
Abstract
Introduction
Glioblastoma is a very aggressive cancer with dismal prognosis despite standard of care including surgical resection, radiation therapy, and chemotherapy. There is interest in applying immunotherapy to glioblastoma as this modality has demonstrated remarkable improvements in the management of several solid tumors including melanoma, renal cell carcinoma, and non-small cell lung cancer. This review aims to provide an overview of the current state of glioblastoma immunotherapy.
Methods
Literature search was performed on PubMed between 1961 and 2020.
Results
Initial clinical trials of checkpoint inhibitors and vaccine therapy for glioblastoma have largely been disappointing for both primary and recurrent glioblastoma. This failure has been attributed to glioblastoma’s highly immunosuppressive environment and multiple mechanisms of therapy resistance including high tumor heterogeneity, low mutational burden, systemic immunosuppression, and local immune dysfunction.
Conclusions
Current clinical trials are exploring combination therapy and novel treatment strategies beyond immune checkpoint therapies and vaccine therapy such as CAR T cells. There is also an effort to establish synergy between immunotherapy and current standard of care. Furthermore, recent advances in personalized neoantigen vaccines suggest a shift towards personalized, patient-specific GBM treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The promise of immunotherapy
The field of cancer immunotherapy arose from the concept of cancer immunosurveillance, first conceived by William Coley in the 1890 s, followed by Ehrlich, and then Thomas and Burnet in the 1950s and 1960s [1,2,3,4]. Cancer immunosurveillance is the notion that the immune system can actively detect and eliminate tumor cells. However, some tumor cells do survive and develop the ability to evade the immune system through a process of immunoediting [5]. Cancer immunotherapy aims to overcome the immunoresistance of tumor cells to promote tumor eradication. This strategy has shown great promise in recent years especially since the development of immune checkpoint inhibitors (ICIs) [6].
Immune checkpoints are an intrinsic feature of the immune system designed to maintain self-tolerance [7]. Cancer cells can exploit this feature by upregulating immune checkpoint pathways such as programmed cell death protein-1 (PD-1)/programmed death ligand-1 (PD-L1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) to prevent an effective anti-tumor immune response. ICIs have revolutionized cancer care for certain solid tissue malignancies and have invigorated immunotherapy research for cancer. ICIs are monoclonal antibodies that block immune checkpoint pathways and prevent tumors from down-regulating the immune response [7]. Anti-PD-1 monoclonal antibodies have shown to improve survival in hepatocellular carcinoma, non-small cell lung cancer (NSCLC), renal cell carcinoma (RCC), melanoma, urothelial cancer, as well as a variety of other solid tumors [8,9,10,11,12,13,14,15]. Anti-CTLA-4 has shown to have a survival benefit for metastatic melanoma and is currently in clinical trials for other tumors including NSCLC, RCC, and prostate cancer [16,17,18].
Despite these promising results, it is important to note that ICIs do not work for all solid tumors, the response rate is low for some tumors, and there are serious associated toxicities. Haslam et al. published a retrospective cross-sectional study in 2019 studying the response rate of six anti-CTLA-4 or anti-PD-1 ICIs [19]. It was found that in 2018, only an estimated 43.6% of cancer patients were eligible for immunotherapy, and the predicted immunotherapy response rate was 12.46% in 2018 with significant variability among cancers. It should be noted that response rate varies significantly with patient characteristics and tumor pathology. Phase III randomized clinical trials between 2015 and 2019 reported response rates between 19 and 60% with the higher response rates observed in trials studying immunotherapy in carefully selected patients and in combination with other treatments such as chemotherapy. [9, 12, 20,21,22]. These varying response rates suggest that there are many roadblocks to effective immunotherapy for solid tumors, and not all patients benefit from this strategy currently. Furthermore, immunotherapy is associated with significant toxicity. Magee et al. conducted a retrospective review of 12,727 patients across 22 studies between 2014 and 2019 and found that grade 3 or greater adverse events were reported in 16.5% of patients that received immunotherapy [23]. It is evident that immunotherapy has revolutionized our management of certain cancers such as melanoma, but it is important to bear in mind that immunotherapy does not benefit all cancer types equally. This review will discuss the past failures of immunotherapy for GBM, approaches currently under investigation, and strategies that hold future promise.
Disappointing initial results with GBM immunotherapy
There is interest in applying immunotherapy to glioblastoma (GBM) given its poor prognosis. The median overall survival (mOS) for GBM is approximately 19 months despite standard of care which includes maximal surgical resection, temozolomide (TMZ) chemotherapy, and radiation therapy (RT) [24, 25]. Despite standard of care, recurrence is common for which treatment options are limited [26].
Immune checkpoint inhibitors
ICIs such as anti-PD-1 have been extensively studied for GBM, given their promising results in many other solid tumors. The first major human clinical trial with anti-PD-1 was CheckMate 143. Phase I safety data from 20 patients with primary GBM showed a tolerable safety profile with only grade 1 or 2 toxicity with anti-PD-1 alone, but 8 of 10 patients in the combination arm of anti-PD-1 plus anti-CTLA-4 had grade 3 or 4 adverse events, leading to discontinuation in 5 of these patients [27]. Another study of the CheckMate 143 trial reported on preliminary safety data from 40 patients with recurrent GBM being treated with anti-PD-1 alone or anti-PD-1 and anti-CTLA-4. Adverse events leading to discontinuation occurred in 10% of patients in the anti-PD-1 only arm and in 20–30% of patients in the anti-PD-1 plus anti-CTLA-4 arms [28]. These studies suggested anti-PD-1 monotherapy is better tolerated than dual ICI therapy, and the dual ICI therapy arm was closed for this trial.
The phase III data from CheckMate 143 have not yet been published in a peer-reviewed journal. Unpublished preliminary data presented at the 2017 World Federation of Neuro-Oncology Societies on anti-PD-1 versus bevacizumab for recurrent GBM showed no significant difference in survival between the two treatment arms (9.8 months with anti-PD-1 vs. 10 months with bevacizumab). Safety analysis from the combination of anti-PD-1 and radiation therapy (RT) with and without temozolomide (TMZ) showed these combinations are well tolerated. Thus, these combinations are still being studied in the ongoing phase III clinical trials Checkmate 498 (NCT02617589) and Checkmate 548 (NCT02667587) [29]. CheckMate 498 is evaluating anti-PD-1 as an alternative to TMZ both in combination with RT in patients with newly-diagnosed O-6-methylguanine-DNA methyltransferase (MGMT)-unmethylated GBM. Although data from CheckMate 498 is unpublished at this time, in May 2019, Bristol-Myers Squibb (BMS) announced that CheckMate 498 did not meet its primary endpoint of overall survival on final analysis [30]. BMS noted that it will complete a full evaluation of the data from this trial and work with clinical investigators on future presentations and publication of the trial results. CheckMate 548 evaluated anti-PD-1 in addition to TMZ plus RT versus TMZ plus RT only in newly diagnosed MGMT-methylated GBM patients. Although data from this trial are also unpublished at this time, BMS announced in September 2019 that CheckMate 548 did not meet one of its primary endpoints, progression-free survival [31]. This trial will continue as planned to allow for the other primary endpoint, overall survival, to mature. To summarize, in unselected patients, there does not appear to be a clear benefit for single checkpoint inhibitor therapy. Therefore, there is a need to develop a better understanding of GBM immunosuppression and determine which patients respond best to immunotherapy.
Vaccine therapy
Another immunotherapy strategy of great interest in GBM is anti-tumor vaccines. The hope is that strengthening the adaptive immune system via vaccination may promote successful anti-tumor responses against GBM. Rindopepimut is a peptide vaccine strategy that targets EGFR variant III (EGFRvIII), a constitutively active mutant EGFR only expressed on GBM cells in 25–30% of patients [32]. The fact that EGFRvIII is only expressed on GBM cells limits off-target toxicity. However, the disadvantage of this approach is that not all patients’ tumors express EGFRvIII, and only the patients with this specific variant would be candidates for this vaccine. Moreover, it is heterogeneously expressed in the tumors that do harbor this variant. In phase II clinical studies, rindopepimut was evaluated in GBM patients following gross total resection and chemoradiotherapy. Of note, mOS was 24 months, a modest improval over historical controls [33,34,35]. Given these encouraging results, rindopepimut was further evaluated in the multicenter phase III trial ACT IV. Patients in this trial were randomized to either rindopepimut or control in combination with TMZ after meeting the predefined criteria for enrollment which included minimal residual disease defined as presence of less than 2 cm2 of contrast-enhancing tumor tissue after surgery and chemoradiotherapy [36]. This study was prematurely terminated after pre-planned interim analysis showed no benefit to treatment (mOS was 20.1 in the rindopepimut arm and 20.0 months in the control arm, p = 0.93). While the ACT IV trial was ongoing, a phase II trial known as ReACT was conducted to explore the efficacy of rindopepimut plus bevacizumab in 72 patients with recurrent GBM [37]. This trial found a mOS of 12.0 months with rindopepimut plus bevacizumab compared to 8.8 months with bevacizumab plus vaccine control (p = 0.0208). Overall, these studies suggest rindopepimut may have some activity in a small, carefully selected cohort of recurrent GBM patients, but further studies are required to determine the optimal patient population and treatment regimen.
