
Abstract
Alzheimer’s disease (AD) is characterized neuropathologically by the deposition of different forms of amyloid β-protein (Aβ) including variable amounts of soluble species that correlate with severity of dementia. The extent of synaptic loss in the brain provides the best morphological correlate of cognitive impairment in clinical AD. Animal research on the pathophysiology of AD has therefore focussed on how soluble Aβ disrupts synaptic mechanisms in vulnerable brain regions such as the hippocampus. Synapic plasticity in the form of persistent activity-dependent increases or decreases in synaptic strength provide a neurophysiological substrate for hippocampal-dependent learning and memory. Acute treatment with human-derived or chemically prepared soluble Aβ that contains certain oligomeric assemblies, potently and selectively disrupts synaptic plasticity causing inhibition of long-term potentiation (LTP) and enhancement of long-term depression (LTD) of glutamatergic transmission. Over time these and related actions of Aβ have been implicated in reducing synaptic integrity. This review addresses the involvement of neurotransmitter intercellular signaling in mediating or modulating the synaptic plasticity disrupting actions of soluble Aβ, with particular emphasis on the different roles of glutamatergic and cholinergic mechanisms. There is growing evidence to support the view that NMDA and possibly nicotinic receptors are critically involved in mediating the disruptive effect of Aβ and that targeting muscarinic receptors can indirectly modulate Aβ’s actions. Such studies should help inform ongoing and future clinical trials of drugs acting through the glutamatergic and cholinergic systems.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In this review, we restrict our discussions largely to the evaluating recent research investigating the effects of amyloid-β protein (Aβ) on excitatory synaptic transmission and plasticity of that transmission in the brain. We focus particularly on neurotransmitter intercellular signaling mechanisms that have already been implicated in providing potential therapeutic effects in patients with Alzheimer’s disease (AD). Previous reviews have covered a large literature on other mechanisms including intracellular and pro-inflammatory pathways (Turner et al. 2003; Lynch 2004; Pena et al. 2006; Rowan et al. 2007; Arendt, 2009).
Amyloid Cascade Hypotheses—from Fibrils to Oligomers and from Neurodegeneration to Synaptic Failure
The amyloid cascade hypothesis of AD, as initially formulated, proposed that the hallmark progressive deposition of insoluble fibrillar Aβ in plaques triggered neurodegeneration which in turn caused the insidious escalation of debilitating symptoms, including progression through the different stages of clinical dementia. Support for this proposal came from the discovery that application of fibril-containing Aβ to cultured neurons was highly toxic in vitro (Lorenzo and Yankner 1996) and that intracerebral injection of fibril-containing Aβ caused a delayed neurodegeneration-associated disruption of performance of cognitive tasks in animals (McDonald et al. 1994; Nitta et al. 1994; Maurice et al. 1996; Stephan et al. 2001). However, the relatively poor correlation between the severity of clinical dementia at the time of death of patients with AD and either the magnitude of fibrillar Aβ load or the extent of neuron loss in the brain provided a major challenge for the original amyloid cascade hypothesis (Roth et al. 1966; Terry, 1996). The hypothesis was substantially revised with the discovery of much stronger correlations between cognitive status and (i) synaptic density rather than neuron loss and (ii) the levels of soluble rather than fibrillar Aβ (Terry 1996; Lue et al. 1999; McLean et al. 1999; Wang et al. 1999). Strong support for a revised amyloid hypothesis incorporating these findings came from reports that certain forms of soluble Aβ can trigger synaptic pruning in cultured neurons and brain slices in vitro (Roselli et al. 2005; Shankar et al. 2007) and cause cognitive impairment in the absence of neurodegeneration in animals (Cleary et al. 2005; Lesne et al. 2006; Haass and Selkoe 2007). Current research investigating the relative importance of the various soluble Aβ assembly states in causing cognitive deficits has emphasized the importance of both low-n oligomers (such as dimers and trimers) and larger oligomers (including some that may form globular structures independent of Aβ aggregation into fibrils).
Vulnerable Networks—Entorhinal Cortex/Hippocampal Pathways
As AD progresses, extensive disruption of connectivity throughout the cortex and many subcortical areas occurs; two of the earliest areas affected are the hippocampus and entorhinal cortex which form a network that is essential for the normal function of episodic memory, thus providing an explanation for why memory problems which rely on this network are a very early and core symptom of AD. This mnemonic function is thought to require a continuous comparison of incoming integrated perceptual content via the entorhinal cortex with information and related predictive schemata initially stored/generated in the hippocampus. The network’s circuitry is mainly comprised of glutamatergic neurons and synapses, which are under tight control from intrinsic GABA-ergic inhibitory interneurons and external inputs including cholinergic neurons. Extensive deposition of Aβ is associated with the disruption of glutamatergic synapses in this network at an early stage of AD (Reitz et al. 2009). Such marked and early deposition of Aβ may be at least partly the result of the relatively high excitatory drive through the network, since Aβ aggregation in the brain has been found to driven by activity at these synapses (Deshpande et al. 2009).
Glutamatergic Mechanisms—Effects of Aβ
Given the initial emphasis of the amyloid cascade hypothesis on neurodegeneration, much early research focused on the ability of Aβ to increase excitotoxicity mediated through glutamate receptors, especially N-methyl-D-aspartate receptors (NMDARs) (Greenamyre and Young 1989; Koh et al. 1990; Lawlor and Davis 1992; Mattson et al. 1992; Hynd et al. 2004). Consistent with these reports, relatively low doses of Aβ were found to exacerbate delayed cognitive impairment caused by activation of NMDARs (Dornan et al. 1993; Nakamura et al. 2006). Possible mechanisms for the Aβ-mediated enhanced excitotoxicity include the ability of Aβ to reduce glutamate uptake (Harris et al. 1995; Keller et al. 1997; Harkany et al. 2000; Fernandez-Tome et al. 2004; Matos et al. 2008), or to increase glutamate release (Arias et al. 1995; Noda et al. 1999; Bobich et al. 2004; Chin et al. 2007; Kabogo et al. 2008; Puzzo et al. 2008).
The important role of glutamatergic mechanisms and in particular NMDARs in causing clinical dementia in AD received validation when the low affinity open channel NMDA receptor antagonist memantine (Lipton 2007; Parsons et al. 2007) was licenced for the treatment of patients. Combined with the realization that mechanisms other than neurodegeneration contribute significantly to the cognitive symptoms of AD, glutamatergic transmission and plasticity of that transmission, rather than solely excitotoxicity, have become a major focus of interest.
Plasticity of Glutamatergic Synaptic Transmission—Disruption of Long-term Potentiation and Long-term Depression by Aβ
Synaptic plasticity mechanisms, including those underlying long-term potentiation (LTP) and long-term depression (LTD) of glutamatergic transmission, provide a neuronal substrate for learning and memory (Morris et al. 2003) and are highly vulnerable to a relatively rapid disruption by soluble Aβ species derived either by chemical synthesis (Cullen et al. 1997; Lambert et al. 1998; Kim et al. 2001; Klyubin et al. 2004; Fig. 1) or from cells that naturally secrete them after the cleavage of amyloid precursor protein by β- and γ-secretases (Walsh et al. 2002). Recent research has lent support for the key role of oligomeric assembly states, and Aβ dimers are believed to be the minimum size that interfere with synaptic plasticity. Thus, synthetic Aβ dimers prepared by covalent linkage and size exclusion chromatography are extremely potent both in vivo and in vitro whereas even relatively high concentrations of Aβ monomers are inactive (Hu et al. 2008; Shankar et al. 2008). Some larger soluble oligomers are also disruptive. Thus, a synthetic 60 kDa Aβ species that forms globular structures can inhibit LTP in hippocampal slices (Barghorn et al. 2005), but not all conformations of Aβ oligomers are active in this model (Ciccotosto et al. 2009; Harmeier et al. 2009). Importantly, human ex vivo samples of cerebrospinal fluid that contained Aβ oligomers but not monomers, potently inhibited LTP (Klyubin et al. 2008). Moreover, post mortem aqueous solutions of cerebral cortex from patients with AD that contained Aβ dimers disrupted LTP and learning (Shankar et al. 2008).

