
Abstract
Activated phosphoinositide 3-kinase delta syndrome (APDS) is a novel primary immunodeficiency (PID) caused by heterozygous gain of function mutations in PI3Kδ catalytic p110δ (PIK3CD) or regulatory p85α (PIK3R1) subunits leading to APDS1 and APDS2, respectively. Patients with APDS present a spectrum of clinical manifestations, particularly recurrent respiratory infections and lymphoproliferation. We searched PubMed, Web of Science, and Scopus databases for APDS patients and screened for eligibility criteria. A total of 243 APDS patients were identified from 55 articles. For all patients, demographic, clinical, immunologic, and molecular data were collected. Overall, 179 APDS1 and 64 APDS2 patients were identified. The most common clinical manifestations were respiratory tract infections (pneumonia (43.6%), otitis media (28.8%), and sinusitis (25.9%)), lymphoproliferation (70.4%), autoimmunity (28%), enteropathy (26.7%), failure to thrive (20.6%), and malignancy (12.8%). The predominant immunologic phenotype was hyper-IgM syndrome (48.1%). Immunologic profiling showed decreased B cells in 74.8% and CD4+ T cells in 64.8% of APDS patients. The c.3061 G>A (p. E1021K) mutation in APDS1 with 85% frequency and c.1425+1 G> (A, C, T) (p.434–475del) mutation in APDS2 with 79% frequency were hotspot mutations. The majority of APDS patients were placed on long-term immunoglobulin replacement therapy. Immunosuppressive agents such as rituximab, tacrolimus, rapamycin, and leniolisib were also administered for autoimmunity and inflammatory complications. In addition, hematopoietic stem cell transplantation (HSCT) was used in 12.8% of patients. APDS has heterogynous clinical manifestations. It should be suspected in patients with history of recurrent respiratory infections, lymphoproliferation, and raised IgM levels. Moreover, HSCT should be considered in patients with severe and complicated clinical manifestations with no or insufficient response to the conventional therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Activated phosphoinositide 3-kinase delta syndrome (APDS) is a recently described primary immunodeficiency (PID) caused by autosomal dominant mutations that increase the activity of the phosphoinositide-3-kinase δ (PI3Kδ) pathway [1, 2]. APDS can be caused by mutations in the PIK3CD gene encoding the p110δ catalytic subunit of PI3Kδ (APDS1) [1, 2] or the PIK3R1 gene that encodes the p85α regulatory subunit of PI3Kδ (APDS2) [3, 4]. Since its first description in 2013, the rapidly increasing number of identified patients indicates that this disorder is a significant cause of primary antibody deficiency (PAD), which should be considered in patients presenting with atypical and severe complications [5].
APDS is clinically heterogeneous and characterized by the early occurrence of recurrent respiratory tract infections, lymphoproliferation, gastrointestinal manifestations, autoimmunity, and increased risk of malignancy [6]. Although most manifestations occurred in the pediatric age, adult-onset and asymptomatic courses were also reported [7]. The immunological phenotypes of APDS patients are also heterogeneous with both B and T cell abnormalities. B cell compartment abnormalities consist of mild B cell lymphopenia, increased transitional B cells, decreased class switched memory B cells and class-switch-recombination defects (CSR-D). Elevated IgM levels have been reported in most of the patients while total IgG and IgA levels can be either normal or markedly decreased. A reduction of naïve CD4+ and CD8+ T cell numbers with increased frequency of effector memory CD8+ T cells and inverted CD4+/CD8+ T cell ratio was also reported [7,8,9].
Considering the increasing number of identified APDS patients and its heterogeneous manifestations, we here aim to systematically summarize and evaluate the existing experience of APDS patients in terms of clinical, immunological, and genetic features.
Materials and Methods
Search Strategy
A comprehensive search limited to English language was performed using PubMed, Web of Science, and Scopus databases applying the following search terms: “APDS,” “Activated PI3K Delta Syndrome,” “Activated PI3Kδ syndrome,” “Activated Phosphoinositide 3 kinase Delta syndrome,” “Activated Phosphoinositide 3 kinase δ syndrome,” “PIK3CD mutation,” and “PIK3R1 mutation” in various combinations. The search was conducted using these terms in the keywords, titles, and abstracts. Reference lists of all full-text articles and major reviews were hand-searched for additional studies.