Another vaccine therapy to reach phase III clinical trials is ICT-107, a multi-peptide vaccine specifically designed for GBM. ICT-107 consists of ex vivo incubation of patient dendritic cells with six peptides found to be over-represented in the gene-expression profile of GBM cells. A phase I study in 17 newly diagnosed GBM patients and 3 recurrent GBM patients confirmed an acceptable safety profile [38]. A phase II study determined some efficacy, at least in HLA-A2 positive patients [39]. A phase III trial was initiated given this promising preliminary data, but was terminated due to insufficient funding. Likewise, DCVax-L, a dendritic cell-based vaccine therapy, also reached phase III clinical trials after showing an acceptable safety profile on phase I testing. Initial results from the phase III trial found that the 331 patients in the intent-to-treat population had a mOS of 23.1 months and a 2.1% grade 3 or 4 adverse event rate [40]. However, this trial was subsequently put on hold indefinitely for unidentified reasons (NCT00045968).
Obstacles in GBM immunotherapy
To date, phase III clinical studies with ICIs and vaccine therapy for GBM have been disappointing. One explanation for these prominent failures is that GBM induces significant systemic and intra-tumoral immunosuppression [41]. Clinicians hoped to overcome this immunosuppression using immunotherapy strategies. However, given the lackluster early results, GBM immunosuppression has proven to be more complex and multi-factorial than initially understood. Indeed, there is a need to better understand the mechanisms of GBM immunosuppression as it could provide insight into effective immunotherapy strategies for GBM [42].
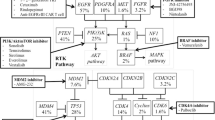
GBM has been described as a heterogeneous tumor, and while our understanding of GBM heterogeneity is still premature, it is thought that this heterogeneity contributes to immunotherapy resistance [43, 44]. The Cancer Genome Atlas Network initially classified GBM into four subtypes based on gene expression analysis: classical, neural, proneural, and mesenchymal [45]. This original classification scheme included transcriptomic analysis of non-tumoral cells in the analysis [46]. Thus, a revised classification scheme after excluding analysis of non-tumoral cells has defined three GBM sub-types: classical, mesenchymal, and proneural [47]. Each subtype has been found to have a distinct gene expression profile, and early proof-of-concept pre-clinical work in a murine GBM xenograft model demonstrates a differential response of each subtype to TMZ and RT [48]. Furthermore, GBM phenotypic plasticity was evaluated in 91 matched patient samples taken prior to treatment and at recurrence. In 55% of patients, subtype of recurrent GBM was different from the subtype of the primary GBM [47]. In addition to this intertumoral heterogeneity, it has been found that GBM also has significant heterogeneity even within the same tumor specimen. Sottoriva et al. analyzed spatially distinct tumor fragments from 11 GBM patients and demonstrated different GBM subtypes within the same tumor [49]. The heterogeneity of GBM results in resistance to treatment modalities including immunotherapy as treatment-resistant clones can allow for persistence of the tumor following targeted elimination of treatment-sensitive clones [50, 51].
Another barrier to mounting an effective anti-tumor immune response in GBM is its low mutational burden [52]. Somatic mutations that accumulate over the course of tumor development lead to the generation of neo-antigens, or novel tumor-specific mutant antigens capable of being recognized by the immune system and evoking a CD8 + T cell mediated anti-tumor response [53]. Goodman et al. showed that higher tumor mutational burden is an independent predictor of immunotherapy response across a large variety of non-CNS tumor types [54]. Furthermore, several case studies have shown patients with hypermutated GBM demonstrate clinical and radiographic response after anti-PD-1 therapy [55, 56]. The low mutational burden of GBM suggests that this cancer has fewer neoantigens available to trigger an anti-tumor immune response. Of note, Indraccolo et al. evaluated GBM samples collected at initial diagnosis and at recurrence for expression of mismatch repair (MMR) proteins. They found that MMR protein expression was partially or completely lost in 25.9% of recurrent GBM samples, and less than 5% of primary GBM samples. The tumor specimens with MMR loss had increased tumor mutational burden [57]. Despite these reports, ICIs as monotherapy for unselected recurrent GBM patients have not shown clinical benefit. Therefore, selecting patients with MMR defects in their tumor may be better candidates for certain immunotherapeutic strategies.
It has been shown in both pre-clinical models and human patients that GBM induces both local immune dysfunction and systemic immunosuppression [58,59,60,61,62,63]. The systemic immune system plays a vital role in mounting an effective immune response within the central nervous system (CNS) [43]. The CNS does have native immune cells, microglia, which differentiate from naïve myeloid cells that migrate to the CNS during fetal development [64]. However, the role of microglia in adaptive immunity is not entirely clear. Alone, microglia are not sufficient to mount an immune response in the CNS. Trafficking of peripheral immune cells across the BBB and into the CNS, mediated by interferon-inducible chemokines in response to inflammation, allows for potent immune responses in the CNS [65]. Systemic immunosuppression in GBM patients prevents the effective trafficking of peripheral immune cells in to the brain tumor.
Several mechanisms have been described for GBM systemic immunosuppression. The presence of GBM has been associated with sequestration of immune cells in the bone marrow. Chongsathidkiet et al. reported that GBM patients and pre-clinical GBM models demonstrated sequestration of T cells in the bone marrow [66]. This existing immune dysfunction is exacerbated by treatment including RT, TMZ and steroids. Hyperfractionated radiation has been shown to contribute to severe systemic immunosuppression. Grossman et al. recorded CD4 + T cell counts before initiating TMZ and hyperfractionated RT in newly-diagnosed GBM patients [67]. They found that the median CD4 T cell count was 664 cells/mm3 prior to treatment initiation, and fell to below 300 cells/mm3 in over 70% of patients 2 months after starting treatment. Patients with CD4 T cell counts below 200 cells/mm3 at 2 months after treatment initiation had significantly shorter survival than patients with greater than 200 cells/mm3 (13.1 months vs 19.7 months, p = 0.002) [67]. In this study, it should be noted that TMZ and dexamethasone could also contribute to lymphopenia. The authors subsequently conducted a study in pancreatic cancer patients and found that stereotactic body radiation had less severe lymphopenia than hyperfractionated radiation therapy [68]. Although this study has not been repeated in GBM, it highlights the lympho-depleting effect of hyperfractionated radiation therapy. Based on these results, studies are evaluating the efficacy of hypo-fractionated radiation therapy for GBM [69]. TMZ has also been shown to negatively impact immunotherapy. Pre-clinical work by Mathios et al. demonstrated that systemic TMZ induces immunosuppression, abrogates the effects of anti-PD-1 therapy in a murine GBM model, and prevents the formation of effective memory T cells [70]. Thus, it could be that immunotherapy is not as effective for GBM as TMZ may be dampening the effect of immunotherapy through iatrogenic immunosuppression. It would be useful to evaluate in future clinical trials the synergy between local chemotherapy and immunotherapy.
Another potential cause of iatrogenic systemic immunosuppression is corticosteroid use, common in GBM patients to control cerebral edema. Giles et al. evaluated lymphocyte proliferation, differentiation, and cytokine production during dexamethasone usage in a murine GBM model. They found that dexamethasone treatment lead to CTLA-4-mediated decrease in naïve T cells proliferation and differentiation [71]. Maxwell et al. found that corticosteroid treatment severely diminished peripheral CD4 + and CD8 + T cell counts. Furthermore, they found that corticosteroid use diminished the efficacy of anti-PD-1 therapy in mice bearing peripheral tumors, but not in mice bearing intra-cranial tumors [72]. The contrasting findings between these two studies suggests that the impact of corticosteroid use on GBM immunotherapy is not fully understood, and clinicians should carefully consider steroid use in patients receiving immunotherapy.
In addition to systemic immunosuppression, GBM’s local immune dysfunction also hampers anti-tumor immune response. One aspect of local immune dysfunction is upregulation of intratumoral regulatory T (Treg) cells. Preclinical GBM models have shown that Treg cells increase within 10 days of brain tumor implantation [73, 74]. Locally, it has been found that there is a high proportion of Treg cells in the tumor microenvironment. It is thought that the upregulation of Treg cells in GBM is, atleast in-part, mediated through tumoral release of the cytokine TGF-beta and upregulation of the enzyme indoleamine 2,3-dioxygenase (IDO) [75,76,77]. Heimberger et al. measured the incidence of Treg cells in 135 glial tumors and found that while Treg cells were rarely present in normal brain tissue, they were significantly more present in many glial tumors, and most frequently in glioblastoma [78]. Fecci et al. found that the increased Treg fraction in GBM patients correlates with impaired patient effector T-cell responsiveness in vitro [62]. Lohr et al. demonstrated a positive correlation between effector T cell infiltration of the tumor and survival in GBM patients [79].