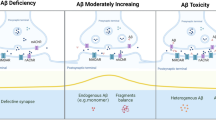
Dose-dependent effects of Aβ on functioning of CA1 glutamatergic synapses in the rat hippocampus in vivo. Anesthetized adult male Wistar rats had a cannula implanted in the lateral cerebral ventricle to enable injection of Aβ and stimulating and recording wire electrodes implanted in the dorsal hippocampus to enable recording of AMPAR-mediated excitatory glutamatergic transmission. In vehicle-injected controls (left hand panels) high frequency conditioning stimulation (arrow, 200 Hz) triggered long-term potentiation (LTP) of synaptic transmission (middle graph) whereas low frequency (bar, 3 Hz) conditioning stimulation failed to induce significant plasticity (bottom graph). In Aβ-treated animals (right hand panels), a high dose (320 pmol in 5 μl, squares), but not a low dose (80 pmol in 5 μl, circles), depresses baseline synaptic transmission (top graphs) whereas low doses selectively modulate synaptic plasticity of this transmission, inhibiting the induction of LTP by 200 Hz conditioning stimulation (80 pmol in 5 μl, middle graph) or enhancing the induction of long-term depression (LTD) by 3 Hz conditioning stimulation (1 pmol in 5 μl, bottom graph). Aβ (right hand panels) or vehicle (left hand panels) was injected intracerebroventricularly (i.c.v.) at the time indicated by the asterisk. Values are the mean ± SEM baseline field excitatory postsynaptic potential (EPSP) (partly based on data in Kim et al. 2001; Hu et al. 2008)
Amyloid β-Protein fragments also facilitate low frequency stimulation-induced LTD over a similar concentration range to that inhibiting LTP (Fig. 1). Thus, acute exposure to synthetic Aβ42 potently enabled the induction of LTD in vivo which was prevented by block of NMDA receptors (Kim et al. 2001; Cheng et al. 2009). In close similarity, cell-derived and human brain-derived Aβ that contained low-n oligomers but not monomers enhanced the induction of an LTD by a sub-threshold low frequency stimulation protocol in vitro (Shankar et al. 2008; Li et al. 2009). However, this Aβ-facilitated LTD was prevented by certain metabotropic glutamate receptor (mGluR) antagonists which can also prevent the inhibition of LTP by Aβ (Wang et al. 2004). Little is known regarding the specific mechanisms involved, but mGluRs have profound effects in regulating many forms of synaptic plasticity (Anwyl 1999; Parsons et al. 2007). Aβ was also found to reduce the sensitivity of a supra-threshold NMDA-dependent LTD to NMDAR antagonists (Li et al. 2009), an effect that was abrogated by a glutamate scavenger system and associated with an Aβ-mediated inhibition of neuronal glutamate uptake.
The extremely high potency of Aβ oligomers in disrupting synaptic plasticity has led to extensive studies into the cellular mechanisms (Pena et al. 2006; Rowan et al. 2007), including putative receptor sites (Verdier and Penke 2004; Wang et al. 2007; Origlia et al. 2008; Yang et al. 2008; Lauren et al. 2009; Yan et al. 2009).
Basal Glutamatergic Transmission
Compared to LTP and LTD, basal glutamatergic transmission through α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), the main receptors mediating fast excitatory postsynaptic potentials (EPSPs), is relatively resistant to disruption by Aβ. Given the ability of Aβ to inhibit glutamate transporters and to cause the release of glutamate, it is somewhat surprising that high concentrations of Aβ or prolonged times are required to affect basal AMPAR-mediated EPSPs (Fig. 1), generally reducing synaptic efficacy, presumably as a result of AMPAR endocytosis (Cullen et al. 1996; Almeida et al. 2005; Hsieh et al. 2006; Rowan et al. 2007; Gu et al. 2009) and receptor desensitization (Li et al. 2009). Interestingly, removal of synaptic AMPARs was reported to require NMDAR activation and LTD-like signaling mechanisms, and was found to be necessary and sufficient for Aβ-induced pruning of dendritic spines (Kamenetz et al. 2003; Hsieh et al. 2006).
Role of NMDARs in the Synaptic Actions of Aβ Oligomers
The use of the NMDAR antagonist memantine in the clinical treatment of AD is somewhat paradoxical, since many forms of learning and related plasticity are dependent on NMDAR activation. We recently tested the proposal that memantine may preferentially target disruptive over physiological NMDAR activation (Parsons et al. 2007; Lipton 2007] by comparing its effects on control NMDAR-dependent LTP and on the inhibitory action of Aβ on such LTP in vitro and in vivo (Klyubin et al. 2009). Memantine reduced the inhibition of LTP induction by Aβ both at medial perforant pathway synapses in the dentate gyrus and at CA3-to-CA1 synapses. However, there was a clear overlap between the concentration range of memantine that inhibited LTP in the absence of Aβ and the range that was protective. This partial selectivity may be analogous to the protection against the inhibition of LTP caused by low Mg2+-induced NMDAR activation afforded by memantine at concentrations that did not significantly affect control LTP (Coan et al. 1989; Frankiewicz et al. 1996; Zajaczkowski et al. 1997; Zorumski and Izumi 1998). Parsons et al. (2007) have proposed that the relative selectivity of memantine against inappropriate background activation of NMDARs is due to its low affinity because it can be rapidly removed from the channel with strong NMDAR activation, as occurs during the induction of LTP. Alternatively, or in addition, memantine may preferentially block extrasynaptic over synaptic NMDARs (Lipton 2007; Léveillé et al. 2008) or GluN2C and -2D over GluN2A and -2B subunit-containing NMDARs (Wrighton et al. 2008; Kotermanski and Johnson 2009); the underlying assumption being that the receptors targeted by memantine are more involved in mediating pathological over physiological functions. Indeed, extrasynaptic NMDARs may preferentially mediate toxic effects of glutamate (Soriano and Hardingham 1997; Lau and Zukin 2007; Zhang et al. 2007).
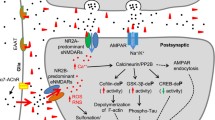
The partial protective action of memantine indicates that the inhibition of LTP by Aβ is NMDAR-dependent (Fig. 2), as is Aβ-induced Ca2+ influx into neurons (Kelly and Ferreira 2006; De Felice et al. 2007; Domingues et al. 2007). Aβ oligomers may bind to a specific site on or adjacent to the NMDAR (Cowburn et al. 1997; De Felice et al. 2007; Lacor et al. 2007). Consistent with an agonist-like action of Aβ, Aβ can selectively increase NMDA-evoked neuronal firing in vivo (Molnar et al. 2004; Szegedi et al. 2005) and rapidly selectively potentiate NMDAR-mediated synaptic currents (Wu et al. 1995a). However at the majority of pathways, Aβ causes no marked change or a reduction in NMDAR-mediated synaptic transmission, the latter being at least partly explained by removal of NMDARs from the synapse (Chen et al. 2002; Raymond et al. 2003; Snyder et al. 2005; Dewachter et al. 2009; Li et al. 2009). Interestingly, the GluN2B subunit has been implicated in Aβ’s effects on NMDARs and in regulating the localization and intracellular actions of Aβ oligomers (Roselli et al. 2005; Abbott et al. 2008; Deshpande et al. 2009; Li et al. 2009). In contrast, the GluN2A subunit may have a more important role in mediating Aβ-induced Ca2+ influx (Domingues et al. 2007).

Schematic outline of putative targets of Aβ-mediated physiological and disruptive actions on synaptic plasticity and hence synaptic integrity. Pathogenic processing of amyloid precursor protein (APP) and Aβ lead to the accumulation of Aβ oligomers which inappropriately enhance activation of certain NMDA receptors (NMDAR), possibly caused by increased extrasynaptic glutamate concentration or a close association between Aβ oligomers and NMDARs. Such actions inhibit LTP induction. Putatively, physiological processing of APP at synapses may release an unknown assembly state of Aβ to activate nicotinic acetylcholine receptors (nAChR) and thereby increase synaptic glutamate release or enhance activation of synaptic NMDARs. Such actions may lower the threshold for the induction of NMDAR-dependent LTP. There is likely to be overlap and cross-talk between these two systems, depending on Aβ concentration and glial and interneuron engagement in addition to direct neuronal targets. LTP-like mechanisms are engaged in memory processing and may, in combination with other forms of plasticity, help maintain synaptic integrity
Some of the strongest support for the hypothesis that Aβ disrupts synaptic plasticity by increasing activation of NMDARs relies on the partial reduction of the Aβ-mediated inhibition of LTP by memantine (Klyubin et al. 2009). Since memantine is not a pure NMDA receptor antagonist and because memantine reduces control LTP over a similar concentration range, caution in such an interpretation is warranted. We have commenced studies that assess the effects of more selective NMDAR antagonists on LTP induction in the absence and presence of Aβ. Our new data are consistent with a role of NMDARs in mediating the disruptive effects of Aβ.
Cholinergic Mechanisms
Decline, disruption, or alterations of cholinergic mechanisms have been implicated in a “cholinergic hypothesis” of AD (Court et al. 2001).
A major neuropathological feature of most patients with clinical AD is the loss of cholinergic neurons in the basal forebrain (Schliebs and Arendt 2006; Geula et al. 2008), and several groups have reported a selective loss of different subtypes of acetylcholine receptors (AChRs) in AD brains (Wevers et al. 2000; Teaktong et al. 2004). By modulating activity-dependent events, AChRs participate in fundamental aspects of synaptic plasticity (Albuquerque et al. 1997; Ge and Dani 2005). The loss of cholinergic projections and decline of AChRs during AD may disrupt normal cholinergic mechanisms that contribute to glutamatergic transmission and synaptic plasticity. Overall, these mechanisms may thus contribute to the cognitive decline observed during the progression of AD. Support for such a view comes from the approval of cholinesterase inhibitors in the treatment of AD and the improvement of cognition in AD patients who receive these drugs.
Cholinergic deficits produced in AD as well as in various animal AD models may be, at least, partly attributable to the suppression of cholinergic functions by Aβ peptides before cholinergic neuron loss in relevant brain areas (Blusztajn and Berse 2000). Aβ peptides negatively alter the cholinergic system at multiple sites, including ACh synthesis, acetylcholine release, and at the receptor level (Auld et al. 2002). Given the cognitive effects of cholinergic interventions, particularly, clinically used cholinesterase inhibitors, in animal models and humans, it is interesting to know if the inhibition of the induction of LTP by Aβ is mediated through disruption of cholinergic transmission. If so, modulation of either nicotinic (nAChRs) or muscarinic (mAChRs) receptors may restore LTP.
