibrillary acidic protein (GFAP) (Müller et al., 2014). These cells also harbored tumor-specific mutations in the forms of distinct cell surface markers and genetic alterations, showing that sampling tumor CTCs could provide valuable information about the primary tumor without direct biopsy. Although 20% is only a minority, the authors acknowledged that the yield may increase when using a marker other than GFAP to isolate CTCs. Sullivan et al. used a “microfluidic device” called the CTC-iChip to negatively select against the leukocyte markers CD45 and CD16 (Sullivan et al., 2014). In this manner, the authors hoped to avoid having to select for a single antigen or differentiating factor among all CTCs, especially since glioblastoma displays inherent heterogeneity. However, the use of the CTC-iChip was only marginally more effective than Muller et al.’s approach where 39% of patients with glioblastoma were detectable. The CTC-iChip was interestingly able to reliably differentiate between patients who had progressive disease versus those who had stable disease. However, other aspects of tumor burden such as small tumor foci occurring around the bulk of the tumor and extent of resection were not able to be distinguished with this method.
To address the low yield exhibited by early attempts at CTC detection, Gao et al. in 2016 employed fluorescence in situ hybridization (FISH) to look for chromosome 8 polyploidy rather than a specific marker to find CTCs (Gao et al., 2016). With this methodology, the authors were able to detect CTCs in 77% of the patients. Interestingly, patients had decreased level of CTCs following 2 years of standard clinical therapy, indicating CTCs could be used as a marker of therapeutic response. MacArthur et al. were also able to detect CTCs in 72% of patients, prior to radiotherapy administration using a probe that stained for human telomerase (MacArthur et al., 2014). Since telomerase has elevated expression in tumor cells vs normal tissue, the probe successfully bound only to tumor cells in peripheral blood. Interestingly, CTCs were only detected in 13% of patients post-radiotherapy treatment. This suggests that CTCs may correlate with overall tumor burden but do not have a high sensitivity of determining tumor presence in smaller, residual tumors.
The detection and isolation of CTCs through these various methods have proved that there is valuable information that can be gleaned from the peripheral blood regarding the nature of a patient’s specific glioblastoma. The aforementioned studies have shown that CTCs can provide an estimate toward pseudoprogression by determining if increase in tumor size seen on imaging correlates with increases in CTC levels post-treatment. However, the sensitivity of CTC detection in glioblastoma remains quite variable and is still in the beginning stages. Furthermore, it has not been shown that there is a linear relationship between tumor burden and CTC level. As such, CTCs remain a novel tool in the potential monitoring of disease progression and therapeutic response that still requires considerable work before it can be used as a sole marker of discerning pseudoprogression.
Circulating Tumor DNA in Plasma
Circulating tumor DNA (ctDNA) was analyzed long after the presence of CTCs was speculated but has been heavily investigated in recent years due to the advent of next-generation sequencing technologies. In the context of oncology, ctDNA is a genetic material that is released by tumor cells through various processes including apoptosis, necrosis, or secretion (Heitzer et al., 2015). Analysis of these DNA fragments can reveal multiple features of the cancer genome including mutations, aneuploidy, rearrangements, and tumor-specific epigenetic modifications (Fig. 1b). The primary challenge in utilizing ctDNA clinically for brain tumors is the very low mutant allele fraction shed in the circulation by these tumors (Diaz & Bardelli, 2014). Given these obstacles, the evolution of ctDNA for diagnostic and prognostic purposes has been largely a recent endeavor. In 2014, Bettegowda et al. examined 640 patients using polymerase chain reaction (PCR) technology to search for the presence of ctDNA (Bettegowda et al., 2014). While the results varied widely between individual tumors, ctDNA was detected in less than 10% of patients with gliomas. Despite the seemingly limited application of ctDNA in glioblastoma, there were two findings of note: Firstly, there were many instances where ctDNA was detectable when there were no discernable CTCs. This suggests that CTCs and ctDNA exist independent from each other in the circulation and that ctDNA may be able to improve upon the sensitivity of CTCs. Secondly, the authors showed that there was a steadily decreasing rate of 2-year survival associated with an increased number of ctDNA mutant fragments. Thus, ctDNA might prove to be a stronger indicator of tumor burden than CTCs. In the context of pseudoprogression, having a peripheral noninvasive marker for tumor burden would clarify the difference between immune infiltration and disease progression seen on imaging.
Several groups have attempted to identify clinicopathological factors associated with low ctDNA shedding by gliomas in the circulation with mixed results. Nabavizadeh et al. assessed the histopathologic and imaging features of patients with gliomas and correlated these tumor features with levels of cell-free DNA (cfDNA) in the circulation (Nabavizadeh et al., 2020). The study showed that tumor size, contrast-enhancing volume, or any other measure of tumor volume did not correlate with baseline cfDNA levels. In contrast, cfDNA concentrations were elevated in patients who had increased metrics corresponding to blood–brain barrier disruption. On histopathology, patients who had increased macrophage staining in the tumor microenvironment also correlated to exhibiting higher ctDNA concentrations. In contrast to perivascular macrophages which may be increasing the integrity of the blood–brain barrier, macrophages within the tumor microenvironment may be liberating DNA from apoptotic and necrotic cells. These findings suggest that future developments in the field of liquid biopsy using circulating genetic material in glioblastoma may involve increasing blood–brain barrier permeability or activating tumor-associated macrophages to increase the yield of detection. However, Mair et al.’s findings using patient-derived xenograft models suggest that blood–brain barrier dynamics do not play a significant role in detection of ctDNA (Mair et al., 2019). In this study, the authors hypothesized that tumor mitochondrial DNA (tmtDNA) would yield greater sensitivity than nuclear ctDNA given its increased abundance in tumor cells. Detection of plasma ctDNA had a sensitivity of 82% in the orthotopic xenograft rat model as compared to nuclear ctDNA’s sensitivity of 24%. The tmtDNA was also found in CSF and urine samples. A principal component analysis revealed that the primary indicators of tmtDNA and ctDNA concentrations corresponded to tumor volume and cellular proliferation. In one of the treatment naïve groups, the authors saw that tmtDNA concentrations were correlated with tumor burden and increased with treatment response due to increased cell death. This suggests that tmtDNA can be involved in monitoring therapeutic progress or looking at disease progression. In contrast to Nabavizadeh et al.’s work, the authors argued that it was not in fact the blood–brain barrier that led to low ctDNA detection in plasma, but the much increased background noise of nontumor cell-free DNA. Thus, there is still no consensus on what leads to sparse ctDNA sensitivity in the plasma. However, there are new possible routes of discovery such as tmtDNA, blood–brain barrier-altering agents, and better sequencing technologies that can be pursued.