An important facet of local immune dysfunction is tumor-associated macrophages (TAMs), which are abundantly expressed in the GBM tumor microenvironment (TME) [80]. Macrophages ingress into the tumor in response to inflammation-mediated chemokines [80]. GBM polarizes these macrophages toward the anti-inflammatory M2 phenotype via metabolites such as kynurenine [81]. These M2-poliarized TAMs contribute to tumor progression through several mechanisms including promotion of genetic instability, suppressing adaptive immunity via expression of immune checkpoint molecules, and supporting cancer stem cells [82]. Bloch et al. found that peripheral blood macrophages from GBM patients had increased PD-L1 expression, the ligand for the immune checkpoint PD-1 [59]. In vitro, these macrophages suppressed T cell activation [59]. Given the abundance of myeloid cells in the TME, therapies targeting this population of cells may prove beneficial for GBM patients.
A hallmark of GBM local immune dysfunction is T cell exhaustion. T cell exhaustion is a state of functional impairment induced by recurrent or prolonged antigen exposure [83, 84]. Woroniecka et al. characterized the T cell exhaustion signature in several tumors by analyzing the TIL and peripheral blood lymphocytes of GBM patients. They found that GBM induces severe T cell exhaustion characterized by upregulation of multiple immune checkpoints such as PD-1, TIM-3, LAG-3, and CTLA-4 [85]. The PD-1 immune checkpoint has been the target of several phase III clinical trial, however, as described above, results thus far have been disappointing. The study by Woroniecka et al. observed that T cells expressing multiple immune checkpoints were more dysfunctional than T cells only expressing the PD-1 checkpoint [85]. This suggests that targeting one immune checkpoint may not be enough, and effective GBM immunotherapy may require combinatorial therapy targeting multiple immune checkpoints.
Current immunotherapy strategies
The characterization of GBM as highly immunosuppressive with multiple mechanisms of immune evasion suggests that targeting only a single immunosuppressive pathway may not improve patient outcomes. Thus, recent immunotherapy strategies place an emphasis on combinatorial strategies that can synergize together to overcome GBM immunoresistance. ICIs are increasingly being studied in a combinatorial context with other therapies.
CheckMate 143 did include an arm of patients who recieved anti-PD-1 and anti-CTLA-4 combination therapy, however, the toxicity was higher with combination therapy and this arm was subsequently discontinued [28]. Several phase I trials are evaluating the safety profile of various dual ICI combinations for GBM patients. The phase I trial NCT02311920 is evaluating anti-CTLA-4 and or anti-PD-1 in combination with temozolomide for patients with newly-diagnosed GBM or gliosarcoma. The phase I trial NCT02794883 is evaluating the safety of anti-CTLA-4 antibody and anti-PD-L1 antibody in recurrent GBM patients. CD-27 is another immune checkpoint as Chahlavi et al. demonstrated that GBM induces T cell apoptosis via binding of the CD70 ligand on tumor cells to the CD27 receptor on T cells [86]. Thus, the phase I/II dose escalation study NCT02335918 is evaluating the combination of anti-CD-27 and anti-PD-1 in patients with various advanced solid tumors including GBM. LAG-3 is an immune checkpoint typically expressed on exhausted T cells [85]. The phase I clinical trial NCT02658981 is evaluating anti-LAG-3 alone and in combination with anti-PD-1 in patients with recurrent GBM. Additionally, IDO has also been characterized as an immune checkpoint. IDO has been shown to play a role in upregulating Treg cells in the tumor microenvironment [77]. In a preclinical murine GBM model, it has been shown that anti-IDO in combination with anti-CTLA-4 and anti-PD-1 is more potent at eradicating GBM than monotherapy alone [87]. Thus, NCT02327078 is evaluating the safety of anti-IDO drug in combination with either anti-CTLA-4 or anti-PD-1 in various tumors including GBM. These clinical trials are summarized below in Table 1.
A novel immunotherapy strategy being explored is the combination of neoadjuvant and adjuvant anti-PD-1 monotherapy with surgical resection. Cloughesy evaluated neoadjuvant and/or adjuvant anti-PD-1 in 35 patients with recurrent, surgically resectable GBM [88]. They found that patients who received neoadjuvant and adjuvant anti-PD-1 had higher overall survival than patients that only received adjuvant anti-PD-1. Similarly, in the phase II trial NCT02550249, Schalper et al. administered neoadjuvant anti-PD-1 and then adjuvant anti-PD-1 after surgical resection of GBM in 30 patients, 27 with recurrent GBM, and 3 with newly-diagnosed [89]. They found that neoadjuvant anti-PD-1 enhanced chemokine transcript expression, increased TCR clonal diversity among TILs and increased overall immune cell infiltration of the tumor, however, no clinical benefit was observed in this study.
Several studies are also studying synergy between radiation therapy and immunotherapy. Evidence for synergy between radiation and immunotherapy arose from the abscopal effect, characterized by Mole et al. in 1953 [90]. The abscopal effect refers to shrinkage of untreated metastasis after local radiation treatment [90]. It is thought that radiation induces tumor necrosis and antigen release, allowing for increased antigen presentation, and a more robust anti-tumor immune response [91]. Additionally, it is thought that radiation may increase the mutational burden of GBM, allowing for the development of more neoantigens that can be recognized by the immune system [92]. Grossman et al. had previously demonstrated that hyperfractionated radiation therapy induces significant immunosuppression, so there is hope that hypofractionated radiation therapy may synergize better with immunotherapy. Pouessel et al. evaluated hypofractionated stereotactic radiotherapy (HFSRT) with anti-PD-1 and found this combination was well-tolerated in the 6 patients studied [93]. NCT0289931 is a phase 1 trial evaluating the safety of HFSRT with anti-PD-1, anti-CTLA-4, and bevacizumab in patients with recurrent GBM. NCT02313272 is a phase 1 trial studying HFSRT with anti-PD-1 and bevacizumab for recurrent high grade glioma. NCT02530502 is evaluating the safety of RT with TMZ and anti-PD-1 in patients with newly-diagnosed GBM. Along the lines of synergy between immunotherapy and radiological approaches to GBM treatment, another interesting combination under evaluation is synergy between anti-PD-1 and MRI-guided laser ablation (MLA) in recurrent GBM (NCT02311582). The aim of MLA is to disrupt the blood–brain barrier, increasing access of tumor antigens to the lymphatic drainage system and immune cells to the brain tumor.
Other immunotherapeutic strategies include oncolytic viruses. Oncolytic viruses infect tumor cells and activates the innate immune system through pattern recognition receptors and pathogen-associated molecular patterns [42]. Once activated, myeloid cells from the innate immune system can upregulate T cell trafficking to the tumor, leading to a stronger anti-tumor immune response [42, 94]. Several early-phase clinical trials are investigating adenovirus, herpes simplex virus, and poliovirus-based oncolytic virus therapies in GBM patients (NCT02798406, NCT0219169, NCT02457845, NCT02062827). The phase III trial ASPECT evaluated administration of the inactivated adenovirus sitimagene ceradenovac with standard of care versus standard of care alone in GBM patients [95]. The ASPECT trial found no difference in mOS, however there was an increased time to death or re-intervention in the cohort that received sitimagene ceradenovac. Toca 5 is a current phase III trial investigating the oncolytic virus Vocimagene amiretrorepvec versus standard of care in recurrent GBM (NCT01470794) after initial results from a phase I dose-escalation showed an acceptable safety profile and a 21.7% durable response rate among 56 patients [96, 97]. Several pre-clinical studies in murine GBM have shown a synergistic benefit to combining oncolytic virus therapy with anti-PD1. As a result, this combination is also being explored in human patients [98,99,100]. The phase II clinical trial NCT02798406 is evaluating the adenovirus-based therapy DNX-2401 in combination with anti-PD-1 for recurrent GBM.
Chimeric antigen receptor (CAR) T cell therapy is a newer strategy which has been approved for B cell lymphoma and leukemia, and is currently being investigated in GBM [101]. CAR T cells are genetically modifying T cells harvested from the patient. These modified CAR T cells are then adoptively transferred back into the patient to elicit an anti-tumor immune response. The CAR T cell consists of an extracellular tumor-specific antigen recognition domain and a T cell activation domain which can be modified to keep the T cell constitutively active [102]. Brown et al. published a case report in which a recurrent GBM patient was treated with CAR T cells against the tumor-specific antigen IL13Rα2 [103]. The patient demonstrated significant clinical and radiographic response, although recurrence occurred 7.5 months after treatment started. O’Rourke et al. evaluated CAR T cells directed against the antigen EGFRvIII in 10 patients with recurrent GBM [104]. This therapy was found to have an acceptable safety profile, and CAR T cell infiltration of the tumor was observed. Unfortunately, this study found no survival benefit to CAR T cell therapy. A phase I trial evaluated a HER-2-targeted CAR T cell therapy and established an acceptable safety profile with 1 of 16 patients achieving partial response and 7 patients demonstrating stable disease for 8 weeks to 29 months [105]. Currently, one of the biggest hindrances to CAR T cell therapy is the heterogeneity in GBM making it difficult to develop a CAR T cell therapy that can target all of the clonal populations [106]. CAR T cells that can recognizer multiple antigens are in development. Pre-clinical data suggests tri-valent CAR T cells that can target the tumor-specific antigens HER2, IL13Rα2, and EphA2 may be more efficacious than bi-valent or mono-valent CAR T cells [107].