Study Selection
The articles were first screened based on the title and abstract to exclude all irrelevant studies and were classified into three categories (include, exclude, or “unclear”); the full-text version of all “unclear” articles was checked and subsequently classified in one of the two categories (include or exclude). All full-text manuscripts were assessed for eligibility criteria: written in English; published in the 2013–2018 period; conducted on human subjects; report of at least one patient with APDS diagnosis; and detailed description of epidemiological, clinical, and immunological features. Studies using animal models, reviews, congress abstracts, and articles in languages other than English were excluded. When necessary, the corresponding authors were contacted.
Data Extraction
In a first step, two researchers extracted the data from all included studies based on the manuscript titles and abstracts. The following data were collected from all identified studies: name of the first author; publication year; number of participants; and demographic, clinical, laboratory, and molecular data. The evaluation of immunologic data quality was based on the age-matched normal ranges included in each article.
Those patients appearing in more than one publication were identified, and overlapping data was deleted. Two reviewers performed the selection process independently, while the third reviewer was consulted to resolve disagreements between two reviewers.
Statistical Analysis
Aggregate data analysis was performed with simple data pooling. By this, an overall summary of subgroup data or data from a number of related studies were combined without weighting. Central and descriptive statistics were reported for quantitative data. For variables with abnormal distribution, median and interquartile ranges (IQR) were calculated. For group comparisons, U Mann–Whitney, chi-square, or Fisher exact tests were applied. A p value < 0.05 was considered statistically significant. All statistical analyses were performed using the SPSS software (v. 25.0, Chicago, IL).
Results
Study Characteristics
The literature search identified a total of 2763 articles. Four hundred and eighty-one articles were duplicated, and 2222 articles were excluded following title and abstract screening. Furthermore, five articles were excluded as they only reported general information regarding APDS patients lacking specific details. As shown in Figure S1, 55 articles fulfilled the inclusion criteria and were subsequently used for data extraction. The total of 292 APDS patients were reported in these 55 articles, and after the removal of duplicate cases, 243 patients remained for data analysis.
Epidemiologic Characteristics of APDS Patients
In this systematic review, we evaluated 243 APDS patients (123 males, 87 females, and 33 unknown gender). The median (IQR) age of diagnosis was 12.0 (6.5–21.5) years with a median (IQR) diagnostic delay of 7.0 (3.4–14.0) years. A positive family history of immunodeficiency was reported in 88 cases (38.6%). Demographic data of all patients are summarized in Table 1. For 103 patients, information regarding their life status was available. Of these, 89 patients (87.9%) are currently alive and 14 (12.1%) have deceased. Malignancies (e.g., lymphoma, and acute myeloid leukemia), cardiopulmonary arrest, bowel perforation, septic shock, multiple organ failure, and pulmonary hemorrhage were the reported causes of death. Infections occurred early in life (< 1 year), while enteropathy, lymphoproliferation, autoimmunity, and malignancy occurred at the median (range) age of 5.0 (1.1–18.0), 3.0 (1.0–6.0), 10.5 (6.0–15.0), and 18.0 (13.2–24.2) years, respectively.
Clinical Spectrum of APDS Patients
Among patients with an established primary diagnosis (n = 39), most patients were diagnosed initially with hyper-IgM (HIGM) syndrome (n = 11, 28.2%) or lymphoma (n = 8, 20.5%). Figure 1 shows the wide range of initial clinical diagnosis of APDS patients. As illustrated in Fig. 2, respiratory tract infections (65%) were the most common first presentation in APDS patients followed by organomegaly (15.8%).

Distribution of available primary clinical diagnosis of 39 APDS patients. Patients with APDS are often diagnosed with HIGM or lymphoma. HIGM hyperimmunoglobulin M, CVID common variable immunodeficiency, CID combined immune deficiency, ALPS autoimmune lymphoproliferative syndrome, XLA X-linked agammaglobulinemia

Type of first clinical manifestations in APDS patients. The most common first presentations in APDS patients reported are respiratory tract infections and organomegaly
Infective Complications
Upper respiratory tract infections (sinusitis (25.9%) and otitis (28.8%)), pneumonia (43.6%), diarrhea (16%), and eye infections (10.7%) were the most common infectious manifestations in APDS patients. Sinusitis was significantly more common in APDS1 (p = 0.028), while pneumonia and eye infection were more frequently reported in APDS2 patients (p < 0.001, and p = 0.004, respectively) (Table 2).