Piccioni et al. revisited the problem of utilizing ctDNA in glioblastoma in 2019 (Piccioni et al., 2019). In the age of personalized medicine, therapies such as tumor vaccines and CAR-T cells require knowledge of individual patient-specific tumoral genetic aberrations. The authors proposed that ctDNA could provide a noninvasive means of achieving this purpose. Utilizing a standard laboratory genetics assay devised for detecting genetic mutations in any solid tumor, Piccioni et al. detected mutations in ctDNA in 50% of the 419 patients tested, a much improved result over prior attempts at discovery of ctDNA in primary brain tumors. This study and all other mutation-based studies that have been performed so far in gliomas have not taken into account the phenomenon called clonal hematopoiesis of indeterminate potential (CHIP) (Chan et al., 2020). CHIP is a result of passenger mutations in the white blood cells (WBCs) that are released in the plasma and can mimic tumor-derived mutations, even ones that occur in hotspot cancer mutations. Therefore, any study that has not accounted for this phenomenon by sequencing at the same sequencing depth in WBCs can overinflate the percent detection of true cancer-derived mutations.
To circumvent the above major confounder in ctDNA analysis and to harness more recent discoveries in genome organization in the blood, a few groups have analyzed the size fragment distribution of cfDNA in the circulation and have found that while circulating DNA in healthy individuals resemble DNA patterns from WBCs, ctDNA has a more variable size distribution and tends to be shorter (Cristiano et al., 2019; Mouliere, Chandrananda, et al. 2018a). By incorporating fragmentation patterns into the analysis, the sensitivity of detecting genomic alterations is increased beyond simply looking at genetic mutations which, at best, occur at a rate of one in a few thousand fragments. Circulating DNA fragments are mapped to the genome and the size and location of the fragments in the genome are taken into account. Differences in circulating DNA between healthy controls and tumor patients have been postulated to arise from differences in organization of genomes regarding open or closed chromatin. Healthy individuals have chromatin and nucleosomal fragmentation patterns that reflected that of healthy WBCs, suggesting that is where the majority of their circulating DNA arises from. Prior work has also suggested a difference between the fragment sizes and positions taken from tumor patients versus healthy controls but has notably disagreed on whether these fragments are larger or smaller (Jiang et al., 2015; Umetani et al., 2006). Mouliere et al. approached the problem by implementing a whole-genome approach rather than relying on specific loci. The authors characterized ctDNA from patients with various tumors as compared to healthy controls and found that ctDNA fragment size shifts to a smaller count of 167 bp in many cancers (Mouliere, Chandrananda, et al. 2018a). Mouliere et al. did also, however, discover that fragments in the 250 to 320 bp range also were enriched for mutated fragments and stated that the origin of these fragments is still unknown. Cristiano et al. built on this work by a shallow sequencing whole-genomic approach combined with machine learning algorithms to account for size differences and coverage in various loci over the genome discovered (Cristiano et al., 2019). The size of the fragments in addition to their mapped location served to create a fragmentation signature that was unique to tumor patients. Interestingly, the fragmentation profiles matched tumor-specific mutant alterations throughout the course of treatment suggesting that fragmentation analysis can be used to monitor therapeutic response. Using the analysis as a prediction tool, the authors found that the ROC curve exhibited an AUC value of 0.94. Subsequently, Zviran et al. have proposed conducting genome-wide sequencing on cfDNA to increase sensitivity of ctDNA detection and monitoring cancer progression, especially in cases of minimal residual disease for non-central nervous system tumors (Zviran et al., 2020). By incorporating aneuploidy detection and algorithmically accounting for sequencing errors, the authors were able to increase detection sensitivity by 2 orders of magnitude, from detection at a concentration of 10–3 to 10–5, at a specificity level of 99.85%. One of the most notable findings was that the whole-genome sequencing was able to track tumor response throughout patient treatment, detect changes in tumor burden earlier than imaging, and was able to detect residual disease after completion of therapy in cases where prior methods of ctDNA detection were unable to do so. This was demonstrated in the ability to detect ctDNA in patients’ status post-resection of tumor. Patients in whom ctDNA was undetectable with the whole-genome sequencing did not exhibit any signs of recurrence in the median 15-month follow-up period. Of note, in a patient who had melanoma and was undergoing checkpoint immunotherapy treatment, ctDNA tracked tumor responses with a greater temporal resolution than CT imaging. The ability to detect low-burden disease and track progress with this method of ctDNA identification could lay the groundwork for the future of discerning progression and pseudoprogression in patients with brain tumors.
DNA Methylation of cfDNA
More recently, Nassiri et al. have utilized cfDNA methylation features in plasma of patients with gliomas and extra-axial CNS tumors and were able to show that gliomas have a distinct methylation signature in the plasma compared to other extra-axial CNS tumors and cancer-free individuals (Nassiri et al., 2020). The performance of their technology in discriminating the presence of a glioma in the plasma of an individual was strikingly higher compared to any other study so far, indicating that cfDNA methylation can be a sensitive marker of the tissue of origin in the blood. The retrospective collection of samples and other confounding factors that could affect cfDNA methylation (age, sex, use of different sequencing platforms, different source of blood samples, timing of collection) necessitates further validation to establish the importance of cfDNA methylation in glioma detection. No study has thus far been performed in the setting of disease monitoring to assess the ability of cfDNA methylation in tracking the tumor dynamics and response to treatment in gliomas.