Role of nAChRs
The hippocampus is an important target for nicotinic influences over memory (Bancroft and Levin 2000; Kenney and Gould 2008). For example, nicotinic antagonists applied within the hippocampus impair memory performance (Levin et al. 2002) and memory deficits produced by lesions of cholinergic projections to the hippocampus can be reversed by nicotine (Yamazaki et al. 2002). These findings are consistent with dense nAChR expression in the hippocampus (Bourin et al. 2003) and with rich cholinergic innervation arising mainly from the medial septum and diagonal band (Woolf 1991).
Nicotine has been postulated to be a possible treatment for AD, improving cognition in humans (Newhouse et al. 2004) and attenuating Aβ-induced amnesia in mice (Maurice et al. 1996). Nicotine enhancement of LTP in vitro and in vivo was previously observed (Fujii et al. 1999; Ji et al. 2001; Mann and Greenfield 2003) and shown to be mediated via α7nAChRs (Matsuyama et al. 2000). We (Welsby et al. 2007) recently studied the effects of nicotine on Aβ-mediated inhibition of LTP. The data suggest that the effect of Aβ could be independent of a direct interaction between Aβ and nicotinic receptors, with Aβ inhibiting control high frequency stimulation (HFS)-induced LTP but not the nicotine-enhanced LTP. Evidence for a lack of an interaction between Aβ and nicotinic receptors was also supported by findings in which Aβ42 inhibition of LTP was not prevented by the selective α7nAChR antagonist methyllycaconitine (Wang et al. 2004). It was, therefore, postulated that the inhibition of HFS-LTP, but not the nicotine-enhanced LTP by Aβ, is due to the two forms of LTP having different underlying induction mechanisms. This is supported by the protein kinase A dependence of nicotine-enhanced LTP but not of HFS-LTP, even though they are both NMDAR-dependent (Welsby et al. 2007). Whereas nicotine-enhanced LTP was dependent on mGluRs and ryanodine receptor-sensitive intracellular calcium stores, but for control LTP, this did not seem to be the case (Welsby et al. 2006). Overall, both acute and chronic nicotine treatments were found to enhance LTP via α7nAChRs consistent with previous research (Matsuyama et al. 2000). Since such enhancement is not blocked by Aβ, these findings support the view that nicotinic agents activating selectively α7nAChR are promising cognitive enhancers for AD. Consistent with potential beneficial effect of nicotine, Srivareerat et al. (2009b) reported recently that 6 weeks of nicotine treatment prevented the Aβ-induced inhibition of basal synaptic transmission and LTP in the hippocampus and Aβ-induced impairment of learning and short-term memory. Interestingly, chronic nicotine also reduced the levels of Aβ40 and β-amyloid precursor protein (APP) converting enzyme (BACE) peptides in the CA1 area and prevented an Aβ-induced reduction of α7- and α4-nAChRs.
The discovery that Aβ42 binds to α7 subunit of nAChRs with high affinity suggested the potential for a role of nAChRs in AD (Oddo and LaFerla 2006). This prospect was supported by the finding that α7nAChRs were found in plaques (Wang et al. 2000), and α7nAChRs positively correlated with neurons that accumulated Aβ and hyperphosphorylated tau in AD brain tissue (Wevers et al. 1999). In a triple transgenic mouse model of AD, which expresses aspects of AD neuropathology and an age-related decline in LTP and cognition, there was a loss of α7nAChRs (Oddo and LaFerla, 2006).
Intriguingly, Aβ has been observed to act as an agonist of α7nAChR (Fodero et al. 2004) mediating the activation of the ERK2MAP kinase signaling cascade (Dineley et al. 2001; Dineley et al. 2002), while other groups have reported inhibitory actions of Aβ peptide on α7nAChR (Pettit et al. 2001; Grassi et al. 2003; Lee and Wang 2003). The different findings may be due to concentration-dependent actions of Aβ, low concentration may activate and higher concentrations desensitize α7nAChR (Dineley et al. 2002) and interacting with other nAChRs subtypes (Oddo and LaFerla 2006). Certain oligomers of Aβ may not bind nAChRs with high affinity (Lauren et al. 2009).
Evidence against a direct interaction of Aβ with α7nAChRs mediating the inhibition of LTP by Aβ was the finding that Aβ12-28, which binds α7nAChRs with high affinity, did not inhibit an α7nAChR-dependent LTP in vivo (Freir and Herron 2003). However, inhibition of an α7nAChR-dependent LTP in hippocampal slices from animals that had received a chronic infusion of Aβ40 was prevented by bath application of a selective agonist for α7nAChRs [3-(2,4-dimethoxybenzylidene)-anabaseine] (DMXB) (Chen et al. 2006). Furthermore, a novel selective α7nAChR-partial agonist which rapidly penetrates into the brain (SSR180711) was found to increase acetylcholine release, glutamatergic neurotransmission, and LTP in rat hippocampus in a dose-dependent manner (Biton et al. 2007).
In apparent contrast to these studies, recent data suggest that blocking α7nAChRs with an antagonist could lessen some of the features of Aβ-mediated deleterious effects (Martin et al. 2004; Mousavi and Hellstrom-Lindahl 2009). This proposal of blocking α7nAChR in a disease characterized for cognitive decline is controversial in view of the evidence of cognitive improvement using α7nAChR agonists (Tietje et al. 2008). However, some effects of α7nAChR agonists can be mimicked by selective α7nAChR antagonists (Ferchmin et al. 2003; Hu et al. 2007). The fast desensitization of α7nAChRs after its activation (Gay et al. 2008) makes it difficult to distinguish between agonistic and antagonistic effects of an α7nAChR targeted drug. In fact, it is not clear whether the cognitive enhancing effects are the result of receptor activation per se or activation-induced receptor desensitization. Somewhat similarly, Dziewczapolski et al. (2009) suggested that interrupting α7nAChR function could be beneficial in the treatment of AD. Using a transgenic mouse model of AD overexpressing a mutated form of the human amyloid precursor protein (APP) and lacking the α7nAChR gene (APPα7KO), they have shown that, despite the presence of high amounts of Aβ, deleting the α7nAChR subunit in the mouse model of AD lead to a protection from the dysfunction of synapses, and learning and memory behavior. Specifically, deleting the α7nAChR subunit preserved the capacity to elicit LTP otherwise deficient in the APP mice.
A link between Aβ-mediated activation of α7nAChR-induced Ca2+ influx and endocytosis of NMDARs has been demonstrated in the cortex, thus bringing together the strands of evidence implicating nAChRs and NMDARs in synaptic dysfunction (Snyder et al. 2005).
Recently, Wu et al. (2008) investigated a possible role of α4β2nAChRs in mediating the impairment of LTP by various forms of Aβ. They reported that intracerebroventricular injection of Aβ40, Aβ25–35, or Aβ31–35 significantly suppressed HFS-induced LTP. Similarly, epibatidine, a specific agonist of α4β2nAChRs, dose dependently suppressed the induction of LTP. Whereas dihydro-beta-erythroidine, a selective antagonist against α4β2 subtype of nAChRs, showed no effect on the induction of LTP, it significantly reversed Aβ31–35-induced LTP impairment, indicating that the α4β2 subtype of nAChRs is required for the suppressive action of Aβ on hippocampal LTP in vivo.
Given the cognitive enhancing effects of cholinesterase inhibitors, their ability to enhance LTP (discussed in Rowan et al. 2003), and the possibility that their enhancing effect may be mediated through nAChRs (Welsby et al. 2009), the role of agonistic and desensitizing actions at these receptors in mediating their facilitatory effects warrants investigation. Moreover, if nAChR-dependent mechanisms are engaged, repeated dosing with cholinesterase inhibitors may exert a similar facilitatory effect to repeated treatment with nicotine (Welsby et al. 2006). If and how mAChRs affect the facilitatory action of cholinesterase inhibitors also is of considerable interest.
Role of mAChRs
Aβ also exerts effects on the cholinergic system by interacting with G-protein-coupled mAChRs. It is generally believed that M2 receptors, most of which are located on presynaptic cholinergic terminals, are reduced in the brains of individuals with AD (Nordberg et al. 1992). The density of postsynaptic M1 receptors remains unaltered, but there is some evidence for disruption of the coupling between the receptors, their G-proteins and second messengers (Warpman et al. 1993).
Selective activation of mAChRs may reduce symptoms and cognitive impairments in individuals suffering from AD. For example, the M1/M4-preferring mAChR agonist xanomeline produced a robust reduction in cognitive deficits and behavioral disturbances in individuals with AD (Bodick et al. 1997). Unfortunately, the clinical effects of xanomeline and other muscarinic agents are associated with adverse side effects attributable to non-specific activation of peripheral M2 and M3 mAChRs (Bymaster et al. 1998). An alternate approach to orthosteric muscarinic agonists is targeting allosteric sites to more selectively activate the receptor by actions at a site removed from acetylcholine binding site. A novel, highly selective allosteric agonist of the M1 subtype (TBPB) was reported to potentiate the NMDAR-mediated currents in hippocampal pyramidal cells (Jones et al. 2008). Activation of M1 mAChRs can enhance NMDAR-dependent LTP (Shinoe et al. 2005).
In contrast, in the medial septum (MS), which participates in memory and learning processes via its cholinergic and GABAergic projections to the hippocampus, oligomeric Aβ-mediated depression of excitatory synaptic transmission was dependent on activation of mAChRs and voltage-dependent Ca2+ channels. Thus, perfusion of MS slices with Aβ40 reduced EPSPs and this effect was blocked by calcicludine (a selective L-type Ca2+ channel antagonist) and also by pirenzepine, a relatively selective M1-receptor antagonist (Santos-Torres et al. 2007).