Given the abundance of TAMs in the GBM microenvironment, there is an interest in pursuing combinatorial immunotherapies that target TAMs [108]. NCT02526017 is a phase I trial evaluating an anti-CSF-1R monoclonal antibody in combination with anti-PD-1 in several cancer types including malignant glioma. CSF-1R is a receptor expressed on myeloid cells that allows for recruitment of TAMs to the tumor microenvironment [109, 110]. The phase 1/II study NCT02829723 is evaluating another anti-CSF-1R agent in combination with anti-PD-1.
Future of GBM immunotherapy
Several trends are emerging in GBM immunotherapy based on our developing understanding of GBM pathophysiology. The first is a focus on combinatorial treatment strategies. It is becoming more evident that single agent immunotherapy may not be enough to overcome GBM’s potent immunosuppression. Therefore, many of the current studies are evaluating combinatorial strategies to find synergy between different immunotherapy strategies as well as between immunotherapy and the current standards of care for GBM: surgical resection, TMZ, and RT.
The second exciting trend is a focus on personalized treatment. The enormous heterogeneity of GBM and individual patient differences makes it difficult to establish one treatment that provides maximal benefit in all GBM patients. Thus, there is an effort to tailor therapy to each individual patient’s unique tumor genetic profile. Personalized neoantigen vaccine therapy was first developed in melanoma patients with very encouraging results [111,112,113]. Briefly, a personalized neoantigen vaccine is developed via an immunogenomics pipeline by first subjecting patient tumor cells and normal cells to whole exome sequencing and RNA sequencing to determine expressed mutations. Next, MHCI prediction algorithms are used to rank candidate neoantigens. The highest ranked neoantigens can be synthesized to generate a neoantigen vaccine [114]. Two phase I trials have been conducted to explore a personalized patient-specific vaccine approach in GBM. Hilf et al. developed personalized vaccines for 15 GBM patients based on tumor transcriptomic and immunopeptidome analysis [115]. Their approach had an acceptable safety profile and elicited a sustained memory CD8 + T cell response as a well as an effector CD4 + T cell response against predicted neoepitopes. Keskin et al. conducted a similar phase I study in which they generated personalized neoantigen-targeting vaccines by comparing whole exome sequencing data and RNA-seq data between the tumor sample and healthy, normal tissue. This study found that patients had neoantigen-specific CD4 + and CD8 + T cells that could migrate into the tumor and elicit an immune response [116]. Based on these studies, NCT03422094 and NCTR02297428 are phase I trials evaluating the safety of a neoantigen-based personalized vaccine in combination with RT or immune checkpoint blockade in GBM patients [114, 117].
Immunotherapy has revolutionized cancer treatment for a variety of solid tumors. There is hope that immunotherapy can also transform the treatment of GBM, a devastating disease with dismal prognosis. Thus far, this has not been the case. The poor response to immunotherapy in GBM is attributed to several factors including high tumor heterogeneity and multiple mechanisms of immunosuppression. Although the results of initial clinical trials are disappointing, they have aided our understanding of how GBM immunosuppression works, and currently on-going trials are building upon the lessons learned from previous trials.
References
Burnet M (1957) Cancer: a biological approach: III. Viruses associated with neoplastic conditions: IV. practical applications. Br Med J 1(5023):841–847. https://doi.org/10.1136/bmj.1.5023.841
Burnet FM (1970) The concept of immunological surveillance. Prog Exp Tumor Res 13:1–27
Thomas L (1961) Cellular and humoral aspects of the hypersensitive states. Acta Medica Scandinavica 170(1):128–128. https://doi.org/10.1111/j.0954-6820.1961.tb00220.x
Dunn GP, Old LJ, Schreiber RD (2004) The immunobiology of cancer immunosurveillance and immunoediting. Immunity 21(2):137–148. https://doi.org/10.1016/j.immuni.2004.07.017
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD (2002) Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3(11):991–998. https://doi.org/10.1038/ni1102-991
Ribas A, Wolchok JD (2018) Cancer immunotherapy using checkpoint blockade. Science 359(6382):1350–1355. https://doi.org/10.1126/science.aar4060
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12(4):252–264. https://doi.org/10.1038/nrc3239
Paz-Ares L, Horn L, Borghaei H, Spigel DR, Steins M, Ready N, Chow LQM, Vokes EE, Felip E, Holgado E, Barlesi F, Kohlhaeufl M, Rodriguez O, Burgio MA, Fayette J, Gettinger SN, Harbison C, Dorange C, Finckenstein FG, Brahmer JR (2015) Phase III, randomized trial (CheckMate 057) of nivolumab (NIVO) versus docetaxel (DOC) in advanced non-squamous cell (non-SQ) non-small cell lung cancer (NSCLC). J Clin Oncol 33((18_suppl)):LBA109–LBA109. https://doi.org/10.1200/jco.2015.33.18_suppl.lba109
Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, Castellano D, Choueiri TK, Gurney H, Donskov F, Bono P, Wagstaff J, Gauler TC, Ueda T, Tomita Y, Schutz FA, Kollmannsberger C, Larkin J, Ravaud A, Simon JS, Xu LA, Waxman IM, Sharma P (2015) Nivolumab versus everolimus in advanced renal-cell carcinoma. New Engl J Med 373(19):1803–1813. https://doi.org/10.1056/NEJMoa1510665
Sharma P, Retz M, Siefker-Radtke A, Baron A, Necchi A, Bedke J, Plimack ER, Vaena D, Grimm M-O, Bracarda S, Arranz JÁ, Pal S, Ohyama C, Saci A, Qu X, Lambert A, Krishnan S, Azrilevich A, Galsky MD (2017) Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol 18(3):312–322. https://doi.org/10.1016/S1470-2045(17)30065-7
El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, Kim T-Y, Choo S-P, Trojan J, Welling TH, Meyer T, Kang Y-K, Yeo W, Chopra A, Anderson J, dela Cruz C, Lang L, Neely J, Tang H, Dastani HB, Melero I (2017) Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. The Lancet 389(10088):2492–2502. https://doi.org/10.1016/S0140-6736(17)31046-2
Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH, Lao CD, Linette GP, Thomas L, Lorigan P, Grossmann KF, Hassel JC, Maio M, Sznol M, Ascierto PA, Mohr P, Chmielowski B, Bryce A, Svane IM, Grob J-J, Krackhardt AM, Horak C, Lambert A, Yang AS, Larkin J (2015) Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 16(4):375–384. https://doi.org/10.1016/S1470-2045(15)70076-8
Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J, Hermes B, Cay Senler F, Csoszi T, Fulop A, Rodriguez-Cid J, Wilson J, Sugawara S, Kato T, Lee KH, Cheng Y, Novello S, Halmos B, Li X, Lubiniecki GM, Piperdi B, Kowalski DM (2018) Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. New Engl J Med 379(21):2040–2051. https://doi.org/10.1056/NEJMoa1810865
Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, Hodi FS, Schachter J, Pavlick AC, Lewis KD, Cranmer LD, Blank CU, O'Day SJ, Ascierto PA, Salama AKS, Margolin KA, Loquai C, Eigentler TK, Gangadhar TC, Carlino MS, Agarwala SS, Moschos SJ, Sosman JA, Goldinger SM, Shapira-Frommer R, Gonzalez R, Kirkwood JM, Wolchok JD, Eggermont A, Li XN, Zhou W, Zernhelt AM, Lis J, Ebbinghaus S, Kang SP, Daud A (2015) Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol 16(8):908–918. https://doi.org/10.1016/S1470-2045(15)00083-2
Fuchs CS, Doi T, Jang RW, Muro K, Satoh T, Machado M, Sun W, Jalal SI, Shah MA, Metges J-P, Garrido M, Golan T, Mandala M, Wainberg ZA, Catenacci DV, Ohtsu A, Shitara K, Geva R, Bleeker J, Ko AH, Ku G, Philip P, Enzinger PC, Bang Y-J, Levitan D, Wang J, Rosales M, Dalal RP, Yoon HH (2018) Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 trialpembrolizumab in advanced gastric and gastroesophageal junction cancerpembrolizumab in advanced gastric and gastroesophageal junction cancer. JAMA Oncol 4 (5): e180013-e180013. doi:10.1001/jamaoncol.2018.0013