Circulating Tumor DNA in CSF
While all the aforementioned studies looked primarily at peripheral blood for isolation of genetic material, the cerebrospinal fluid (CSF) is another compartment that shows promise. The CNS is bathed by CSF and has been shown to be a reservoir for brain or spinal cord tumors, especially those of which lie adjacent to a CSF cavity (Wang et al., 2015). Mouliere et al. used a shallow whole-genome sequencing approach to detect and characterize ctDNA present in the CSF (Mouliere, Mair, et al. 2018b). Their method was able to detect ctDNA without the prior need of an intracranial biopsy to search for exact somatic mutations. As such, the detection rate for ctDNA in the CSF was similar to prior studies that used expensive whole-exome sequencing. One interesting finding was that ctDNA in CSF had a different fragmentation pattern than ctDNA in the plasma. Such a fragmentation signature could be used to further identify circulating tumor genetic material in the CSF in the future without any prior mutational or genetic knowledge. Miller et al. furthered the work by showing that the ctDNA detected in patient CSF had the same somatic mutations as those found in the original tumor (Miller et al., 2019). Analysis of longitudinal CSF samples highlighted the gradual evolution of the genetic landscape over the time course of a tumor. Thus, CSF ctDNA highlights the advantages of a liquid biopsy; in that, a peripheral, noninvasive method is able to monitor genetic changes in the primary tumor over sequential timepoints. In a similar vein, other forms of genetic material besides DNA have proven to be attractive candidates for isolation. The release of RNA through biological activity of tumor cells into the extracellular milieu suggests its utility as a clinical biomarker (Everaert et al., 2019). Akers et al. examined miRNA in the CSF to evaluate its role as a biomarker for glioblastoma (Akers et al., 2017). The authors found that 30–50% of miRNAs detected in patient glioblastoma samples were seen in CSF samples but at a decreased abundance. Using cutoff criteria for expression fold changes, 29 miRNAs were identified of which 9 were chosen to create a glioblastoma signature for diagnosis through CSF analysis. The signature resulted in a sensitivity of 80%, identifying 8 out of 10 glioblastoma patients, and a specificity of 67%, distinguishing 8 out of 12 healthy patients when profiling cisternal CSF. The study was thus able to establish that CSF could be used to diagnose patients with glioblastoma. Further work needs to be done to elucidate whether tumor burden affects the level of miRNA secretion and if it can be used to track progression and pseudoprogression in patients.
While these techniques continue to be developed in the context of glioblastoma, circulating genetic material has been trialed in the context of discerning pseudoprogression when treated with immunotherapy in other tumors. Lee et al. in 2018 explored the efficacy of determining pseudoprogression with ctDNA in patients with metastatic melanoma when given anti-PD-1 treatment (Lee et al., 2018). 29 of 125 patients were determined to have “disease progression” as denoted by RECIST criteria. Of the 29 patients, 9 experienced pseudoprogression according to immune-related response criteria (iiRC) guidelines. All 9 patients were classified as having a “favorable” ctDNA assessment involving parameters that indicated minimal to no detection of ctDNA. Overall, the ctDNA’s rate of pseudoprogression prediction was at a sensitivity of 90% and a specificity of 100%. Anagnostou et al. analyzed longitudinal plasma samples of patients with non-small cell lung cancer treated with immune checkpoint inhibitors (Anagnostou et al., 2019). 9 patients exhibited a molecular response in which ctDNA dropped to completely undetectable levels on average 9 weeks after therapy. The second category was patients who had molecular resistance in which ctDNA levels did not decrease or rose after beginning therapy. The third pattern was in a subset of the molecular responder category in which ctDNA response displayed clonal selection indicative of immune escape mechanisms in response to PD-1 blockade. The authors report that early drops in ctDNA levels were stronger predictors for progression-free survival (PFS) and overall survival (OS) as compared to radiographic imaging and tumor burden. Bratman et al. confirmed these findings in an expanded cohort in patients with solid tumors treated with anti-PD-1 therapy (Bratman et al., 2020). In their sample of 94 patients, the authors found that baseline ctDNA levels correlated with overall survival and progression-free survival. Similar to Anagnostou et al., the authors found that the change in ctDNA levels following treatment allowed patients to be grouped into response categories that correlated well with survival. Thus, ctDNA analysis can help quickly identify whether a patient is responding to immunotherapy and whether they should be advanced to other therapies that may be more effective. Moding et al. explored this idea of whether all patients should be treated similarly based on their ctDNA profile after chemoradiation treatment (Moding et al., 2020). Patients with non-small cell lung cancer who had undetectable ctDNA following chemoradiation treatment did not show significant benefit when receiving additional immune checkpoint inhibition therapy, while patients who continued to have detectable levels of ctDNA following chemoradiation exhibited increased survival benefit with follow-up immune checkpoint inhibition therapy. Therefore, ctDNA has shown the ability to act as a potential biomarker not only for tumor burden but also as a predictor of treatment response.
As the technology for next-generation sequencing improves for analysis of ctDNA, similar methods can be applied to RNA, potentially providing another avenue of exploration. Given that tumor cells are constantly undergoing necrosis and apoptosis, particularly so in response to chemotherapy and radiation treatment, analysis of circulating genetic material released from tumors shows promise. As discoveries into depth of sequencing to increase fidelity and breadth of sequencing to increase sensitivity are explored, isolation of ctDNA and RNA may be able to provide accurate monitoring of tumor burden relative to the patient’s baseline with high temporal resolution. Such methods would be valuable in distinguishing between disease progression vs pseudoprogression on imaging without having to wait for set periods of time to wait for clinical improvement or deterioration.
Extracellular Vesicles
One aspect of the study conducted on miRNA by Akers et al. that ultimately was not pursued by the authors was the incorporation of extracellular vesicles. Extracellular vesicles (EVs) broadly are membrane-bound vesicles secreted by cells from endosomal compartments or the plasma membrane that have been found to be involved in intercellular communication, tumor angiogenesis and growth, and tumor immune response (Raposo & Stoorvogel, 2013) (Fig. 1c). EVs carry many different biological molecules including proteins, genetic material, lipids, and signaling molecules (Han et al., 2019; Raposo & Stoorvogel, 2013). Some advantages that EVs provide over CTCs and ctDNA are their robustness in the periphery and their specific function as signaling molecules. While DNA and RNA degradation are significant concerns during detection and isolation, the fact that genetic material in EVs are bound in a membrane makes them much more resistant to degradation by proteases and DNases. Furthermore, due to their role in intercellular communication, genetic material and proteins involved in crosstalk can be found in greater abundance within these vesicles than unbound in peripheral blood. In the Akers et al. study, the authors examined whether the miRNA glioblastoma signature they were developing would be more enriched in EVs (Akers et al., 2017). However, they found that almost all the miRNAs they identified within EVs were isolated free-floating in CSF. Therefore, with this particular biomarker, the authors felt there was no need to take the extra step of isolating EVs. However, this is not always the case as certain molecules are significantly more upregulated in EVs and biomarkers present within the membranes themselves can be crucial for evaluating tumor presence (Han et al., 2019).