Interestingly, transgenic mice overexpressing mutant APP and presenilin-1 display synaptic dysfunction which was associated with a reduction in the ability of endogenous mAChR activation to reduce basal glutamatergic transmission in the CA1 area of the hippocampus (Goto et al. 2008). Both choline acetyltransferase activity and muscarinic receptor binding is also reduced in these mice, explaining the impairment of mAChR-mediated effects (Machova et al. 2008). These results indicate that cholinergic modulation of glutamatergic transmission is already impaired at the onset of the formation of Aβ deposits, and that muscarinic receptor dysfunction is one of the causes of functional impairment.
Putative Physiological Role of Aβ in Synaptic Plasticity
Despite well-established deleterious effects of certain Aβ species, there is growing evidence that APP and its fragments, including Aβ itself, may play a role in normal neuronal functioning (Bishop and Robinson 2004; Senechal et al. 2006; Wasling et al. 2009). Indeed, APP-deficient mice show compromised neuronal function, reduction in synaptic markers and deficits in learning and memory as well as synaptic plasticity, although there is some lack of agreement as to the relative importance of different APP products and the role of compensatory changes (Muller et al. 1994; Zheng et al. 1995; Dawson et al. 1999; Phinney et al. 1999; Seabrook et al. 1999; Ring et al. 2007; Senechal et al. 2008). Somewhat similar considerations arise in the case of BACE knockout mice (Ma et al. 2007; Wang et al. 2008a). Remarkably, endogenous Aβ has been implicated even in neuronal survival in cultured neurons (Plant et al., 2003). Of particular relevance to synaptic mechanisms, Kamenetz et al. (2003) suggested that Aβ may serve as a normal negative feedback mechanism in the regulation of synaptic activity. Since several potential therapeutic approaches of AD treatment are designed to target APP, it is important to understand the physiological functions of the different APP breakdown products in synaptic plasticity.
Aβ itself may mediate mechanisms of synaptic plasticity under normal physiological conditions (Fig. 2). Firstly, studies of the direct effect of relatively low concentrations of Aβ have found that exogenous application of Aβ40 or Aβ42 can enhance hippocampal HFS-induced LTP in vitro (Wu et al. 1995b; Puzzo et al. 2008). Puzzo et al. (2008) provided evidence that the facilitation of LTP was mediated through a presynaptic enhancement of glutamate release. Secondly, as outlined above, similar low concentrations of exogenously added Aβ fragments can facilitate low frequency stimulation-induced LTD in vitro and in vivo (Kim et al. 2001; Shankar et al. 2008; Cheng et al. 2009; Li et al. 2009). Thirdly, in unpublished work we have found that intracerebroventricular administration of the anti-Aβ antibody 4G8 can inhibit HFS-induced LTP in vivo, although in this case, we cannot rule out the possibility that 4G8-mediated neutralization sAPPα is responsible for the failure to induce LTP. Indeed, sAPPα is another candidate for a physiological role in the regulation of synaptic plasticity. Exogenous application of sAPPα modulated LTD and enhanced LTP in vitro and in vivo (Ishida et al. 1997; Taylor et al. 2008).
The mechanisms underlying opposite effects of Aβ on synaptic plasticity are still poorly understood. One possible scenario is that different Aβ species act via different receptors and as a result produce different synaptic effects. Ramsden et al. (2001) found that the Aβ-induced increases in K+ currents, in cultured neurons, depend on Aβ aggregation state. In addition, it has been shown that protofibrils may activate neurons differently than fibrils (Ye et al. 2004). Moreover, an in vitro model of Aβ toxicity demonstrated that integrin-mediated polymerization of Aβ on neurons caused toxicity (Wright et al. 2007). Blocking the same (αV) integrin subunit can prevent Aβ-mediated inhibition of LTP (Wang et al. 2008b).
Another proposed explanation of the opposite synaptic effects of Aβ is that exposure to excessive levels of Aβ can turn Aβ-mediated physiological regulation to pathology (Kim and Tsai 2009). The existence of several parallels between Aβ-induced impairment of and apparent physiological regulation of synaptic plasticity supports this idea. For example, activation of NMDA receptors are implicated in inhibition of LTP (Chen et al. 2002; Wang et al. 2004; Hu et al. 2008; Klyubin et al. 2009) and the facilitation of LTD induction (Kim et al. 2001) by Aβ. Similarly, the α7nAChR has been shown to be involved in facilitation of LTP by Aβ in low concentration (Puzzo et al. 2008) and inhibition of LTP in transgenic mice overexpressing human APP (Dziewczapolski et al. 2009).
Finally, even prolonged uncontrolled augmentation of LTP by low concentrations of Aβ may lead to saturation of synaptic processes and to negative effects on learning and memory. Interestingly, increased LTP was reported in the hippocampus of mice overexpressing some forms of mutant human APP (Jolas et al. 2002), even though there is memory impairment in the same mutant mice (Janus et al. 2000; Chishti et al. 2001).
Aβ-Associated Network-level Disruption of Function
Although transgenic APP models do not easily allow the isolation of a specific action of Aβ recent research has provided a tantalizing glimpse of how Aβ may cause widespread neuronal and glial dysfunction in these models. In the hippocampal network, the activity pattern of pyramidal neurons preferentially tuned to fire depending on the spatial location of the animal in the recording arena, so-called “place-cells”, is changed dramatically such that in older animals with extensive amyloid plaques there is a degradation of the neuronal representation of the environment (Cacucci et al. 2008). In the cortex, the activity of neuronal and glial cells changes in relation to their distance from plaques in vivo, prompting the suggestion that mobile Aβ oligomers or pro-inflammatory and oxidative stress mediators may be responsible. Thus, neurons near plaques tend to be hyperactive whereas those neurons further away tend to have reduced activity, as measured by Ca2+ imaging (Busche et al. 2008). Unfortunately it was not possible to distinguish between the activity of excitatory and inhibitory neurons, although the authors provided evidence that the increased neuronal activity may be caused by increased glutamatergic drive following reduced synaptic inhibition from GABAergic inputs, rather than being due to increased intrinsic excitability. These data indicate that Aβ may not trigger a uniform reduction of synaptic activity in the cortex. Intriguingly, intercellular waves of Ca2+ are seen to spread across groups of astrocytes over relatively long distances apparently independent of neuronal activity, often starting near plaques, but with approximately a quarter of astrocytes showing intrinsic hyperactivity (Kuchibhotla et al. 2009). Such findings emphasise the importance of examining functional changes in neuronal and glial cells together.
Conclusion
Future studies need to directly assess and evaluate the role of adaptive and maladaptive changes at the intercellular level in mediating and modulating Aβ-induced modification of synaptic function and plasticity with a view to integrating apparently conflicting findings (Small 2008; Savioz et al. 2009). Research that examines how environmental and systemic factors such as stress affect the threshold and direction of the effects of Aβ on synaptic plasticity is only beginning (Kang et al. 2007; Li et al. 2007; Srivareerat et al. 2009a) and should advance our understanding of clinically important variables.
Overall (Fig. 2), current data support the view that certain NMDARs are critically involved in mediating the disruptive effect of Aβ oligomers on synaptic plasticity. Whether or not selective NMDAR antagonists are as, or more, protective than memantine warrants thorough investigation. The role of nicotinic and particularly muscarinic receptors may be more indirect, mediating physiological antagonism of the effects of Aβ. How these receptor subtypes contribute to the facilitatory effects of cholinesterase inhibitors on synaptic plasticity, especially the role of receptor desensitization, needs to be assessed as a priority. Given the interplay between APP/Aβ processing and specific transmitter receptor subtypes, hopefully, it will be possible to combine disease modifying and symptomatic treatment aspects of these approaches in future therapies.
References
Abbott, J. J., Howlett, D. R., Francis, P. T., & Williams, R. J. (2008). Abeta(1–42) modulation of Akt phosphorylation via alpha7 nAChR and NMDA receptors. Neurobiology of Aging, 29, 992–1001.
Albuquerque, E. X., Alkondon, M., Pereira, E. F., et al. (1997). Properties of neuronal nicotinic acetylcholine receptors: Pharmacological characterization and modulation of synaptic function. Journal of Pharmacology and Experimental Therapeutics, 280, 1117–1136.
Almeida, C. G., Tampellini, D., Takahashi, R. H., et al. (2005). Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiology of Diseases, 20, 187–198.
Anwyl, R. (1999). Metabotropic glutamate receptors: Electrophysiological properties and role in plasticity. Brain Research. Brain Research Reviews, 29, 83–120.
Arendt, T. (2009). Synaptic degeneration in Alzheimer’s disease. Acta Neuropathologica, 118, 167–179.
Arias, C., Arrieta, I., & Tapia, R. (1995). Beta-amyloid peptide fragment 25–35 potentiates the calcium-dependent release of excitatory amino acids from depolarized hippocampal slices. Journal of Neuroscience Research, 41, 561–566.
Auld, D. S., Kornecook, T. J., Bastianetto, S., & Quirion, R. (2002). Alzheimer’s disease and the basal forebrain cholinergic system: Relations to beta-amyloid peptides, cognition, and treatment strategies. Progress in Neurobiology, 68, 209–245.