Pathogenic Organisms
The most common infection-causing agents identified in APDS patients were viruses (Epstein–Barr virus (EBV), cytomegalovirus (CMV), Varicella zoster virus (VZV), and Human papillomavirus reported in 28.1%, 14.7%, 6.9%, and 5.6% of patients, respectively), bacteria (Streptococcus pneumoniae (12.1%), Haemophilus influenza (10.4%) and Staphylococcus aureus (1.7%)), and fungal pathogens (Candida albicans (6.5%)). In general, Streptococcus pneumonia and Haemophilus influenza were significantly more frequent in APDS1 than in APDS2 (p = 0.002 and p = 0.005, respectively) (Table S1).
Non-infective Complications
Non-neoplastic Lymphoproliferation
Among patients with lymphoproliferative diseases, lymphadenopathy was the most frequent manifestation (58.4%), followed by splenomegaly (43.7%) and hepatomegaly (24.2%). As illustrated in Table 2, lymphadenopathy was more common in APDS2, whereas hepatomegaly was reported more frequently in APDS1 patients (p = 0.004 and p = 0.001, respectively). Benign lymphoid proliferations including reactive hyperplasia and lymphadenitis in the gastrointestinal and respiratory tract were reported in 60 (24.7%) patients and more frequently in APDS1 patients (p < 0.001). Tonsillar hypertrophy was observed in 21 (8.6%) patients with a predominant representation of APDS2 patients (p < 0.001).
Autoimmune and Inflammatory Disease
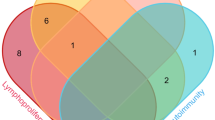
A variety of autoimmune conditions, including hematological, rheumatologic, gastrointestinal, endocrine, and dermatologic disorders, were reported in about one-fourth of patients (28%). As shown in Fig. 3, autoimmune hematologic disorders including autoimmune hemolytic anemia (AIHA) and immune thrombocytopenic purpura (ITP) were the most frequent autoimmune complications (76.2%) among reported autoimmunity conditions. Figure 3 also illustrates the coexistence of different autoimmune diseases in APDS, as 6.5% of patients had multiorgan autoimmunity. Enteropathy was reported in 26.7% of APDS patients, and it was more frequent in APDS1 (28.5%) than APDS2 (21.9%); although, this difference was not statistically significant.

Venn diagram showing multiorgan autoimmune manifestations in APDS patients. Autoimmune manifestations were reported in 68 patients (although in 9 patients no details were available). Hematologic manifestations (immune thrombocytopenic purpura, autoimmune hemolytic anemia, neutropenia, Evans syndrome), gastrointestinal manifestations (autoimmune hepatitis and inflammatory bowel disease), renal manifestations (glomerulonephritis and lupus-like nephritis), rheumatologic diseases (Sjögren’s syndrome, juvenile idiopathic arthritis and SLE-like disease), and other manifestations (diabetes mellitus type 1, autoimmune thyroiditis, and psoriasis) have been observed in APDS patients. The combination gastrointestinal symptoms with autoimmune hematologic manifestations were the most common
Malignancies
Thirty-one patients (12.8%) developed malignant diseases (Table S2) with a median age of onset of 18.0 years (range, 13.2–24.2 years). Diffuse large B cell lymphoma (DLBCL) was diagnosed in 14 (5.7%), classical Hodgkin lymphoma (CHL) in 9 (3.7%), and marginal zone B cell lymphoma in 5 (2.0%) patients. Among patients with malignancy, 8 (25.8%) patients had multiple lymphomas. The frequency of malignancy was higher in patients with a history of (chronic) viral infections than those without previous viral infections (19.5% vs. 7.4%, p = 0.006). Sixty-seven patients (27.6%) had a positive EBV infection history, and among these, 12 patients (17.9%) developed a lymphoma. EBV infection was reported in about half (n = 12, 44.4%) of the patients who developed lymphoma (n = 27, 11.1%). The lymphoma risk was significantly higher in patients with a previous EBV infection history (17.9% vs. 8.5%, p = 0.037).
Other Complications
Bronchiectasis was the most common structural complication in 28.4% of patients, and history of recurrent pneumonia was reported in about half of these patients (52.2%). Despite the observed higher frequency of pneumonia in APDS2, bronchiectasis was more common in APDS1 than in APDS2 patients (p = 0.001).