The role of miRNAs in serum exosomes as a potential biomarker was re-evaluated by Santangelo et al. in 2017 (Santangelo et al., 2018). Exosomes are a class of EVs specifically in the size range of 40–150 nm that originate from cellular endosomes. The authors settled on examining the clinical utility of 3 pre-selected miRNAs (miR-21, miR-222, and miR-124-3p) found in exosomes to act as a diagnostic biomarker of glioma. Results showed that expression levels according to real-time qPCR of all 3 of the miRNAs individually and cumulatively were significantly higher in glioblastoma patients than control populations. The AUC value for cumulative miRNA was 0.87, showing potential for diagnostic accuracy. Interestingly, this genetic signature was specific for high-grade gliomas seeing as miRNA expression was significantly higher in high-grade gliomas as compared to both low-grade gliomas and healthy controls. The authors concluded that based on their findings, certain individual miRNAs or their cumulative expression values in combination with pre-operative MRIs could help determine tumor presence vs brain metastases and possibly predict glioma grading without the need of pathological analysis. They next showed that the expression levels were dramatically reduced in high-grade gliomas after surgical resection, indicating that the miRNAs may be able to ascertain tumor presence post-resection when imaging is unclear. The authors then examined whether the miRNA levels could be used to discern pseudoprogression vs progression. Of the 2 patients that were examined, clinical progress correlated well with miRNA expression levels after resection. When the miRNA levels decreased, the patient showed clinical improvement. In the other patient, pseudoprogression was considered but ultimately the patient had an increase in miRNA expression levels after resection and later had clinical progression. Thus, the study proved that exosomes were a reliable source of genetic information that could potentially be used to predict a patient’s disease course after surgical resection. Lan et al. followed a similar route by examining the clinical utility of miR-301a in exosomes as a peripheral biomarker. The authors first provided evidence that the exosomes were not simply static entities but themselves acted on low-grade glioma cells and increased their tumorigenicity. They then found that miR-301a expression had significantly increased expression in patients with gliomas versus healthy controls. Similar to Santangelo et al.’s findings, the miR-301a expression levels corresponded to the grade of the tumor as well and were specific for gliomas when compared to other cancers. Thus, in both studies examining exosomal miRNAs, there was a difference in expression level between tumor grades, showing an increased pathological resolution when comparing exosomal biomarkers to CTCs and ctDNA. Similarly, miR-301a levels decreased after surgery and rebounded after recurrence. The AUC for miR-301a expression was 0.937 conferring a more accurate diagnostic biomarker accuracy when compared to Santangelo et al.’s study. mIR-301a was not examined in a patient to determine whether progression vs pseudoprogression could be discerned but given its similarity to Santangelo et al.’s results, further work should be done in following patient levels, particularly through an immunotherapy course. Lan et al. revisited their miRNA exosomal approach to diagnose and predict patient glioblastoma course more recently in 2020 (Lan et al., 2020). They were able to show similar findings to novel miRNA biomarkers exhibited in previous papers such as significantly higher miR-210 levels in patients with glioma, a correlation with tumor grade, abrogation of expression levels with resection and an increase with recurrence, and evidence that expression levels of miR-210 were independently associated with patient prognosis. As the field continues to expand and sequencing technologies become cheaper and more accurate, genetic biomarkers within exosomes will continue to develop as potential diagnostic and prognostic biomarkers.
Another avenue of approach in identifying exosomal biomarkers relevant to glioblastoma is determining glioblastoma signatures within the tumor and then assessing whether they are present within exosomes. Furthermore, exosomes exhibit multiple facets of tumor pathology. They can simultaneously act as a messenger by being a peripherally accessible source of information about the tumor microenvironment, while actively participating in increased tumorigenesis, as shown by Huang et al.’s study (Huang et al., 2018). While the authors did not examine specifically whether tumor burden could be examined using exosome-derived nucleic acids, and thus be applied to discernment of pseudoprogression, the study showed that exosomal biomarkers can be a valuable source of information about the tumor microenvironment and have the potential to track patient course.
Future Directions
More research needs to be conducted in the study of utilizing multiple methods and techniques to determine treatment response in GBM patients receiving immunotherapy. Basler et al. created a model that incorporated both blood biomarkers (S100, LDH) and radiomics analyses (PET/CT) to help differentiate pseudoprogression from disease progression in patients with metastatic melanoma (Basler et al., 2020). They assessed a cohort of 112 patients undergoing immune checkpoint inhibitor therapy. A combined blood biomarkers and radiomics model was developed with an AUC = 0.82, which supports the notion that a multi-modality approach will be most efficacious in identifying treatment response in cancer patients receiving immunotherapy.
References
Akers, J. C., Hua, W., Li, H., Ramakrishnan, V., Yang, Z., Quan, K., et al. (2017). A cerebrospinal fluid microRNA signature as biomarker for glioblastoma. Oncotarget, 8(40), 68769–68779. https://doi.org/10.18632/oncotarget.18332
Anagnostou, V., Forde, P. M., White, J. R., Niknafs, N., Hruban, C., Naidoo, J., et al. (2019). Dynamics of tumor and immune responses during immune checkpoint blockade in non-small cell lung cancer. Cancer Research, 79(6), 1214–1225. https://doi.org/10.1158/0008-5472.CAN-18-1127
Artzi, M., Bokstein, F., Blumenthal, D. T., Aizenstein, O., Liberman, G., Corn, B. W., & Bashat, D. B. (2014). Differentiation between vasogenic-edema versus tumor-infiltrative area in patients with glioblastoma during bevacizumab therapy: A longitudinal MRI study. European Journal of Radiology, 83(7), 1250–1256.