Bancroft, A., & Levin, E. D. (2000). Ventral hippocampal alpha4beta2 nicotinic receptors and chronic nicotine effects on memory. Neuropharmacology, 39, 2770–2778.
Barghorn, S., Nimmrich, V., Striebinger, A., et al. (2005). Globular amyloid beta-peptide oligomer—a homogenous and stable neuropathological protein in Alzheimer’s disease. Journal of Neurochemistry, 95, 834–847.
Bishop, G. M., & Robinson, S. R. (2004). Physiological roles of amyloid-beta and implications for its removal in Alzheimer’s disease. Drugs and Aging, 21, 621–630.
Biton, B., Bergis, O. E., Galli, F., et al. (2007). SSR180711, a novel selective alpha7 nicotinic receptor partial agonist: (1) Binding and functional profile. Neuropsychopharmacology, 32, 1–16.
Blusztajn, J. K., & Berse, B. (2000). The cholinergic neuronal phenotype in Alzheimer’s disease. Metabolic Brain Disease, 15, 45–64.
Bobich, J. A., Zheng, Q., & Campbell, A. (2004). Incubation of nerve endings with a physiological concentration of Abeta1-42 activates CaV2.2(N-Type)-voltage operated calcium channels and acutely increases glutamate and noradrenaline release. Journal of Alzheimer’s Disease, 6, 243–255.
Bodick, N. C., Offen, W. W., Levey, A. I., et al. (1997). Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Archives of Neurology, 54, 465–473.
Bourin, M., Ripoll, N., & Dailly, E. (2003). Nicotinic receptors and Alzheimer’s disease. Current Medical Research and Opinion, 19, 169–177.
Busche, M. A., Eichhoff, G., Adelsberger, H., et al. (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science, 321, 1686–1689.
Bymaster, F. P., Shannon, H. E., Rasmussen, K., et al. (1998). Unexpected antipsychotic-like activity with the muscarinic receptor ligand (5R, 6R)6-(3-propylthio-1, 2, 5-thiadiazol-4-yl)-1-azabicyclo[3.2.1]octane. European Journal of Pharmacology, 356, 109–119.
Cacucci, F., Yi, M., Wills, T. J., Chapman, P., & O’Keefe, J. (2008). Place cell firing correlates with memory deficits and amyloid plaque burden in Tg2576 Alzheimer mouse model. Proceedings of the National Academy of Sciences of the United States of America, 105, 7863–7868.
Chen, Q. S., Wei, W. Z., Shimahara, T., & Xie, C. W. (2002). Alzheimer amyloid beta-peptide inhibits the late phase of long-term potentiation through calcineurin-dependent mechanisms in the hippocampal dentate gyrus. Neurobiology of Learning and Memory, 77, 354–371.
Chen, L., Yamada, K., Nabeshima, T., & Sokabe, M. (2006). Alpha7 nicotinic acetylcholine receptor as a target to rescue deficit in hippocampal LTP induction in beta-amyloid infused rats. Neuropharmacology, 50, 254–268.
Cheng, L., Yin, W. J., Zhang, J. F., & Qi, J. S. (2009). Amyloid beta-protein fragments 25-35 and 31-35 potentiate long-term depression in hippocampal CA1 region of rats in vivo. Synapse, 63, 206–214.
Chin, J. H., Ma, L., MacTavish, D., & Jhamandas, J. H. (2007). Amyloid beta protein modulates glutamate-mediated neurotransmission in the rat basal forebrain: Involvement of presynaptic neuronal nicotinic acetylcholine and metabotropic glutamate receptors. Journal of Neuroscience, 27, 9262–9269.
Chishti, M. A., Yang, D. S., Janus, C., et al. (2001). Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. Journal of Biological Chemistry, 276, 21562–21570.
Ciccotosto, G. D., Tew, D. J., Drew, S. C., et al. (2009). Stereospecific interactions are necessary for Alzheimer disease amyloid-beta toxicity. Neurobiology of Aging. doi:10.1016/j.neurobiolaging.2009.02.018.
Cleary, J. P., Walsh, D. M., Hofmeister, J. J., et al. (2005). Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nature Neuroscience, 8, 79–84.
Coan, E. J., Irving, A. J., & Collingridge, G. L. (1989). Low-frequency activation of the NMDA receptor system can prevent the induction of LTP. Neuroscience Letters, 105, 205–210.
Court, J., Martin-Ruiz, C., Piggott, M., Spurden, D., Griffiths, M., & Perry, E. (2001). Nicotinic receptor abnormalities in Alzheimer’s disease. Biological Psychiatry, 49, 175–184.
Cowburn, R. F., Wiehager, B., Trief, E., Li-Li, M., & Sundstrom, E. (1997). Effects of beta-amyloid-(25-35) peptides on radioligand binding to excitatory amino acid receptors and voltage-dependent calcium channels: Evidence for a selective affinity for the glutamate and glycine recognition sites of the NMDA receptor. Neurochemical Research, 22, 1437–1442.
Cullen, W. K., Wu, J., Anwyl, R., & Rowan, M. J. (1996). Beta-amyloid produces a delayed NMDA receptor-dependent reduction in synaptic transmission in rat hippocampus. NeuroReport, 8, 87–92.
Cullen, W. K., Suh, Y. H., Anwyl, R., & Rowan, M. J. (1997). Block of LTP in rat hippocampus in vivo by β-amyloid precursor protein fragments. NeuroReport, 8, 3213–3217.
Dawson, G. R., Seabrook, G. R., Zheng, H., et al. (1999). Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience, 90, 1–13.
De Felice, F. G., Velasco, P. T., Lambert, M. P., et al. (2007). Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. Journal of Biological Chemistry, 282, 11590–11601.
Deshpande, A., Kawai, H., Metherate, R., Glabe, C. G., & Busciglio, J. (2009). A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. Journal of Neuroscience, 29, 4004–4015.
Dewachter, I., Filipkowski, R. K., Priller, C., et al. (2009). Deregulation of NMDA-receptor function and down-stream signaling in APP[V717I] transgenic mice. Neurobiology of Aging, 30, 241–256.
Dineley, K. T., Westerman, M., Bui, D., Bell, K., Ashe, K. H., & Sweatt, J. D. (2001). Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer’s disease. Journal of Neuroscience, 21, 4125–4133.
Dineley, K. T., Bell, K. A., Bui, D., & Sweatt, J. D. (2002). Beta-amyloid peptide activates alpha 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. Journal of Biological Chemistry, 277, 25056–25061.
Domingues, A., Almeida, S., da Cruz e Silva, E. F., Oliveira, C. R., & Rego, A. C. (2007). Toxicity of beta-amyloid in HEK293 cells expressing NR1/NR2A or NR1/NR2B N-methyl-D-aspartate receptor subunits. Neurochemistry International, 50, 872–880.
Dornan, W. A., Kang, D. E., McCampbell, A., & Kang, E. E. (1993). Bilateral injections of beta A(25-35) + IBO into the hippocampus disrupts acquisition of spatial learning in the rat. NeuroReport, 5, 165–168.
Dziewczapolski, G., Glogowski, C. M., Masliah, E., & Heinemann, S. F. (2009). Deletion of the alpha7 nicotinic acetylcholine receptor gene improves cognitive deficits and synaptic pathology in a mouse model of Alzheimer’s disease. Journal of Neuroscience, 29, 8805–8815.
Ferchmin, P. A., Perez, D., Eterovic, V. A., & de Vellis, J. (2003). Nicotinic receptors differentially regulate N-methyl-D-aspartate damage in acute hippocampal slices. Journal of Pharmacology and Experimental Therapeutics, 305, 1071–1078.
Fernandez-Tome, P., Brera, B., Arevalo, M. A., & de Ceballos, M. L. (2004). Beta-amyloid25–35 inhibits glutamate uptake in cultured neurons and astrocytes: Modulation of uptake as a survival mechanism. Neurobiology of Diseases, 15, 580–589.
Fodero, L. R., Mok, S. S., Losic, D., et al. (2004). Alpha7-nicotinic acetylcholine receptors mediate an Abeta(1-42)-induced increase in the level of acetylcholinesterase in primary cortical neurones. Journal of Neurochemistry, 88, 1186–1193.
Frankiewicz, T., Potier, B., Bashir, Z. I., Collingridge, G. L., & Parsons, C. G. (1996). Effects of memantine and MK-801 on NMDA-induced currents in cultured neurones and on synaptic transmission and LTP in area CA1 of rat hippocampal slices. British Journal of Pharmacology, 117, 689–697.
Freir, D. B., & Herron, C. E. (2003). Nicotine enhances the depressive actions of A beta 1-40 on long-term potentiation in the rat hippocampal CA1 region in vivo. Journal of Neurophysiology, 89, 2917–2922.
Fujii, S., Ji, Z., Morita, N., & Sumikawa, K. (1999). Acute and chronic nicotine exposure differentially facilitate the induction of LTP. Brain Research, 846, 137–143.
Gay, E. A., Giniatullin, R., Skorinkin, A., & Yakel, J. L. (2008). Aromatic residues at position 55 of rat alpha7 nicotinic acetylcholine receptors are critical for maintaining rapid desensitization. Journal of Physiology, 586, 1105–1115.
Ge, S., & Dani, J. A. (2005). Nicotinic acetylcholine receptors at glutamate synapses facilitate long-term depression or potentiation. Journal of Neuroscience, 25, 6084–6091.