McDermott D, Haanen J, Chen TT, Lorigan P, O'Day S (2013) Efficacy and safety of ipilimumab in metastatic melanoma patients surviving more than 2 years following treatment in a phase III trial (MDX010-20). Ann Oncol: Off J Eur Soc Med Oncol 24(10):2694–2698. https://doi.org/10.1093/annonc/mdt291
Weber J, Thompson JA, Hamid O, Minor D, Amin A, Ron I, Ridolfi R, Assi H, Maraveyas A, Berman D, Siegel J, O'Day SJ (2009) A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res: Off J Am Assoc Cancer Res 15(17):5591–5598. https://doi.org/10.1158/1078-0432.ccr-09-1024
Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ (2010) Improved survival with ipilimumab in patients with metastatic melanoma. New Engl J Med 363(8):711–723. https://doi.org/10.1056/NEJMoa1003466
Haslam A, Prasad V (2019) Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open 2(5):e192535–e192535. https://doi.org/10.1001/jamanetworkopen.2019.2535
Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, Savage KJ, Hernberg MM, Lebbe C, Charles J, Mihalcioiu C, Chiarion-Sileni V, Mauch C, Cognetti F, Arance A, Schmidt H, Schadendorf D, Gogas H, Lundgren-Eriksson L, Horak C, Sharkey B, Waxman IM, Atkinson V, Ascierto PA (2015) Nivolumab in previously untreated melanoma without BRAF mutation. New Engl J Med 372(4):320–330. https://doi.org/10.1056/NEJMoa1412082
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, Barlesi F, Kohlhaufl M, Arrieta O, Burgio MA, Fayette J, Lena H, Poddubskaya E, Gerber DE, Gettinger SN, Rudin CM, Rizvi N, Crino L, Blumenschein GR Jr, Antonia SJ, Dorange C, Harbison CT, Graf Finckenstein F, Brahmer JR (2015) Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. New Engl J Med 373(17):1627–1639. https://doi.org/10.1056/NEJMoa1507643
Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, Blank CU, Hamid O, Mateus C, Shapira-Frommer R, Kosh M, Zhou H, Ibrahim N, Ebbinghaus S, Ribas A (2015) Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 372(26):2521–2532. https://doi.org/10.1056/NEJMoa1503093
Magee DE, Hird AE, Klaassen Z, Sridhar SS, Nam RK, Wallis CJD, Kulkarni GS (2020) Adverse event profile for immunotherapy agents compared with chemotherapy in solid organ tumors: a systematic review and meta-analysis of randomized clinical trials. Ann Oncol: Off J Eur Soc Med Oncol 31(1):50–60. https://doi.org/10.1016/j.annonc.2019.10.008
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl J Med 352(10):987–996. https://doi.org/10.1056/NEJMoa043330
Di Carlo DT, Cagnazzo F, Benedetto N, Morganti R, Perrini P (2019) Multiple high-grade gliomas: epidemiology, management, and outcome: a systematic review and meta-analysis. Neurosurg Rev 42(2):263–275. https://doi.org/10.1007/s10143-017-0928-7
Cohen MH, Shen YL, Keegan P, Pazdur R (2009) FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 14(11):1131–1138. https://doi.org/10.1634/theoncologist.2009-0121
Sampson JH, Vlahovic G, Sahebjam S, Omuro AMP, Baehring JM, Hafler DA, Voloschin AD, Paliwal P, Grosso J, Coric V, Cloughesy TF, Lim M, Reardon DA (2015) Preliminary safety and activity of nivolumab and its combination with ipilimumab in recurrent glioblastoma (GBM): CHECKMATE-143. J Clin Oncol 33((15_suppl)):3010–3010. https://doi.org/10.1200/jco.2015.33.15_suppl.3010
Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, Voloschin A, Ramkissoon SH, Ligon KL, Latek R, Zwirtes R, Strauss L, Paliwal P, Harbison CT, Reardon DA, Sampson JH (2018) Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol 20(5):674–686. https://doi.org/10.1093/neuonc/nox208
Lim M, Omuro A, Vlahovic G, Reardon DA, Sahebjam S, Cloughesy T, Baehring J, Butowski N, Potter V, Zwirtes R, Paliwal P, Carleton M, Sampson J, Brandes AA (2017) 325ONivolumab (nivo) in combination with radiotherapy (RT) ± temozolomide (TMZ): updated safety results from CheckMate 143 in pts with methylated or unmethylated newly diagnosed glioblastoma (GBM). Ann Oncol 28 (suppl_5). doi:10.1093/annonc/mdx366
, 2020. B-MSB-MSMhnbcp-rc-nb-m-s-a-p--c--s-dAJ Bristol-Myers squibb announces phase 3 CheckMate -498 study did not meet primary endpoint of overall survival with opdivo (nivolumab) plus radiation in patients with newly diagnosed MGMT-unmethylated glioblastoma multiforme
Squibb B-M Bristol-Myers squibb provides update on phase 3 opdivo (nivolumab) CheckMate -548 trial in patients with newly diagnosed MGMT-methylated glioblastoma multiforme. Bristol-Myers Squibb
Weller M, Kaulich K, Hentschel B, Felsberg J, Gramatzki D, Pietsch T, Simon M, Westphal M, Schackert G, Tonn JC, von Deimling A, Davis T, Weiss WA, Loeffler M, Reifenberger G (2014) Assessment and prognostic significance of the epidermal growth factor receptor vIII mutation in glioblastoma patients treated with concurrent and adjuvant temozolomide radiochemotherapy. Int J Cancer 134(10):2437–2447. https://doi.org/10.1002/ijc.28576
Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, Gilbert MR, Herndon JE 2nd, McLendon RE, Mitchell DA, Reardon DA, Sawaya R, Schmittling RJ, Shi W, Vredenburgh JJ, Bigner DD (2010) Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol: Off J Am Soc Clin Oncol 28(31):4722–4729. https://doi.org/10.1200/jco.2010.28.6963
Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, Mrugala MM, Jensen R, Baehring JM, Sloan A, Archer GE, Bigner DD, Cruickshank S, Green JA, Keler T, Davis TA, Heimberger AB, Sampson JH (2015) A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol 17(6):854–861. https://doi.org/10.1093/neuonc/nou348
Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, Friedman HS, Gilbert MR, Herndon JE, McLendon RE, Mitchell DA, Reardon DA, Sawaya R, Schmittling R, Shi W, Vredenburgh JJ, Bigner DD, Heimberger AB (2011) Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol 13(3):324–333. https://doi.org/10.1093/neuonc/noq157
Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, Ashby L, Mechtler L, Goldlust SA, Iwamoto F, Drappatz J, O'Rourke DM, Wong M, Hamilton MG, Finocchiaro G, Perry J, Wick W, Green J, He Y, Turner CD, Yellin MJ, Keler T, Davis TA, Stupp R, Sampson JH (2017) Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol 18(10):1373–1385. https://doi.org/10.1016/s1470-2045(17)30517-x
Reardon DA, Schuster J, Tran DD, Fink KL, Nabors LB, Li G, Bota DA, Lukas RV, Desjardins A, Ashby LS, Duic JP, Mrugala MM, Werner A, Hawthorne T, He Y, Green JA, Yellin MJ, Turner CD, Davis TA, Sampson JH, Group TRS (2015) ReACT: overall survival from a randomized phase II study of rindopepimut (CDX-110) plus bevacizumab in relapsed glioblastoma. J Clin Oncol 33((15_suppl)):2009–2009. https://doi.org/10.1200/jco.2015.33.15_suppl.2009
Phuphanich S, Wheeler CJ, Rudnick JD, Mazer M, Wang H, Nuno MA, Richardson JE, Fan X, Ji J, Chu RM, Bender JG, Hawkins ES, Patil CG, Black KL, Yu JS (2013) Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother: CII 62(1):125–135. https://doi.org/10.1007/s00262-012-1319-0
Wen PY, Reardon DA, Phuphanich S, Aiken R, Landolfi JC, Curry WT, Zhu J-J, Glantz MJ, Peereboom DM, Markert J, LaRocca RV, O'Rourke D, Fink KL, Kim LJ, Gruber ML, Lesser GJ, Pan E, Kesari S, Hawkins ES, Yu J (2014) A randomized, double-blind, placebo-controlled phase 2 trial of dendritic cell (DC) vaccination with ICT-107 in newly diagnosed glioblastoma (GBM) patients. J Clin Oncol 32((15_suppl)):2005–2005. https://doi.org/10.1200/jco.2014.32.15_suppl.2005
Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, Heth JA, Salacz M, Taylor S, D'Andre SD, Iwamoto FM, Dropcho EJ, Moshel YA, Walter KA, Pillainayagam CP, Aiken R, Chaudhary R, Goldlust SA, Bota DA, Duic P, Grewal J, Elinzano H, Toms SA, Lillehei KO, Mikkelsen T, Walbert T, Abram SR, Brenner AJ, Brem S, Ewend MG, Khagi S, Portnow J, Kim LJ, Loudon WG, Thompson RC, Avigan DE, Fink KL, Geoffroy FJ, Lindhorst S, Lutzky J, Sloan AE, Schackert G, Krex D, Meisel H-J, Wu J, Davis RP, Duma C, Etame AB, Mathieu D, Kesari S, Piccioni D, Westphal M, Baskin DS, New PZ, Lacroix M, May S-A, Pluard TJ, Tse V, Green RM, Villano JL, Pearlman M, Petrecca K, Schulder M, Taylor LP, Maida AE, Prins RM, Cloughesy TF, Mulholland P, Bosch ML (2018) First results on survival from a large phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med 16(1):142–142. https://doi.org/10.1186/s12967-018-1507-6
Razavi S-M, Lee KE, Jin BE, Aujla PS, Gholamin S, Li G (2016) Immune evasion strategies of glioblastoma. Front Surg 3 (11). doi:10.3389/fsurg.2016.00011
Lim M, Xia Y, Bettegowda C, Weller M (2018) Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol 15(7):422–442. https://doi.org/10.1038/s41571-018-0003-5
Jackson CM, Choi J, Lim M (2019) Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol 20(9):1100–1109. https://doi.org/10.1038/s41590-019-0433-y
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suva ML, Regev A, Bernstein BE (2014) Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344(6190):1396–1401. https://doi.org/10.1126/science.1254257
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O'Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17(1):98–110. https://doi.org/10.1016/j.ccr.2009.12.020
Sidaway P (2017) CNS cancer: Glioblastoma subtypes revisited. Nat Rev Clin Oncol 14(10):587. https://doi.org/10.1038/nrclinonc.2017.122
Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, deCarvalho AC, Lyu S, Li P, Li Y, Barthel F, Cho HJ, Lin Y-H, Satani N, Martinez-Ledesma E, Zheng S, Chang E, Sauvé C-EG, Olar A, Lan ZD, Finocchiaro G, Phillips JJ, Berger MS, Gabrusiewicz KR, Wang G, Eskilsson E, Hu J, Mikkelsen T, DePinho RA, Muller F, Heimberger AB, Sulman EP, Nam D-H, Verhaak RGW (2017) Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32(1):42–56.e46. https://doi.org/10.1016/j.ccell.2017.06.003
Teo W-Y, Sekar K, Seshachalam P, Shen J, Chow W-Y, Lau CC, Yang H, Park J, Kang S-G, Li X, Nam D-H, Hui KM (2019) Relevance of a TCGA-derived glioblastoma subtype gene-classifier among patient populations. Sci Rep 9(1):7442. https://doi.org/10.1038/s41598-019-43173-y
Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavare S (2013) Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA 110(10):4009–4014. https://doi.org/10.1073/pnas.1219747110
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suvà ML, Regev A, Bernstein BE (2014) Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344(6190):1396–1401. https://doi.org/10.1126/science.1254257
Mahlokozera T, Vellimana AK, Li T, Mao DD, Zohny ZS, Kim DH, Tran DD, Marcus DS, Fouke SJ, Campian JL, Dunn GP, Miller CA, Kim AH (2017) Biological and therapeutic implications of multisector sequencing in newly diagnosed glioblastoma. Neuro-Oncol 20(4):472–483. https://doi.org/10.1093/neuonc/nox232
Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, Huse JT, de Groot J, Li S, Overwijk WW, Spetzler D, Heimberger AB (2017) Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol 19(8):1047–1057. https://doi.org/10.1093/neuonc/nox026
Gubin MM, Artyomov MN, Mardis ER, Schreiber RD (2015) Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest 125(9):3413–3421. https://doi.org/10.1172/jci80008
Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA, Kurzrock R (2017) Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther 16(11):2598–2608. https://doi.org/10.1158/1535-7163.MCT-17-0386
Johanns TM, Miller CA, Dorward IG, Tsien C, Chang E, Perry A, Uppaluri R, Ferguson C, Schmidt RE, Dahiya S, Ansstas G, Mardis ER, Dunn GP (2016) Immunogenomics of Hypermutated Glioblastoma: A patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov 6(11):1230–1236. https://doi.org/10.1158/2159-8290.cd-16-0575
Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, Durno C, Krueger J, Cabric V, Ramaswamy V, Zhukova N, Mason G, Farah R, Afzal S, Yalon M, Rechavi G, Magimairajan V, Walsh MF, Constantini S, Dvir R, Elhasid R, Reddy A, Osborn M, Sullivan M, Hansford J, Dodgshun A, Klauber-Demore N, Peterson L, Patel S, Lindhorst S, Atkinson J, Cohen Z, Laframboise R, Dirks P, Taylor M, Malkin D, Albrecht S, Dudley RWR, Jabado N, Hawkins CE, Shlien A, Tabori U (2016) Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol 34(19):2206–2211. https://doi.org/10.1200/JCO.2016.66.6552
Indraccolo S, Lombardi G, Fassan M, Pasqualini L, Giunco S, Marcato R, Gasparini A, Candiotto C, Nalio S, Fiduccia P, Fanelli GN, Pambuku A, Della Puppa A, D'Avella D, Bonaldi L, Gardiman MP, Bertorelle R, De Rossi A, Zagonel V (2019) Genetic, epigenetic, and immunologic profiling of MMR-deficient relapsed glioblastoma. Clin Cancer Res 25(6):1828–1837. https://doi.org/10.1158/1078-0432.ccr-18-1892
Roszman T, Elliott L, Brooks W (1991) Modulation of T-cell function by gliomas. Immunol Today 12(10):370–374. https://doi.org/10.1016/0167-5699(91)90068-5
Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT (2013) Gliomas promote immunosuppression through induction of B7–H1 expression in tumor-associated macrophages. Clin Cancer Res: Official J Am Assoc Cancer Res 19(12):3165–3175. https://doi.org/10.1158/1078-0432.ccr-12-3314
Chae M, Peterson TE, Balgeman A, Chen S, Zhang L, Renner DN, Johnson AJ, Parney IF (2015) Increasing glioma-associated monocytes leads to increased intratumoral and systemic myeloid-derived suppressor cells in a murine model. Neuro Oncol 17(7):978–991. https://doi.org/10.1093/neuonc/nou343
Elliott LH, Brooks WH, Roszman TL (1987) Activation of immunoregulatory lymphocytes obtained from patients with malignant gliomas. J Neurosurg 67(2):231–236. https://doi.org/10.3171/jns.1987.67.2.0231
Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, Herndon JE 2nd, Bigner DD, Dranoff G, Sampson JH (2006) Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Can Res 66(6):3294–3302. https://doi.org/10.1158/0008-5472.can-05-3773
Dunn GP, Fecci PE, Curry WT (2012) Cancer immunoediting in malignant glioma. Neurosurgery 71 (2):201–222; discussion 222-203. doi:10.1227/NEU.0b013e31824f840d
Hutter G, Theruvath J, Graef CM, Zhang M, Schoen MK, Manz EM, Bennett ML, Olson A, Azad TD, Sinha R, Chan C, Assad Kahn S, Gholamin S, Wilson C, Grant G, He J, Weissman IL, Mitra SS, Cheshier SH (2019) Microglia are effector cells of CD47-SIRPalpha antiphagocytic axis disruption against glioblastoma. Proc Natl Acad Sci USA 116(3):997–1006. https://doi.org/10.1073/pnas.1721434116
Klein RS, Izikson L, Means T, Gibson HD, Lin E, Sobel RA, Weiner HL, Luster AD (2004) IFN-Inducible protein 10/CXC chemokine ligand 10-independent induction of experimental autoimmune encephalomyelitis. J Immunol 172(1):550–559. https://doi.org/10.4049/jimmunol.172.1.550
Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, Woroniecka K, Elsamadicy AA, Dechant CA, Kemeny HR, Sanchez-Perez L, Cheema TA, Souders NC, Herndon JE, Coumans JV, Everitt JI, Nahed BV, Sampson JH, Gunn MD, Martuza RL, Dranoff G, Curry WT, Fecci PE (2018) Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med 24(9):1459–1468. https://doi.org/10.1038/s41591-018-0135-2
Grossman SA, Ye X, Lesser G, Sloan A, Carraway H, Desideri S, Piantadosi S (2011) Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin Cancer Res: Off J Am Assoc Cancer Res 17(16):5473–5480. https://doi.org/10.1158/1078-0432.ccr-11-0774
Wild AT, Herman JM, Dholakia AS, Moningi S, Lu Y, Rosati LM, Hacker-Prietz A, Assadi RK, Saeed AM, Pawlik TM, Jaffee EM, Laheru DA, Tran PT, Weiss MJ, Wolfgang CL, Ford E, Grossman SA, Ye X, Ellsworth SG (2016) Lymphocyte-sparing effect of stereotactic body radiation therapy in patients with unresectable pancreatic cancer. Int J Radiat Oncol Biol Phys 94(3):571–579. https://doi.org/10.1016/j.ijrobp.2015.11.026