Basler, L., Gabryś, H. S., Hogan, S. A., Pavic, M., Bogowicz, M., Vuong, D., et al. (2020). Radiomics, tumor volume, and blood biomarkers for early prediction of pseudoprogression in patients with metastatic melanoma treated with immune checkpoint inhibition. Clinical Cancer Research. https://doi.org/10.1158/1078-0432.CCR-20-0020
Bettegowda, C., Sausen, M., Leary, R. J., Kinde, I., Wang, Y., Agrawal, N., et al. (2014). Detection of circulating tumor DNA in early- and late-stage human malignancies. Science Translational Medicine, 6(224), 224ra24. https://doi.org/10.1126/scitranslmed.3007094
Bratman, S. V., Yang, S. Y. C., Iafolla, M. A. J., Liu, Z., Hansen, A. R., Bedard, P. L., et al. (2020). Personalized circulating tumor DNA analysis as a predictive biomarker in solid tumor patients treated with pembrolizumab. Nature Cancer, 1(9), 873–881. https://doi.org/10.1038/s43018-020-0096-5
Carthon, B. C., Wolchok, J. D., Yuan, J., Kamat, A., Ng Tang, D. S., Sun, J., et al. (2010). Preoperative CTLA-4 blockade: Tolerability and immune monitoring in the setting of a presurgical clinical trial. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 16(10), 2861–2871. https://doi.org/10.1158/1078-0432.CCR-10-0569
Chaddad, A., Sabri, S., Niazi, T., & Abdulkarim, B. (2018). Prediction of survival with multi-scale radiomic analysis in glioblastoma patients. Medical & Biological Engineering & Computing, 56(12), 2287–2300. https://doi.org/10.1007/s11517-018-1858-4
Chamberlain, M. C. (2008). Pseudoprogression in glioblastoma. Journal of Clinical Oncology, 26(26), 4359–4359. https://doi.org/10.1200/JCO.2008.18.4440
Chan, H. T., Chin, Y. M., Nakamura, Y., & Low, S.-K. (2020). Clonal hematopoiesis in liquid biopsy: From biological noise to valuable clinical implications. Cancers, 12(8), 2277. https://doi.org/10.3390/cancers12082277
Chistiakov, D. A., & Chekhonin, V. P. (2018). Circulating tumor cells and their advances to promote cancer metastasis and relapse, with focus on glioblastoma multiforme. Experimental and Molecular Pathology, 105(2), 166–174. https://doi.org/10.1016/j.yexmp.2018.07.007
Cloughesy, T. F., Mochizuki, A. Y., Orpilla, J. R., Hugo, W., Lee, A. H., Davidson, T. B., et al. (2019). Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nature Medicine, 25(3), 477–486. https://doi.org/10.1038/s41591-018-0337-7
Coppin, C., Porzsolt, F., Autenrieth, M., Kumpf, J., Coldman, A., & Wilt, T. (2004). Immunotherapy for advanced renal cell cancer. Cochrane Database of Systematic Reviews. https://doi.org/10.1002/14651858.CD001425.pub2
Crane, C. A., Han, S. J., Ahn, B., Oehlke, J., Kivett, V., Fedoroff, A., et al. (2013). Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 19(1), 205–214. https://doi.org/10.1158/1078-0432.CCR-11-3358
Cristiano, S., Leal, A., Phallen, J., Fiksel, J., Adleff, V., Bruhm, D. C., et al. (2019). Genome-wide cell-free DNA fragmentation in patients with cancer. Nature, 570(7761), 385–389. https://doi.org/10.1038/s41586-019-1272-6
Crowley, E., Di Nicolantonio, F., Loupakis, F., & Bardelli, A. (2013). Liquid biopsy: Monitoring cancer-genetics in the blood. Nature Reviews Clinical Oncology, 10(8), 472–484. https://doi.org/10.1038/nrclinonc.2013.110
Davis, M. E. (2016). Glioblastoma: Overview of disease and treatment. Clinical Journal of Oncology Nursing, 20(5), S2–S8. https://doi.org/10.1188/16.CJON.S1.2-8
Dawson, S.-J., Tsui, D. W. Y., Murtaza, M., Biggs, H., Rueda, O. M., Chin, S.-F., et al. (2013). Analysis of circulating tumor DNA to monitor metastatic breast cancer. The New England Journal of Medicine, 368(13), 1199–1209. https://doi.org/10.1056/NEJMoa1213261
Delgado-López, P. D., & Corrales-García, E. M. (2016). Survival in glioblastoma: A review on the impact of treatment modalities. Clinical and Translational Oncology, 18(11), 1062–1071. https://doi.org/10.1007/s12094-016-1497-x
Desjardins, A., Gromeier, M., Herndon, J. E., Beaubier, N., Bolognesi, D. P., Friedman, A. H., et al. (2018). Recurrent glioblastoma treated with recombinant poliovirus. New England Journal of Medicine, 379(2), 150–161. https://doi.org/10.1056/NEJMoa1716435
Di Meo, A., Bartlett, J., Cheng, Y., Pasic, M. D., & Yousef, G. M. (2017). Liquid biopsy: A step forward towards precision medicine in urologic malignancies. Molecular Cancer, 16(1), 80. https://doi.org/10.1186/s12943-017-0644-5
Diaz, L. A., & Bardelli, A. (2014). Liquid biopsies: Genotyping circulating tumor DNA. Journal of Clinical Oncology, 32(6), 579–586. https://doi.org/10.1200/JCO.2012.45.2011
Diehl, F., Schmidt, K., Choti, M. A., Romans, K., Goodman, S., Li, M., et al. (2008). Circulating mutant DNA to assess tumor dynamics. Nature Medicine, 14(9), 985–990. https://doi.org/10.1038/nm.1789
Elshafeey, N., Kotrotsou, A., Hassan, A., Elshafei, N., Hassan, I., Ahmed, S., et al. (2019). Multicenter study demonstrates radiomic features derived from magnetic resonance perfusion images identify pseudoprogression in glioblastoma. Nature Communications, 10(1), 1–9.
Everaert, C., Helsmoortel, H., Decock, A., Hulstaert, E., Van Paemel, R., Verniers, K., et al. (2019). Performance assessment of total RNA sequencing of human biofluids and extracellular vesicles. Scientific Reports, 9(1), 17574. https://doi.org/10.1038/s41598-019-53892-x
Farina, M. S., Lundgren, K. T., & Bellmunt, J. (2017). Immunotherapy in urothelial cancer: Recent results and future perspectives. Drugs, 77(10), 1077–1089. https://doi.org/10.1007/s40265-017-0748-7
Frelaut, M., du Rusquec, P., de Moura, A., Le Tourneau, C., & Borcoman, E. (2020). Pseudoprogression and hyperprogression as new forms of response to immunotherapy. BioDrugs: Clinical Immunotherapeutics, Biopharmaceuticals and Gene Therapy, 34, 463–476.