Geula, C., Nagykery, N., Nicholas, A., & Wu, C. K. (2008). Cholinergic neuronal and axonal abnormalities are present early in aging and in Alzheimer disease. Journal of Neuropathology and Experimental Neurology, 67, 309–318.
Goto, Y., Niidome, T., Hongo, H., Akaike, A., Kihara, T., & Sugimoto, H. (2008). Impaired muscarinic regulation of excitatory synaptic transmission in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. European Journal of Pharmacology, 583, 84–91.
Grassi, F., Palma, E., Tonini, R., Amici, M., Ballivet, M., & Eusebi, F. (2003). Amyloid beta(1-42) peptide alters the gating of human and mouse alpha-bungarotoxin-sensitive nicotinic receptors. Journal of Physiology, 547, 147–157.
Greenamyre, J. T., & Young, A. B. (1989). Excitatory amino acids and Alzheimer’s disease. Neurobiology of Aging, 10, 593–602.
Gu, Z., Liu, W., & Yan, Z. (2009). Beta-amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. Journal of Biological Chemistry, 284, 10639–10649.
Haass, C., & Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nature Reviews. Molecular Cell Biology, 8, 101–112.
Harkany, T., Abraham, I., Timmerman, W., et al. (2000). Beta-amyloid neurotoxicity is mediated by a glutamate-triggered excitotoxic cascade in rat nucleus basalis. European Journal of Neuroscience, 12, 2735–2745.
Harmeier, A., Wozny, C., Rost, B. R., et al. (2009). Role of amyloid-beta glycine 33 in oligomerization, toxicity, and neuronal plasticity. Journal of Neuroscience, 29, 7582–7590.
Harris, M. E., Carney, J. M., Cole, P. S., et al. (1995). Beta-amyloid peptide-derived, oxygen-dependent free radicals inhibit glutamate uptake in cultured astrocytes: Implications for Alzheimer’s disease. NeuroReport, 6, 1875–1879.
Hsieh, H., Boehm, J., Sato, C., et al. (2006). AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron, 52, 831–843.
Hu, M., Schurdak, M. E., Puttfarcken, P. S., El Kouhen, R., Gopalakrishnan, M., & Li, J. (2007). High content screen microscopy analysis of A beta 1-42-induced neurite outgrowth reduction in rat primary cortical neurons: Neuroprotective effects of alpha 7 neuronal nicotinic acetylcholine receptor ligands. Brain Research, 1151, 227–235.
Hu, N. W., Smith, I. M., Walsh, D. M., & Rowan, M. J. (2008). Soluble amyloid-beta peptides potently disrupt hippocampal synaptic plasticity in the absence of cerebrovascular dysfunction in vivo. Brain, 131, 2414–2424.
Hynd, M. R., Scott, H. L., & Dodd, P. R. (2004). Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochemistry International, 45, 583–595.
Ishida, A., Furukawa, K., Keller, J. N., & Mattson, M. P. (1997). Secreted form of beta-amyloid precursor protein shifts the frequency dependency for induction of LTD, and enhances LTP in hippocampal slices. NeuroReport, 8, 2133–2137.
Janus, C., Pearson, J., McLaurin, J., et al. (2000). A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature, 408, 979–982.
Ji, D., Lape, R., & Dani, J. A. (2001). Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron, 31, 131–141.
Jolas, T., Zhang, X. S., Zhang, Q., et al. (2002). Long-term potentiation is increased in the CA1 area of the hippocampus of APP(swe/ind) CRND8 mice. Neurobiology of Diseases, 11, 394–409.
Jones, C. K., Brady, A. E., Davis, A. A., et al. (2008). Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. Journal of Neuroscience, 28, 10422–10433.
Kabogo, D., Rauw, G., Amritraj, A., Baker, G., & Kar, S. (2008). Beta-amyloid-related peptides potentiate K(+)-evoked glutamate release from adult rat hippocampal slices. Neurobiology of Aging. doi:10.1016/j.neurobiolaging.2008.1008.1009.
Kamenetz, F., Tomita, T., Hsieh, H., et al. (2003). APP processing and synaptic function. Neuron, 37, 925–937.
Kang, J. E., Cirrito, J. R., Dong, H., Csernansky, J. G., & Holtzman, D. M. (2007). Acute stress increases interstitial fluid amyloid-beta via corticotropin-releasing factor and neuronal activity. Proceedings of the National Academy of Sciences of the United States of America, 104, 10673–10678.
Keller, J. N., Pang, Z., Geddes, J. W., et al. (1997). Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide: Role of the lipid peroxidation product 4-hydroxynonenal. Journal of Neurochemistry, 69, 273–284.
Kelly, B. L., & Ferreira, A. (2006). Beta-amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. Journal of Biological Chemistry, 281, 28079–28089.
Kenney, J. E., & Gould, T. W. (2008). Modulation of hippocampus-dependent learning and synaptic plasticity by nicotine. Molecular Neurobiology, 38, 101–121.
Kim, D., & Tsai, L. H. (2009). Bridging physiology and pathology in AD. Cell, 137, 997–1000.
Kim, J. H., Anwyl, R., Suh, Y. H., Djamgoz, M. B., & Rowan, M. J. (2001). Use-dependent effects of amyloidogenic fragments of (beta)-amyloid precursor protein on synaptic plasticity in rat hippocampus in vivo. Journal of Neuroscience, 21, 1327–1333.
Klyubin, I., Walsh, D. M., Cullen, W. K., et al. (2004). Soluble Arctic amyloid beta protein inhibits hippocampal long-term potentiation in vivo. European Journal of Neuroscience, 19, 2839–2846.
Klyubin, I., Betts, V., Welzel, A. T., et al. (2008). Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: Prevention by systemic passive immunization. Journal of Neuroscience, 28, 4231–4237.
Klyubin, I., Wang, Q., Reed, M. N., et al. (2009). Protection against Aβ-mediated rapid disruption of synaptic plasticity and memory by memantine. Neurobiology of Aging. doi:10.1016/j.neurobiolaging.2009.04.005.
Koh, J. Y., Yang, L. L., & Cotman, C. W. (1990). Beta-amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damage. Brain Research, 533, 315–320.
Kotermanski, S. E., & Johnson, J. W. (2009). Mg2+ imparts NMDA receptor subtype selectivity to the Alzheimer’s drug memantine. Journal of Neuroscience, 29, 2774–2779.
Kuchibhotla, K. V., Lattarulo, C. R., Hyman, B. T., & Bacskai, B. J. (2009). Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science, 323, 1211–1215.
Lacor, P. N., Buniel, M. C., Furlow, P. W., et al. (2007). Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. Journal of Neuroscience, 27, 796–807.
Lambert, M. P., Barlow, A. K., Chromy, B. A., et al. (1998). Diffusible, nonfibrillar ligands derived from A beta(1-42) are potent central nervous system neurotoxins. Proceedings of the National Academy of Sciences of the United States of America, 95, 6448–6453.
Lau, C. G., & Zukin, R. S. (2007). NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nature Reviews Neuroscience, 8, 413–426.
Lauren, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., & Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature, 457, 1128–1132.
Lawlor, B. A., & Davis, K. L. (1992). Does modulation of glutamatergic function represent a viable therapeutic strategy in Alzheimer’s disease? Biological Psychiatry, 31, 337–350.
Lee, D. H., & Wang, H. Y. (2003). Differential physiologic responses of alpha7 nicotinic acetylcholine receptors to beta-amyloid-40 and beta-amyloid1-42. Journal of Neurobiology, 55, 25–30.
Lesne, S., Koh, M. T., Kotilinek, L., et al. (2006). A specific amyloid-beta protein assembly in the brain impairs memory. Nature, 440, 352–357.
Léveillé, F., El Gaamouch, F., Gouix, E., et al. (2008). Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. FASEB Journal, 22, 4258–4271.
Levin, E. D., Bradley, A., Addy, N., & Sigurani, N. (2002). Hippocampal alpha 7 and alpha 4 beta 2 nicotinic receptors and working memory. Neuroscience, 109, 757–765.
Li, S., Feig, L. A., & Hartley, D. M. (2007). A brief, but repeated, swimming protocol is sufficient to overcome amyloid beta-protein inhibition of hippocampal long-term potentiation. European Journal of Neuroscience, 26, 1289–1298.
Li, S., Hong, S., Shepardson, N. E., Walsh, D. M., Shankar, G. M., & Selkoe, D. (2009). Soluble oligomers of amyloid beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron, 62, 788–801.
Lipton, S. A. (2007). Pathologically activated therapeutics for neuroprotection. Nature Reviews Neuroscience, 8, 803–808.
Lorenzo, A., & Yankner, B. A. (1996). Amyloid fibril toxicity in Alzheimer’s disease and diabetes. Annals of the New York Academy of Sciences, 77, 89–95.
Lue, L. F., Kuo, Y. M., Roher, A. E., et al. (1999). Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. American Journal of Pathology, 155, 853–862.
Lynch, M. A. (2004). Long-term potentiation and memory. Physiological Reviews, 84, 87–136.
Ma, H., Lesne, S., Kotilinek, L., et al. (2007). Involvement of beta-site APP cleaving enzyme 1 (BACE1) in amyloid precursor protein-mediated enhancement of memory and activity-dependent synaptic plasticity. Proceedings of the National Academy of Sciences of the United States of America, 104, 8167–8172.