Failure to thrive was reported in 20.6% and was significantly more prevalent in APDS2 when compared to APDS1 (51.6% vs. 9.5%, p = < 0.001). Ear nose and throat problems (e.g., adenoid and tonsillar hypertrophy, otitis, hearing impairment, parotitis) were observed in 35.7% of APDS individuals. Neurologic/learning disorders were repeatedly reported in APDS patients (14%) and were significantly more frequent in those with APDS2 mutations (p = 0.001).
Immunological Findings of APDS Patients
Immunological data is summarized in Table 3. Of the evaluated APDS patients, 34.6% had lymphopenia. Low B cell counts (74.8%) were the most common, followed by low numbers of T cells (28.4%) and NK cells (18.1%). CD4+ T cell deficiency was reported in 64.8% of the APDS patients. In addition, 88.3% of the patients showed reduced naive CD4+ T cells (CD4+CD45RA+). Although 14.5% of the patients had low CD8+ T cells, 30.9% showed increased numbers and 76.5% had raised numbers of activated CD8+ T cells (CD8+CD45RO+). Overall, APDS1 individuals had lower T cell counts than APDS2 (p = < 0.001) andthe CD4/CD8 index was inverted in both APDS1 (68.8%) and APDS2 (66%) patients. Evaluation of the B cell compartment revealed low class-switched memory B cells in 32 (53.3%) of 60 patients and expansion of transitional B cells in 52 (83.9%) of 62 patients. Adjusted to the age-matched normal ranges, low IgG, IgA serum levels, and raised IgM levels were observed in 102 (57.6%), 98 (57.3%), and 107 (60.5%) patients, respectively, and 78 patients (48.1%) showed a HIGM phenotype. This HIGM immunophenotype was more prevalent in the APDS2 group when compared to the APDS1 group (63% vs. 40.7%, p < 0.001). Overall, 15.4% showed normal IgG, IgA, and IgM levels; 4.3% had selective IgA deficiency; 15.4% hypogammaglobulinemia; and 16.6% IgG subclass deficiency.
Molecular Findings of APDS Patients
Among 243 patients reported with APDS in 191 families, 179 patients had mutations in the PIK3CD gene and 64 patients in the PIK3R1 gene. We did not have access to the mutations of four patients, as in one APDS1 case, the mutation was not reported and in three APDS2 cases the family relationships were not declared [8, 10]. Altogether, 23 different DNA mutations (11 in PIK3CD and 12 in PIK3R1 genes) were reported. Despite the different mutations at the coding DNA level, these 23 different mutations resulted in 11 (PIK3CD) and 2 (PIK3R1) types of protein changes (Table 4). This data suggests that the APDS1 causing mutation c.3061 G>A (p. E1021K) with a frequency of 85% and the APDS2 causing mutation c.1425+1 G> (A, C, T) (p.434–475del) with a frequency of 79% were hotspot mutations. Recurrent de novo mutations were frequently observed in the APDS1 [1, 2, 11,12,13,14,15,16,17,18,19] and APDS2 [4, 8, 20,21,22,23,24,25], and this does not seem to be due to a founder effect [1]. All mutations in PIK3CD gene were missense, while nearly all mutations in PIK3R1 gene were splice site mutation except one missense mutation. Of the 11 splice site mutations, only one affected the splice acceptor site and 10 affected the splice donor site (Fig. 4).

Schematic diagram of the p110δ and p85α protein domains and positions of mutations. Adaptor-binding domain (ABD); Ras-binding domain (RBD); N-terminal lobe (N-lobe); C-terminal lobe (C-lobe); SRC homology domain (SH); Rho GTPase-activating protein (RhoGAP); N-terminal SH2 (cSH2); interSH2 (iSH2); C-terminal SH2 (cSH2)
The Therapeutic Approach in APDS Patients
Following the diagnosis of PID or as a symptomatic treatment approach, intravenous Ig (IVIg) replacement therapy was initiated in 151 (62.9%) patients. High dose IVIg (hdIVIg) was used to treat ITP and AIHA in 38 (84.4%) patients. Anti-inflammatory drugs such as corticosteroids were often first-line therapy for autoimmunity and inflammation. Immunosuppressive drugs such as mycophenolate mofetil and rituximab were also frequently prescribed. The emerging new targeted ADPS treatment option such as rapamycin (sirolimus) or leniolisib, which is a PI3Kδ inhibitor, was used in 29 (11.9%) patients. Furthermore, allogeneic hematopoietic stem cell transplantation (HSCT) with HLA identical donors was performed in 31 (12.8%) patients at the median (IQR) age of 9.5 (8.0–14.7) years. Among HSCT complications, acute or chronic GVHD occurred in about 45% of the transplants. Other organ transplantations (kidney and liver) were reported in four (1.6%) cases. Moreover, tonsillectomy and/or adenoidectomy were performed in 21 (8.6%) patients, and 10 (4.1%) patients had undergone splenectomy.