Gao, F., Cui, Y., Jiang, H., Sui, D., Wang, Y., Jiang, Z., et al. (2016). Circulating tumor cell is a common property of brain glioma and promotes the monitoring system. Oncotarget, 7(44), 71330–71340. https://doi.org/10.18632/oncotarget.11114
Gao, X., Zhang, M., Tang, Y., & Liang, X. (2017). Cancer cell dormancy: Mechanisms and implications of cancer recurrence and metastasis. Oncotargets and Therapy. https://doi.org/10.2147/OTT.S140854
Gevaert, O., Mitchell, L. A., Achrol, A. S., Xu, J., Echegaray, S., Steinberg, G. K., et al. (2014). Glioblastoma multiforme: Exploratory radiogenomic analysis by using quantitative image features. Radiology, 273(1), 168–174.
Gubin, M. M., Esaulova, E., Ward, J. P., Malkova, O. N., Runci, D., Wong, P., et al. (2018). High-dimensional analysis delineates myeloid and lymphoid compartment remodeling during successful immune-checkpoint cancer therapy. Cell, 175(4), 1014-1030.e19. https://doi.org/10.1016/j.cell.2018.09.030
Han, L., Lam, E.W.-F., & Sun, Y. (2019). Extracellular vesicles in the tumor microenvironment: Old stories, but new tales. Molecular Cancer, 18(1), 59. https://doi.org/10.1186/s12943-019-0980-8
Hanif, F., Muzaffar, K., Perveen, K., Malhi, S. M., & Simjee, S. U. (2017). Glioblastoma multiforme: A review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pacific Journal of Cancer Prevention: APJCP, 18(1), 3–9. https://doi.org/10.22034/APJCP.2017.18.1.3
Heitzer, E., Ulz, P., & Geigl, J. B. (2015). Circulating tumor DNA as a liquid biopsy for cancer. Clinical Chemistry, 61(1), 112–123. https://doi.org/10.1373/clinchem.2014.222679
Huang, K., Fang, C., Yi, K., Liu, X., Qi, H., Tan, Y., et al. (2018). The role of PTRF/Cavin1 as a biomarker in both glioma and serum exosomes. Theranostics, 8(6), 1540–1557. https://doi.org/10.7150/thno.22952
Jackson, C. M., Choi, J., & Lim, M. (2019). Mechanisms of immunotherapy resistance: Lessons from glioblastoma. Nature Immunology, 20(9), 1100–1109. https://doi.org/10.1038/s41590-019-0433-y
Jamshidi, N., Diehn, M., Bredel, M., & Kuo, M. D. (2014). Illuminating radiogenomic characteristics of glioblastoma multiforme through integration of MR imaging, messenger RNA expression, and DNA copy number variation. Radiology, 270(1), 1–2. https://doi.org/10.1148/radiol.13130078
Jiang, P., Chan, C. W. M., Chan, K. C. A., Cheng, S. H., Wong, J., Wong, V.W.-S., et al. (2015). Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proceedings of the National Academy of Sciences of the United States of America, 112(11), E1317-1325. https://doi.org/10.1073/pnas.1500076112
Kasten, B. B., Udayakumar, N., Leavenworth, J. W., Wu, A. M., Lapi, S. E., McConathy, J. E., et al. (2019). Current and future imaging methods for evaluating response to immunotherapy in neuro-oncology. Theranostics, 9(17), 5085–5104. https://doi.org/10.7150/thno.34415
Korpics, M. C., Bhave, S. R., Redler, G., Pitroda, S., Luke, J. J., & Chmura, S. J. (2019). A validated radiomics T-cell score predicts response to multi-site SBRT combined with pembrolizumab. International Journal of Radiation Oncology Biology Physics, 105(1), S74.
Kumar, V., Gu, Y., Basu, S., Berglund, A., Eschrich, S. A., Schabath, M. B., et al. (2012). Radiomics: The process and the challenges. Magnetic Resonance Imaging, 30(9), 1234–1248.
Lan, F., Yue, X., & Xia, T. (2020). Exosomal microRNA-210 is a potentially non-invasive biomarker for the diagnosis and prognosis of glioma. Oncology Letters, 19(3), 1967–1974. https://doi.org/10.3892/ol.2020.11249
Lee, J. H., Long, G. V., Menzies, A. M., Lo, S., Guminski, A., Whitbourne, K., et al. (2018). Association between circulating tumor DNA and pseudoprogression in patients with metastatic melanoma treated with anti-programmed cell death 1 antibodies. JAMA Oncology, 4(5), 717. https://doi.org/10.1001/jamaoncol.2017.5332
Lee, J., Narang, S., Martinez, J., Rao, G., & Rao, A. (2015). Spatial habitat features derived from multiparametric magnetic resonance imaging data are associated with molecular subtype and 12-month survival status in glioblastoma multiforme. PLoS ONE, 10(9), e0136557.