Liao G, Zhao Z, Yang H, Li X (2019) Efficacy and safety of hypofractionated radiotherapy for the treatment of newly diagnosed glioblastoma multiforme: a systematic review and meta-analysis. Front Oncol 9 (1017). doi:10.3389/fonc.2019.01017
Mathios D, Kim JE, Mangraviti A, Phallen J, Park CK, Jackson CM, Garzon-Muvdi T, Kim E, Theodros D, Polanczyk M, Martin AM, Suk I, Ye X, Tyler B, Bettegowda C, Brem H, Pardoll DM, Lim M (2016) Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci Transl Med 8 (370):370ra180. doi:10.1126/scitranslmed.aag2942
Giles AJ, Hutchinson M-KND, Sonnemann HM, Jung J, Fecci PE, Ratnam NM, Zhang W, Song H, Bailey R, Davis D, Reid CM, Park DM, Gilbert MR (2018) Dexamethasone-induced immunosuppression: mechanisms and implications for immunotherapy. J Immunother Cancer 6(1):51. https://doi.org/10.1186/s40425-018-0371-5
Maxwell R, Luksik AS, Garzon-Muvdi T, Hung AL, Kim ES, Wu A, Xia Y, Belcaid Z, Gorelick N, Choi J, Theodros D, Jackson CM, Mathios D, Ye X, Tran PT, Redmond KJ, Brem H, Pardoll DM, Kleinberg LR, Lim M (2018) Contrasting impact of corticosteroids on anti-PD-1 immunotherapy efficacy for tumor histologies located within or outside the central nervous system. Oncoimmunology 7(12):e1500108. https://doi.org/10.1080/2162402x.2018.1500108
Sugihara AQ, Rolle CE, Lesniak MS (2009) Regulatory T cells actively infiltrate metastatic brain tumors. Int J Oncol 34(6):1533–1540. https://doi.org/10.3892/ijo_00000282
Kennedy BC, Maier LM, D'Amico R, Mandigo CE, Fontana EJ, Waziri A, Assanah MC, Canoll P, Anderson RCE, Anderson DE, Bruce JN (2009) Dynamics of central and peripheral immunomodulation in a murine glioma model. BMC Immunol 10(1):11. https://doi.org/10.1186/1471-2172-10-11
Ueda R, Fujita M, Zhu X, Sasaki K, Kastenhuber ER, Kohanbash G, McDonald HA, Harper J, Lonning S, Okada H (2009) Systemic inhibition of transforming growth factor-beta in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin Cancer Res: Off J Am Assoc Cancer Res 15(21):6551–6559. https://doi.org/10.1158/1078-0432.ccr-09-1067
Kaminska B, Kocyk M, Kijewska M (2013) TGF beta signaling and its role in glioma pathogenesis. Adv Exp Med Biol 986:171–187. https://doi.org/10.1007/978-94-007-4719-7_9
Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon K-S, Auffinger B, Tobias AL, Han Y, Lesniak MS (2012) IDO Expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res 18(22):6110–6121. https://doi.org/10.1158/1078-0432.ccr-12-2130
Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, Yang DS, Sun W, Qiao W, Hiraoka N, Fuller GN (2008) Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin Cancer Res: Off J Am Assoc Cancer Res 14(16):5166–5172. https://doi.org/10.1158/1078-0432.ccr-08-0320
Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, Grau S, Hiraoka N, Eckstein V, Ecker RC, Korff T, von Deimling A, Unterberg A, Beckhove P, Herold-Mende C (2011) Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin Cancer Res: Off J Am Assoc Cancer Res 17(13):4296–4308. https://doi.org/10.1158/1078-0432.ccr-10-2557
Chen Z, Hambardzumyan D (2018) Immune microenvironment in glioblastoma subtypes. Front Immunol 9:1004. https://doi.org/10.3389/fimmu.2018.01004
Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao CC, Gutierrez-Vazquez C, Kenison J, Tjon EC, Barroso A, Vandeventer T, de Lima KA, Rothweiler S, Mayo L, Ghannam S, Zandee S, Healy L, Sherr D, Farez MF, Prat A, Antel J, Reardon DA, Zhang H, Robson SC, Getz G, Weiner HL, Quintana FJ (2019) Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci 22(5):729–740. https://doi.org/10.1038/s41593-019-0370-y
Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P (2017) Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 14(7):399–416. https://doi.org/10.1038/nrclinonc.2016.217
Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R (1998) Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 188(12):2205–2213. https://doi.org/10.1084/jem.188.12.2205
Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R (2003) Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol 77(8):4911–4927. https://doi.org/10.1128/jvi.77.8.4911-4927.2003
Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, Elsamadicy AA, Cui X, Koyama S, Jackson C, Hansen LJ, Johanns TM, Sanchez-Perez L, Chandramohan V, Yu YA, Bigner DD, Giles A, Healy P, Dranoff G, Weinhold KJ, Dunn GP, Fecci PE (2018) T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res: Off J Am Assoc Cancer Res 24(17):4175–4186. https://doi.org/10.1158/1078-0432.ccr-17-1846
Chahlavi A, Rayman P, Richmond AL, Biswas K, Zhang R, Vogelbaum M, Tannenbaum C, Barnett G, Finke JH (2005) Glioblastomas induce T-lymphocyte death by two distinct pathways involving gangliosides and CD70. Can Res 65(12):5428–5438. https://doi.org/10.1158/0008-5472.can-04-4395
Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, Cheng Y, Kim JW, Qiao J, Zhang L, Han Y, Lesniak MS (2014) Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res: Off J Am Assoc Cancer Res 20(20):5290–5301. https://doi.org/10.1158/1078-0432.ccr-14-0514
Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, Wang AC, Ellingson BM, Rytlewski JA, Sanders CM, Kawaguchi ES, Du L, Li G, Yong WH, Gaffey SC, Cohen AL, Mellinghoff IK, Lee EQ, Reardon DA, O'Brien BJ, Butowski NA, Nghiemphu PL, Clarke JL, Arrillaga-Romany IC, Colman H, Kaley TJ, de Groot JF, Liau LM, Wen PY, Prins RM (2019) Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med 25(3):477–486. https://doi.org/10.1038/s41591-018-0337-7
Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA, Inogés S, de Andrea C, López-Diaz de Cerio A, Tejada S, Berraondo P, Villarroel-Espindola F, Choi J, Gúrpide A, Giraldez M, Goicoechea I, Gallego Perez-Larraya J, Sanmamed MF, Perez-Gracia JL, Melero I (2019) Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med 25(3):470–476. https://doi.org/10.1038/s41591-018-0339-5
Mole RH (1953) Whole body irradiation; radiobiology or medicine? Br J Radiol 26(305):234–241. https://doi.org/10.1259/0007-1285-26-305-234
Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, Camphausen K, Luiten RM, de Ru AH, Neijssen J, Griekspoor A, Mesman E, Verreck FA, Spits H, Schlom J, van Veelen P, Neefjes JJ (2006) Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med 203(5):1259–1271. https://doi.org/10.1084/jem.20052494
Germano G, Lamba S, Rospo G, Barault L, Magri A, Maione F, Russo M, Crisafulli G, Bartolini A, Lerda G, Siravegna G, Mussolin B, Frapolli R, Montone M, Morano F, de Braud F, Amirouchene-Angelozzi N, Marsoni S, D'Incalci M, Orlandi A, Giraudo E, Sartore-Bianchi A, Siena S, Pietrantonio F, Di Nicolantonio F, Bardelli A (2017) Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 552(7683):116–120. https://doi.org/10.1038/nature24673
Pouessel D, Mervoyer A, Larrieu-Ciron D, Cabarrou B, Attal J, Robert M, Frenel J-S, Olivier P, Poublanc M, Mounier M, Moyal E (2018) Hypofractionnated stereotactic radiotherapy and anti-PDL1 durvalumab combination in recurrent glioblastoma: results of the phase I part of the phase I/II STERIMGLI trial. J Clin Oncol 36((15_suppl)):2046–2046. https://doi.org/10.1200/JCO.2018.36.15_suppl.2046
Martikainen M, Essand M (2019) Virus-based immunotherapy of glioblastoma Cancers (Basel) 11:11
Westphal M, Yla-Herttuala S, Martin J, Warnke P, Menei P, Eckland D, Kinley J, Kay R, Ram Z (2013) Adenovirus-mediated gene therapy with sitimagene ceradenovec followed by intravenous ganciclovir for patients with operable high-grade glioma (ASPECT): a randomised, open-label, phase 3 trial. Lancet Oncol 14(9):823–833. https://doi.org/10.1016/s1470-2045(13)70274-2