Machova, E., Jakubik, J., Michal, P., et al. (2008). Impairment of muscarinic transmission in transgenic APPswe/PS1dE9 mice. Neurobiology of Aging, 29, 368–378.
Mann, E. O., & Greenfield, S. A. (2003). Novel modulatory mechanisms revealed by the sustained application of nicotine in the guinea-pig hippocampus in vitro. Journal of Physiology, 551, 539–550.
Martin, S. E., de Fiebre, N. E., & de Fiebre, C. M. (2004). The alpha7 nicotinic acetylcholine receptor-selective antagonist, methyllycaconitine, partially protects against beta-amyloid1–42 toxicity in primary neuron-enriched cultures. Brain Research, 1022, 254–256.
Matos, M., Augusto, E., Oliveira, C. R., & Agostinho, P. (2008). Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: Involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience, 156, 898–910.
Matsuyama, S., Matsumoto, A., Enomoto, T., & Nishizaki, T. (2000). Activation of nicotinic acetylcholine receptors induces long-term potentiation in vivo in the intact mouse dentate gyrus. European Journal of Neuroscience, 12, 3741–3747.
Mattson, M. P., Cheng, B., Davis, D., Bryant, K., Lieberburg, I., & Rydel, R. E. (1992). Beta-amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. Journal of Neuroscience, 12, 376–389.
Maurice, T., Lockhart, B. P., & Privat, A. (1996). Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Research, 706, 181–193.
McDonald, M. P., Dahl, E. E., Overmier, J. B., Mantyh, P., & Cleary, J. (1994). Effects of an exogenous beta-amyloid peptide on retention for spatial learning. Behavioral and Neural Biology, 62, 60–67.
McLean, C. A., Cherny, R. A., Fraser, F. W., et al. (1999). Soluble pool of A beta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Annals of Neurology, 46, 860–866.
Molnar, Z., Soos, K., Lengyel, I., Penke, B., Szegedi, V., & Budai, D. (2004). Enhancement of NMDA responses by beta-amyloid peptides in the hippocampus in vivo. NeuroReport, 15, 1649–1652.
Morris, R. G., Moser, E. I., Riedel, G., et al. (2003). Elements of a neurobiological theory of the hippocampus: The role of activity-dependent synaptic plasticity in memory. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 358, 773–786.
Mousavi, M., & Hellstrom-Lindahl, E. (2009). Nicotinic receptor agonists and antagonists increase sAPPalpha secretion and decrease Abeta levels in vitro. Neurochemistry International, 54, 237–244.
Muller, U., Cristina, N., Li, Z. W., et al. (1994). Behavioral and anatomical deficits in mice homozygous for a modified beta-amyloid precursor protein gene. Cell, 79, 755–765.
Nakamura, S., Murayama, N., Noshita, T., Katsuragi, R., & Ohno, T. (2006). Cognitive dysfunction induced by sequential injection of amyloid-beta and ibotenate into the bilateral hippocampus; protection by memantine and MK-801. European Journal of Pharmacology, 548, 115–122.
Newhouse, P. A., Potter, A., & Singh, A. (2004). Effects of nicotinic stimulation on cognitive performance. Current Opinion in Pharmacology, 4, 36–46.
Nitta, A., Itoh, A., Hasegawa, T., & Nabeshima, T. (1994). Beta-amyloid protein-induced Alzheimer’s disease animal model. Neuroscience Letters, 170, 63–66.
Noda, M., Nakanishi, H., & Akaike, N. (1999). Glutamate release from microglia via glutamate transporter is enhanced by amyloid-beta peptide. Neuroscience, 92, 1465–1474.
Nordberg, A., Alafuzoff, I., & Winblad, B. (1992). Nicotinic and muscarinic subtypes in the human brain: Changes with aging and dementia. Journal of Neuroscience Research, 31, 103–111.
Oddo, S., & LaFerla, F. M. (2006). The role of nicotinic acetylcholine receptors in Alzheimer’s disease. Journal of Physiology (Paris), 99, 172–179.
Origlia, N., Righi, M., Capsoni, S., et al. (2008). Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-beta-mediated cortical synaptic dysfunction. Journal of Neuroscience, 28, 3521–3530.
Parsons, C. G., Stoffler, A., & Danysz, W. (2007). Memantine: A NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system—too little activation is bad, too much is even worse. Neuropharmacology, 53, 699–723.
Pena, F., Gutierrez-Lerma, A., Quiroz-Baez, R., & Arias, C. (2006). The role of beta-amyloid protein in synaptic function: Implications for Alzheimer’s disease therapy. Current Neuropharmacology, 4, 149–163.
Pettit, D. L., Shao, Z., & Yakel, J. L. (2001). Beta-amyloid(1-42) peptide directly modulates nicotinic receptors in the rat hippocampal slice. Journal of Neuroscience, 21, RC120.
Phinney, A. L., Calhoun, M. E., Wolfer, D. P., Lipp, H. P., Zheng, H., & Jucker, M. (1999). No hippocampal neuron or synaptic bouton loss in learning-impaired aged beta-amyloid precursor protein-null mice. Neuroscience, 90, 1207–1216.
Plant, L. D., Boyle, J. P., Smith, I. F., Peers, C., & Pearson, H. A. (2003). The production of amyloid beta peptide is a critical requirement for the viability of central neurons. Journal of Neuroscience, 23, 5531–5535.
Puzzo, D., Privitera, L., Leznik, E., et al. (2008). Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. Journal of Neuroscience, 28, 14537–14545.
Ramsden, M., Plant, L. D., Webster, N. J., Vaughan, P. F., Henderson, Z., & Pearson, H. A. (2001). Differential effects of unaggregated and aggregated amyloid beta protein (1-40) on K(+) channel currents in primary cultures of rat cerebellar granule and cortical neurones. Journal of Neurochemistry, 79, 699–712.
Raymond, C. R., Ireland, D. R., & Abraham, W. C. (2003). NMDA receptor regulation by amyloid-beta does not account for its inhibition of LTP in rat hippocampus. Brain Research, 968, 263–272.
Reitz, C., Honig, L., Vonsattel, J. P., Tang, M. X., & Mayeux, R. (2009). Memory performance is related to amyloid and tau pathology in the hippocampus. Journal of Neurology, Neurosurgery and Psychiatry, 80, 715–721.
Ring, S., Weyer, S. W., Kilian, S. B., et al. (2007). The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. Journal of Neuroscience, 27, 7817–7826.
Roselli, F., Tirard, M., Lu, J., et al. (2005). Soluble beta-amyloid1-40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. Journal of Neuroscience, 25, 11061–11070.
Roth, M., Tomlinson, B. E., & Blessed, G. (1966). Correlation between scores for dementia and counts of ‘senile plaques’ in cerebral grey matter of elderly subjects. Nature, 209, 109–110.
Rowan, M. J., Klyubin, I., Cullen, W. K., & Anwyl, R. (2003). Synaptic plasticity in animal models of early Alzheimer’s disease. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 358, 821–828.
Rowan, M. J., Klyubin, I., Wang, Q., Hu, N. W., & Anwyl, R. (2007). Synaptic memory mechanisms: Alzheimer’s disease amyloid beta-peptide-induced dysfunction. Biochemical Society Transactions, 35, 1219–1223.
Santos-Torres, J., Fuente, A., Criado, J. M., Riolobos, A. S., Heredia, M., & Yajeya, J. (2007). Glutamatergic synaptic depression by synthetic amyloid beta-peptide in the medial septum. Journal of Neuroscience Research, 85, 634–648.
Savioz, A., Leuba, G., Vallet, P. G., & Walzer, C. (2009). Contribution of neural networks to Alzheimer disease’s progression. Brain Research Bulletin. doi:10.1016/j.brainresbull.2009.06.006.
Schliebs, R., & Arendt, T. (2006). The significance of the cholinergic system in the brain during aging and in Alzheimer’s disease. Journal of Neural Transmission, 113, 1625–1644.
Seabrook, G. R., Smith, D. W., Bowery, B. J., et al. (1999). Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein. Neuropharmacology, 38, 349–359.
Senechal, Y., Larmet, Y., & Dev, K. K. (2006). Unraveling in vivo functions of amyloid precursor protein: Insights from knockout and knockdown studies. Neurodegenerative Diseases, 3, 134–147.
Senechal, Y., Kelly, P. H., & Dev, K. K. (2008). Amyloid precursor protein knockout mice show age-dependent deficits in passive avoidance learning. Behavioural Brain Research, 186, 126–132.
Shankar, G. M., Bloodgood, B. L., Townsend, M., Walsh, D. M., Selkoe, D. J., & Sabatini, B. L. (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. Journal of Neuroscience, 27, 2866–2875.
Shankar, G. M., Li, S., Mehta, T. H., et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nature Medicine, 14, 837–842.
Shinoe, T., Matsui, M., Taketo, M. M., & Manabe, T. (2005). Modulation of synaptic plasticity by physiological activation of M1 muscarinic acetylcholine receptors in the mouse hippocampus. Journal of Neuroscience, 25, 11194–11200.
Small, D. H. (2008). Network dysfunction in Alzheimer’s disease: Does synaptic scaling drive disease progression? Trends in Molecular Medicine, 14, 103–108.
Snyder, E. M., Nong, Y., Almeida, C. G., et al. (2005). Regulation of NMDA receptor trafficking by amyloid-beta. Nature Neuroscience, 8, 1051–1058.
Soriano, F. X., & Hardingham, G. E. (1997). Compartmentalized NMDA receptor signalling to survival and death. Journal of Physiology, 584, 381–387.