Lim, M., Xia, Y., Bettegowda, C., & Weller, M. (2018). Current state of immunotherapy for glioblastoma. Nature Reviews Clinical Oncology, 15(7), 422–442. https://doi.org/10.1038/s41571-018-0003-5
Limkin, E. J., & Sun, R. (2020). Radiomics to predict response to immunotherapy: An imminent reality? Future Oncology. https://doi.org/10.2217/fon-2020-0015
Luke, J. J., Flaherty, K. T., Ribas, A., & Long, G. V. (2017). Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nature Reviews Clinical Oncology, 14(8), 463–482. https://doi.org/10.1038/nrclinonc.2017.43
Luksik, A. S., Maxwell, R., Garzon-Muvdi, T., & Lim, M. (2017). The role of immune checkpoint inhibition in the treatment of brain tumors. Neurotherapeutics, 14(4), 1049–1065. https://doi.org/10.1007/s13311-017-0513-3
Lun, M., Lok, E., Gautam, S., Wu, E., & Wong, E. T. (2011). The natural history of extracranial metastasis from glioblastoma multiforme. Journal of Neuro-Oncology, 105(2), 261–273. https://doi.org/10.1007/s11060-011-0575-8
MacArthur, K. M., Kao, G. D., Chandrasekaran, S., Alonso-Basanta, M., Chapman, C., Lustig, R. A., et al. (2014). Detection of brain tumor cells in the peripheral blood by a telomerase promoter-based assay. Cancer Research, 74(8), 2152–2159. https://doi.org/10.1158/0008-5472.CAN-13-0813
Mair, R., Mouliere, F., Smith, C. G., Chandrananda, D., Gale, D., Marass, F., et al. (2019). Measurement of plasma cell-free mitochondrial tumor DNA improves detection of glioblastoma in patient-derived orthotopic xenograft models. Cancer Research, 79(1), 220–230. https://doi.org/10.1158/0008-5472.CAN-18-0074
Markovic, S. N., Galli, F., Suman, V. J., Nevala, W. K., Paulsen, A. M., Hung, J. C., et al. (2018). Non-invasive visualization of tumor infiltrating lymphocytes in patients with metastatic melanoma undergoing immune checkpoint inhibitor therapy: A pilot study. Oncotarget, 9(54), 30268–30278. https://doi.org/10.18632/oncotarget.25666
Mathai, R. A., Vidya, R. V. S., Reddy, B. S., Thomas, L., Udupa, K., Kolesar, J., & Rao, M. (2019). Potential utility of liquid biopsy as a diagnostic and prognostic tool for the assessment of solid tumors: Implications in the precision oncology. Journal of Clinical Medicine, 8(3), 373. https://doi.org/10.3390/jcm8030373
Medikonda, R., Dunn, G., Rahman, M., Fecci, P., & Lim, M. (2020). A review of glioblastoma immunotherapy. Journal of Neuro-Oncology. https://doi.org/10.1007/s11060-020-03448-1
Mieghem, E. V., Wozniak, A., Geussens, Y., Menten, J., Vleeschouwer, S. D., Calenbergh, F. V., et al. (2013). Defining pseudoprogression in glioblastoma multiforme. European Journal of Neurology, 20(10), 1335–1341. https://doi.org/10.1111/ene.12192
Miller, A. M., Shah, R. H., Pentsova, E. I., Pourmaleki, M., Briggs, S., Distefano, N., et al. (2019). Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature, 565(7741), 654–658. https://doi.org/10.1038/s41586-019-0882-3
Moding, E. J., Liu, Y., Nabet, B. Y., Chabon, J. J., Chaudhuri, A. A., Hui, A. B., et al. (2020). Circulating tumor DNA dynamics predict benefit from consolidation immunotherapy in locally advanced non-small-cell lung cancer. Nature Cancer, 1(2), 176–183. https://doi.org/10.1038/s43018-019-0011-0
Moldenhauer, G., Momburg, F., Möller, P., Schwartz, R., & Hämmerling, G. J. (1987). Epithelium-specific surface glycoprotein of Mr 34,000 is a widely distributed human carcinoma marker. British Journal of Cancer, 56(6), 714–721.
Mouliere, F., Chandrananda, D., Piskorz, A. M., Moore, E. K., Morris, J., Ahlborn, L. B., et al. (2018a). Enhanced detection of circulating tumor DNA by fragment size analysis. Science Translational Medicine, 10(466), 4921. https://doi.org/10.1126/scitranslmed.aat4921
Mouliere, F., Mair, R., Chandrananda, D., Marass, F., Smith, C. G., Su, J., et al. (2018b). Detection of cell-free DNA fragmentation and copy number alterations in cerebrospinal fluid from glioma patients. EMBO Molecular Medicine, 10(12), e9323. https://doi.org/10.15252/emmm.201809323
Müller, C., Holtschmidt, J., Auer, M., Heitzer, E., Lamszus, K., Schulte, A., et al. (2014). Hematogenous dissemination of glioblastoma multiforme. Science Translational Medicine, 6(247), 247ra101. https://doi.org/10.1126/scitranslmed.3009095
Nabavizadeh, S. A., Ware, J. B., Guiry, S., Nasrallah, M. P., Mays, J. J., Till, J. E., et al. (2020). Imaging and histopathologic correlates of plasma cell-free DNA concentration and circulating tumor DNA in adult patients with newly diagnosed glioblastoma. Neuro-Oncology Advances, 2(1), 16. https://doi.org/10.1093/noajnl/vdaa016
Nassiri, F., Chakravarthy, A., Feng, S., Shen, S. Y., Nejad, R., Zuccato, J. A., et al. (2020). Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nature Medicine, 26(7), 1044–1047. https://doi.org/10.1038/s41591-020-0932-2
Nelson, B. (2009). Empty archives: Most researchers agree that open access to data is the scientific ideal, so what is stopping it happening? Bryn Nelson investigates why many researchers choose not to share. Nature, 461(7261), 160–164.
Nicholas, S., Mathios, D., Ruzevick, J., Jackson, C., Yang, I., & Lim, M. (2013). Current trends in glioblastoma multiforme treatment: Radiation therapy and immune checkpoint inhibitors. Brain Tumor Research and Treatment, 1(1), 2–8. https://doi.org/10.14791/btrt.2013.1.1.2
Okada, H., Weller, M., Huang, R., Finocchiaro, G., Gilbert, M. R., Wick, W., et al. (2015). Immunotherapy Response Assessment in Neuro-Oncology (iRANO): A report of the RANO Working Group. The Lancet Oncology, 16(15), e534–e542. https://doi.org/10.1016/S1470-2045(15)00088-1
Pantel, K., & Alix-Panabières, C. (2013). Real-time liquid biopsy in cancer patients: Fact or fiction? Cancer Research, 73(21), 6384–6388. https://doi.org/10.1158/0008-5472.CAN-13-2030
Pantel, K., & Alix-Panabières, C. (2019). Liquid biopsy and minimal residual disease—latest advances and implications for cure. Nature Reviews Clinical Oncology, 16(7), 409–424. https://doi.org/10.1038/s41571-019-0187-3
Piccioni, D. E., Achrol, A. S., Kiedrowski, L. A., Banks, K. C., Boucher, N., Barkhoudarian, G., et al. (2019). Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncology, 8(2), CNS34. https://doi.org/10.2217/cns-2018-0015