Cloughesy TF, Landolfi J, Vogelbaum MA, Ostertag D, Elder JB, Bloomfield S, Carter B, Chen CC, Kalkanis SN, Kesari S, Lai A, Lee IY, Liau LM, Mikkelsen T, Nghiemphu P, Piccioni D, Accomando W, Diago OR, Hogan DJ, Gammon D, Kasahara N, Kheoh T, Jolly DJ, Gruber HE, Das A, Walbert T (2018) Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro Oncol 20(10):1383–1392. https://doi.org/10.1093/neuonc/noy075
Cloughesy TF, Landolfi J, Hogan DJ, Bloomfield S, Carter B, Chen CC, Elder JB, Kalkanis SN, Kesari S, Lai A, Lee IY, Liau LM, Mikkelsen T, Nghiemphu PL, Piccioni D, Walbert T, Chu A, Das A, Diago OR, Gammon D, Gruber HE, Hanna M, Jolly DJ, Kasahara N, McCarthy D, Mitchell L, Ostertag D, Robbins JM, Rodriguez-Aguirre M, Vogelbaum MA (2016) Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci Transl Med 8(341):341–375. https://doi.org/10.1126/scitranslmed.aad9784
Cockle JV, Rajani K, Zaidi S, Kottke T, Thompson J, Diaz RM, Shim K, Peterson T, Parney IF, Short S, Selby P, Ilett E, Melcher A, Vile R (2016) Combination viroimmunotherapy with checkpoint inhibition to treat glioma, based on location-specific tumor profiling. Neuro Oncol 18(4):518–527. https://doi.org/10.1093/neuonc/nov173
Jiang H, Rivera-Molina Y, Gomez-Manzano C, Clise-Dwyer K, Bover L, Vence LM, Yuan Y, Lang FF, Toniatti C, Hossain MB, Fueyo J (2017) Oncolytic adenovirus and tumor-targeting immune modulatory therapy improve autologous cancer vaccination. Can Res 77(14):3894–3907. https://doi.org/10.1158/0008-5472.can-17-0468
Chen CY, Hutzen B, Wedekind MF, Cripe TP (2018) Oncolytic virus and PD-1/PD-L1 blockade combination therapy. Oncol Virother 7:65–77. https://doi.org/10.2147/ov.s145532
Maus MV, Grupp SA, Porter DL, June CH (2014) Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 123(17):2625–2635. https://doi.org/10.1182/blood-2013-11-492231
Finney HM, Akbar AN, Lawson AD (2004) Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol 172(1):104–113. https://doi.org/10.4049/jimmunol.172.1.104
Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, Kurien A, Priceman SJ, Wang X, Harshbarger TL, D'Apuzzo M, Ressler JA, Jensen MC, Barish ME, Chen M, Portnow J, Forman SJ, Badie B (2016) Regression of glioblastoma after chimeric antigen receptor T-cell therapy. New Engl J Med 375(26):2561–2569. https://doi.org/10.1056/NEJMoa1610497
O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, Isaacs R, Mohan S, Plesa G, Lacey SF, Navenot JM, Zheng Z, Levine BL, Okada H, June CH, Brogdon JL, Maus MV (2017) A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 9(399):984. https://doi.org/10.1126/scitranslmed.aaa0984
Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, Robertson C, Gray TL, Diouf O, Wakefield A, Ghazi A, Gerken C, Yi Z, Ashoori A, Wu M-F, Liu H, Rooney C, Dotti G, Gee A, Su J, Kew Y, Baskin D, Zhang YJ, New P, Grilley B, Stojakovic M, Hicks J, Powell SZ, Brenner MK, Heslop HE, Grossman R, Wels WS, Gottschalk S (2017) HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol 3(8):1094–1101. https://doi.org/10.1001/jamaoncol.2017.0184
Fecci PE, Sampson JH (2019) The current state of immunotherapy for gliomas: an eye toward the future. J Neurosurg 131(3):657–666. https://doi.org/10.3171/2019.5.jns181762
Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, Wu M-F, Orange JS, Sumazin P, Man T-K, Joseph SK, Hegde M, Ahmed N (2017) Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology 20(4):506–518. https://doi.org/10.1093/neuonc/nox182
Ding AS, Routkevitch D, Jackson C, Lim M (2019) Targeting myeloid cells in combination treatments for glioma and other tumors. Front Immunol 10:1715. https://doi.org/10.3389/fimmu.2019.01715
Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, Segall JE (2012) Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med (Cambridge, Mass) 18:519–527. https://doi.org/10.2119/molmed.2011.00217
Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, Setty M, Leslie CS, Oei Y, Pedraza A, Zhang J, Brennan CW, Sutton JC, Holland EC, Daniel D, Joyce JA (2013) CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 19(10):1264–1272. https://doi.org/10.1038/nm.3337
Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, Chen C, Olive O, Carter TA, Li S, Lieb DJ, Eisenhaure T, Gjini E, Stevens J, Lane WJ, Javeri I, Nellaiappan K, Salazar AM, Daley H, Seaman M, Buchbinder EI, Yoon CH, Harden M, Lennon N, Gabriel S, Rodig SJ, Barouch DH, Aster JC, Getz G, Wucherpfennig K, Neuberg D, Ritz J, Lander ES, Fritsch EF, Hacohen N, Wu CJ (2017) An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547(7662):217–221. https://doi.org/10.1038/nature22991
Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, Linette GP (2015) Cancer immunotherapy: a dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348(6236):803–808. https://doi.org/10.1126/science.aaa3828
Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrors B, Omokoko T, Vormehr M, Albrecht C, Paruzynski A, Kuhn AN, Buck J, Heesch S, Schreeb KH, Muller F, Ortseifer I, Vogler I, Godehardt E, Attig S, Rae R, Breitkreuz A, Tolliver C, Suchan M, Martic G, Hohberger A, Sorn P, Diekmann J, Ciesla J, Waksmann O, Bruck AK, Witt M, Zillgen M, Rothermel A, Kasemann B, Langer D, Bolte S, Diken M, Kreiter S, Nemecek R, Gebhardt C, Grabbe S, Holler C, Utikal J, Huber C, Loquai C, Tureci O (2017) Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547(7662):222–226. https://doi.org/10.1038/nature23003
Johanns TM, Miller CA, Liu CJ, Perrin RJ, Bender D, Kobayashi DK, Campian JL, Chicoine MR, Dacey RG, Huang J, Fritsch EF, Gillanders WE, Artyomov MN, Mardis ER, Schreiber RD, Dunn GP (2019) Detection of neoantigen-specific T cells following a personalized vaccine in a patient with glioblastoma. OncoImmunology 8(4):e1561106. https://doi.org/10.1080/2162402X.2018.1561106
Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C, Platten M, Tabatabai G, Dutoit V, van der Burg SH, Thor Straten P, Martinez-Ricarte F, Ponsati B, Okada H, Lassen U, Admon A, Ottensmeier CH, Ulges A, Kreiter S, von Deimling A, Skardelly M, Migliorini D, Kroep JR, Idorn M, Rodon J, Piro J, Poulsen HS, Shraibman B, McCann K, Mendrzyk R, Lower M, Stieglbauer M, Britten CM, Capper D, Welters MJP, Sahuquillo J, Kiesel K, Derhovanessian E, Rusch E, Bunse L, Song C, Heesch S, Wagner C, Kemmer-Bruck A, Ludwig J, Castle JC, Schoor O, Tadmor AD, Green E, Fritsche J, Meyer M, Pawlowski N, Dorner S, Hoffgaard F, Rossler B, Maurer D, Weinschenk T, Reinhardt C, Huber C, Rammensee HG, Singh-Jasuja H, Sahin U, Dietrich PY, Wick W (2019) Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 565(7738):240–245. https://doi.org/10.1038/s41586-018-0810-y
Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, Shukla SA, Hu Z, Li L, Le PM, Allesoe RL, Richman AR, Kowalczyk MS, Abdelrahman S, Geduldig JE, Charbonneau S, Pelton K, Iorgulescu JB, Elagina L, Zhang W, Olive O, McCluskey C, Olsen LR, Stevens J, Lane WJ, Salazar AM, Daley H, Wen PY, Chiocca EA, Harden M, Lennon NJ, Gabriel S, Getz G, Lander ES, Regev A, Ritz J, Neuberg D, Rodig SJ, Ligon KL, Suva ML, Wucherpfennig KW, Hacohen N, Fritsch EF, Livak KJ, Ott PA, Wu CJ, Reardon DA (2019) Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565(7738):234–239. https://doi.org/10.1038/s41586-018-0792-9
Johanns TM, Bowman-Kirigin JA, Liu C, Dunn GP (2017) Targeting neoantigens in glioblastoma: an overview of cancer immunogenomics and translational implications. Neurosurgery 64 (CN_suppl_1):165–176. https://doi.org/10.1093/neuros/nyx321
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The corresponding author receives research support from Arbor, BMS, Accuray, DNAtrix, Tocagen, Biohaven, and Kyrin-Kyowa. He is a consultant for Tocagen, VBI, and Stryker.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Medikonda, R., Dunn, G., Rahman, M. et al. A review of glioblastoma immunotherapy. J Neurooncol 151, 41–53 (2021). https://doi.org/10.1007/s11060-020-03448-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-020-03448-1