Srivareerat, M., Tran, T. T., Alzoubi, K. H., & Alkadhi, K. A. (2009a). Chronic psychosocial stress exacerbates impairment of cognition and long-term potentiation in beta-amyloid rat model of Alzheimer’s disease. Biological Psychiatry, 65, 918–926.
Srivareerat, M., Tran, T. T., Salim, S., Aleisa, A. M., & Alkadhi, K. A. (2009b). Chronic nicotine restores normal Abeta levels and prevents short-term memory and E-LTP impairment in Abeta rat model of Alzheimer’s disease. Neurobiology of Aging. doi:10.1016/j.neurobiolaging.2009.04.015.
Stephan, A., Laroche, S., & Davis, S. (2001). Generation of aggregated beta-amyloid in the rat hippocampus impairs synaptic transmission and plasticity and causes memory deficits. Journal of Neuroscience, 21, 5703–5714.
Szegedi, V., Juhasz, G., Budai, D., & Penke, B. (2005). Divergent effects of Abeta1-42 on ionotropic glutamate receptor-mediated responses in CA1 neurons in vivo. Brain Research, 1062, 120–126.
Taylor, C. J., Ireland, D. R., Ballagh, I., et al. (2008). Endogenous secreted amyloid precursor protein-alpha regulates hippocampal NMDA receptor function, long-term potentiation and spatial memory. Neurobiology of Diseases, 31, 250–260.
Teaktong, T., Graham, A. J., Court, J. A., et al. (2004). Nicotinic acetylcholine receptor immunohistochemistry in Alzheimer’s disease and dementia with Lewy bodies: Differential neuronal and astroglial pathology. Journal of the Neurological Sciences, 225, 39–49.
Terry, R. D. (1996). The pathogenesis of Alzheimer disease: An alternative to the amyloid hypothesis. Journal of Neuropathology and Experimental Neurology, 55, 1023–1025.
Tietje, K. R., Anderson, D. J., Bitner, R. S., et al. (2008). Preclinical characterization of A-582941: A novel alpha7 neuronal nicotinic receptor agonist with broad spectrum cognition-enhancing properties. CNS Neuroscience and Therapeutics, 14, 65–82.
Turner, P. R., O’Connor, K., Tate, W. P., & Abraham, W. C. (2003). Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Progress in Neurobiology, 70, 1–32.
Verdier, Y., & Penke, B. (2004). Binding sites of amyloid beta-peptide in cell plasma membrane and implications for Alzheimer’s disease. Current Protein and Peptide Science, 5, 19–31.
Walsh, D. M., Klyubin, I., Fadeeva, J. V., et al. (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature, 416, 535–539.
Wang, J., Dickson, D. W., Trojanowski, J. Q., & Lee, V. M. (1999). The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Experimental Neurology, 158, 328–337.
Wang, H. Y., Lee, D. H., D’Andrea, M. R., Peterson, P. A., Shank, R. P., & Reitz, A. B. (2000). Beta-amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. Journal of Biological Chemistry, 275, 5626–5632.
Wang, Q., Walsh, D. M., Rowan, M. J., Selkoe, D. J., & Anwyl, R. (2004). Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. Journal of Neuroscience, 24, 3370–3378.
Wang, Q., Klyubin, I., Wright, S., Griswold-Prenner, I., Rowan, M. J., & Anwyl, R. (2007). Alpha v integrins mediate beta-amyloid induced inhibition of long-term potentiation. Neurobiology of Aging, 29, 1485–1493
Wang, H., Song, L., Laird, F., Wong, P. C., & Lee, H. K. (2008a). BACE1 knock-outs display deficits in activity-dependent potentiation of synaptic transmission at mossy fiber to CA3 synapses in the hippocampus. Journal of Neuroscience, 28, 8677–8681.
Wang, Q., Klyubin, I., Wright, S., Griswold-Prenner, I., Rowan, M. J., & Anwyl, R. (2008b). Alpha v integrins mediate beta-amyloid induced inhibition of long-term potentiation. Neurobiology of Aging, 29, 1485–1493.
Warpman, U., Alafuzoff, I., & Nordberg, A. (1993). Coupling of muscarinic receptors to GTP proteins in postmortem human brain—alterations in Alzheimer’s disease. Neuroscience Letters, 150, 39–43.
Wasling, P., Daborg, J., Riebe, I., et al. (2009). Synaptic retrogenesis and amyloid-beta in Alzheimer’s disease. Journal of Alzheimers Disease, 16, 1–14.
Welsby, P., Rowan, M., & Anwyl, R. (2006). Nicotinic receptor-mediated enhancement of long-term potentiation involves activation of metabotropic glutamate receptors and ryanodine-sensitive calcium stores in the dentate gyrus. European Journal of Neuroscience, 24, 3109–3118.
Welsby, P. J., Rowan, M. J., & Anwyl, R. (2007). Beta-amyloid blocks high frequency stimulation induced LTP but not nicotine enhanced LTP. Neuropharmacology, 53, 188–195.
Welsby, P. J., Rowan, M. J., & Anwyl, R. (2009). Intracellular mechanisms underlying the nicotinic enhancement of LTP in the rat dentate gyrus. European Journal of Neuroscience, 29, 65–75.
Wevers, A., Monteggia, L., Nowacki, S., et al. (1999). Expression of nicotinic acetylcholine receptor subunits in the cerebral cortex in Alzheimer’s disease: Histotopographical correlation with amyloid plaques and hyperphosphorylated-tau protein. European Journal of Neuroscience, 11, 2551–2565.
Wevers, A., Burghaus, L., Moser, N., et al. (2000). Expression of nicotinic acetylcholine receptors in Alzheimer’s disease: Postmortem investigations and experimental approaches. Behavioural Brain Research, 113, 207–215.
Woolf, N. J. (1991). Cholinergic systems in mammalian brain and spinal cord. Progress in Neurobiology, 37, 475–524.
Wright, S., Malinin, N. L., Powell, K. A., Yednock, T., Rydel, R. E., & Griswold-Prenner, I. (2007). Alpha2beta1 and alphaVbeta1 integrin signaling pathways mediate amyloid-beta-induced neurotoxicity. Neurobiology of Aging, 28, 226–237.
Wrighton, D. C., Baker, E. J., Chen, P. E., & Wyllie, D. J. A. (2008). Mg2+ and memantine block of rat recombinant NMDA receptors containing chimeric NR2A/2D subunits expressed in Xenopus laevis oocytes. Journal of Physiology, 586, 211–225.
Wu, J., Anwyl, R., & Rowan, M. J. (1995a). Beta-amyloid selectively augments NMDA receptor-mediated synaptic transmission in rat hippocampus. NeuroReport, 6, 2409–2413.
Wu, J., Anwyl, R., & Rowan, M. J. (1995b). Beta-amyloid-(1-40) increases long-term potentiation in rat hippocampus in vitro. European Journal of Pharmacology, 284, R1–R3.
Wu, M. N., He, Y. X., Guo, F., & Qi, J. S. (2008). Alpha4beta2 nicotinic acetylcholine receptors are required for the amyloid beta protein-induced suppression of long-term potentiation in rat hippocampal CA1 region in vivo. Brain Research Bulletin, 77, 84–90.
Yamazaki, Y., Hamaue, N., & Sumikawa, K. (2002). Nicotine compensates for the loss of cholinergic function to enhance long-term potentiation induction. Brain Research, 946, 148–152.
Yan, S. D., Bierhaus, A., Nawroth, P. P., & Stern, D. M. (2009). RAGE and Alzheimer’s disease: A progression factor for amyloid-beta-induced cellular perturbation? Journal of Alzheimers Disease, 16, 833–843.
Yang, T., Knowles, J. K., Lu, Q., et al. (2008). Small molecule, non-peptide p75 ligands inhibit Abeta-induced neurodegeneration and synaptic impairment. PLoS ONE, 3, e3604.
Ye, C., Walsh, D. M., Selkoe, D. J., & Hartley, D. M. (2004). Amyloid beta-protein induced electrophysiological changes are dependent on aggregation state: N-methyl-D-aspartate (NMDA) versus non-NMDA receptor/channel activation. Neuroscience Letters, 366, 320–325.
Zajaczkowski, W., Frankiewicz, T., Parsons, C. G., & Danysz, W. (1997). Uncompetitive NMDA receptor antagonists attenuate NMDA-induced impairment of passive avoidance learning and LTP. Neuropharmacology, 36, 961–971.
Zhang, S. J., Steijaert, M. N., Lau, D., et al. (2007). Decoding NMDA receptor signaling: Identification of genomic programs specifying neuronal survival and death. Neuron, 53, 549–562.
Zheng, H., Jiang, M., Trumbauer, M. E., et al. (1995). Beta-amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell, 81, 525–531.
Zorumski, C. F., & Izumi, Y. (1998). Modulation of LTP induction by NMDA receptor activation and nitric oxide release. Progress in Brain Research, 118, 173–182.
Acknowledgments
The authors wish to acknowledge the support of Science Foundation Ireland, the Health Research Board of Ireland, IRCSET, GSK and the Irish Development Authority.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ondrejcak, T., Klyubin, I., Hu, NW. et al. Alzheimer’s Disease Amyloid β-Protein and Synaptic Function. Neuromol Med 12, 13–26 (2010). https://doi.org/10.1007/s12017-009-8091-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-009-8091-0