Preusser, M., Lim, M., Hafler, D. A., Reardon, D. A., & Sampson, J. H. (2015). Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nature Reviews Neurology, 11(9), 504–514. https://doi.org/10.1038/nrneurol.2015.139
Raposo, G., & Stoorvogel, W. (2013). Extracellular vesicles: Exosomes, microvesicles, and friends. Journal of Cell Biology, 200(4), 373–383. https://doi.org/10.1083/jcb.201211138
Reardon, D. A., Brandes, A. A., Omuro, A., Mulholland, P., Lim, M., Wick, A., et al. (2020). Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: The CheckMate 143 phase 3 randomized clinical trial. JAMA Oncology, 6(7), 1003–1010. https://doi.org/10.1001/jamaoncol.2020.1024
Sampson, J. H., Heimberger, A. B., Archer, G. E., Aldape, K. D., Friedman, A. H., Friedman, H. S., et al. (2010). Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 28(31), 4722–4729. https://doi.org/10.1200/JCO.2010.28.6963
Santangelo, A., Imbrucè, P., Gardenghi, B., Belli, L., Agushi, R., Tamanini, A., et al. (2018). A microRNA signature from serum exosomes of patients with glioma as complementary diagnostic biomarker. Journal of Neuro-Oncology, 136(1), 51–62. https://doi.org/10.1007/s11060-017-2639-x
Schumacher, T., Bunse, L., Pusch, S., Sahm, F., Wiestler, B., Quandt, J., et al. (2014). A vaccine targeting mutant IDH1 induces antitumour immunity. Nature, 512(7514), 324–327. https://doi.org/10.1038/nature13387
Selby, M. J., Engelhardt, J. J., Quigley, M., Henning, K. A., Chen, T., Srinivasan, M., & Korman, A. J. (2013). Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunology Research, 1(1), 32–42. https://doi.org/10.1158/2326-6066.CIR-13-0013
Sherriff, J., Tamangani, J., Senthil, L., Cruickshank, G., Spooner, D., Jones, B., et al. (2013). Patterns of relapse in glioblastoma multiforme following concomitant chemoradiotherapy with temozolomide. The British Journal of Radiology, 86(1022), 20120414. https://doi.org/10.1259/bjr.20120414
Simpson, T. R., Li, F., Montalvo-Ortiz, W., Sepulveda, M. A., Bergerhoff, K., Arce, F., et al. (2013). Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. The Journal of Experimental Medicine, 210(9), 1695–1710. https://doi.org/10.1084/jem.20130579
Soeda, A., Hara, A., Kunisada, T., Yoshimura, S., Iwama, T., & Park, D. M. (2015). The evidence of glioblastoma heterogeneity. Scientific Reports, 5(1), 7979. https://doi.org/10.1038/srep07979
Sullivan, J. P., Nahed, B. V., Madden, M. W., Oliveira, S. M., Springer, S., Bhere, D., et al. (2014). Brain tumor cells in circulation are enriched for mesenchymal gene expression. Cancer Discovery, 4(11), 1299–1309. https://doi.org/10.1158/2159-8290.CD-14-0471
Sun, R., Limkin, E. J., Vakalopoulou, M., Dercle, L., Champiat, S., Han, S. R., et al. (2018). A radiomics approach to assess tumour-infiltrating CD8 cells and response to anti-PD-1 or anti-PD-L1 immunotherapy: An imaging biomarker, retrospective multicohort study. The Lancet Oncology, 19(9), 1180–1191.
Trebeschi, S., Drago, S. G., Birkbak, N. J., Kurilova, I., Cǎlin, A. M., Delli Pizzi, A., et al. (2019). Predicting response to cancer immunotherapy using noninvasive radiomic biomarkers. Annals of Oncology, 30(6), 998–1004.
Umetani, N., Giuliano, A. E., Hiramatsu, S. H., Amersi, F., Nakagawa, T., Martino, S., & Hoon, D. S. B. (2006). Prediction of breast tumor progression by integrity of free circulating DNA in serum. Journal of Clinical Oncology, 24(26), 4270–4276. https://doi.org/10.1200/JCO.2006.05.9493
Vansteenkiste, J., Zielinski, M., Linder, A., Dahabreh, J., Gonzalez, E. E., Malinowski, W., et al. (2013). Adjuvant MAGE-A3 immunotherapy in resected non–small-cell lung cancer: Phase II randomized study results. Journal of Clinical Oncology, 31(19), 2396–2403. https://doi.org/10.1200/JCO.2012.43.7103
Wang, Y., Zhou, Y., & Hu, Z. (2017). The functions of circulating tumor cells in early diagnosis and surveillance during cancer advancement. Journal of Translational Internal Medicine, 5(3), 135–138. https://doi.org/10.1515/jtim-2017-0029
Wang, Y., Springer, S., Zhang, M., McMahon, K. W., Kinde, I., Dobbyn, L., et al. (2015). Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proceedings of the National Academy of Sciences of the United States of America, 112(31), 9704–9709. https://doi.org/10.1073/pnas.1511694112
Wen, P. Y., Macdonald, D. R., Reardon, D. A., Cloughesy, T. F., Sorensen, A. G., Galanis, E., et al. (2010). Updated response assessment criteria for high-grade gliomas: Response assessment in Neuro-Oncology Working Group. Journal of Clinical Oncology, 28(11), 1963–1972. https://doi.org/10.1200/JCO.2009.26.3541
Wolchok, J. D., Hoos, A., O’Day, S., Weber, J. S., Hamid, O., Lebbé, C., et al. (2009). Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clinical Cancer Research, 15(23), 7412–7420. https://doi.org/10.1158/1078-0432.CCR-09-1624
Zhang, W., Xia, W., Lv, Z., Xin, Y., Ni, C., & Yang, L. (2017). Liquid biopsy for cancer: Circulating tumor cells, circulating free DNA or exosomes? Cellular Physiology and Biochemistry, 41(2), 755–768. https://doi.org/10.1159/000458736
Zhang, Y., Mudgal, P., Wang, L., Wu, H., Huang, N., Alexander, P. B., et al. (2020). T cell receptor repertoire as a prognosis marker for heat shock protein peptide complex-96 vaccine trial against newly diagnosed glioblastoma. Oncoimmunology, 9(1), 1749476. https://doi.org/10.1080/2162402X.2020.1749476
Zviran, A., Schulman, R. C., Shah, M., Hill, S. T. K., Deochand, S., Khamnei, C. C., et al. (2020). Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nature Medicine, 26(7), 1114–1124. https://doi.org/10.1038/s41591-020-0915-3
Author information
Authors and Affiliations
Contributions
Conceptualization: DM, CB, ML; literature search and data analysis: DM, SS, and TK. The first draft of the manuscript was written by DM, SS, and TK. All authors commented, critically revised, and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mathios, D., Srivastava, S., Kim, T. et al. Emerging Technologies for Non-invasive Monitoring of Treatment Response to Immunotherapy for Brain Tumors. Neuromol Med 24, 74–87 (2022). https://doi.org/10.1007/s12017-021-08677-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-021-08677-9