Discussion
The present study was designed to systematically review the clinical, immunological, and molecular characteristics of APDS patients. Most of the patients were initially diagnosed as HIGM syndrome. Impaired cellular immunity was observed more frequently in APDS1 patients, whereas the humoral arm of the immune system including B cell and subsequent antibody production was mostly affected in APDS2 individuals. To date, more than 240 APDS patients have been reported in the literature with a broad spectrum of clinical manifestations including recurrent respiratory infections, lymphoproliferation, autoimmunity, enteropathy, and increased risk of malignancies (particularly lymphoma); however, chronic infections, especially of the respiratory tract, in the first year of life are the most common clinical manifestations.
The predominant immunologic phenotype suggested a HIGM syndrome in 48%, and not surprisingly, 28% of the patients were initially diagnosed with a HIGM syndrome as many of these patients also had progressive B- and T cell lymphopenia or CSR defects following disturbed germinal center architecture [8]. This is in line with previous cohort studies reporting increased IgM levels in APDS patients [7, 8, 11, 20]. We, therefore, suggest to add PIK3CD and PIK3R1 mutations to the differential diagnosis in patients presenting respiratory infections, lymphadenopathy, and an immunological phenotype compatible with a HIGM syndrome, along with the list of genes known to be associated with CSR defects and somatic hypermutation (AICDA, CD40, CD40L, and NEMO).
As noted in the result section, the bronchiectasis prevalence was higher in patients with APDS1 in spite of lower prevalence of pneumonia in this group. On the other hand, about half of all APDS patients who developed bronchiectasis had no history of pneumonia.
Since most of the bronchiectasis in immunodeficiency is expected to be post-infectious, this may be due to issues with pneumonia diagnosis and/or reporting. However, it can be proposed that other etiologies may have predisposed these patients to the development of airway damages such as bronchial obstruction secondary to respiratory tract lymphoid proliferation and inflammatory processes involved in autoimmunity, both of which were more frequent in APDS1 patients. In addition, p110δ overexpression may itself contribute to airway remodeling in APDS1 patients as previous studies have shown that inhibition of p110δ isoform via IC87114 reduces the production of contractile proteins a-SMA and calponin and release of proinflammatory cytokine IL-6 [26, 27]. Altogether, we suggest assessment of patients with chest computed tomography (CT) scan during early stages of disease as a considerable number of patients develop bronchiectasis without preceding pneumonia.
As with previous studies, we found that growth disorders were significantly more frequent in APDS2 comparing to APDS1. Moreover, three APDS2 patients also suffered from SHORT syndrome [28, 29] while no association between SHORT syndrome and APDS1 was reported. As some of the APDS2 patients presented features such as poor growth, learning disability, microcephaly, and glucose metabolism impairment, under-diagnosis of SHORT syndrome in this group is possible. The role of PIK3R1 in growth disorders has been investigated previously [30]. It has been proposed that high amounts of monomeric P85α compete with the p85-p110 dimer for the binding to the tyrosine-phosphorylated IRS-1, leading to ineffective activation of the insulin and insulin-like growth factor (IGF)-1 [31]. In addition, the role of other catalytic PI3K subunits such as p110α and negative feedback mechanisms due to chronic overactivation of the PI3K pathway are also feasible mechanisms that would explain the growth restriction in APDS patients [28]. However, growth hormone therapy is not suggested, as growth hormone receptor activation further stimulates the PI-3 kinase pathway [32].
Malignancy is one of the major complications of APDS. Thirty-one malignancies were reported in 243 APDS patients, and lymphoma (in particular diffuse large B cell lymphoma (DLBCL)) was the most common type of malignancy (89%). This is in line with the findings of the previous studies [7,8,9, 33]. The PI3K/Akt/mTOR pathway has a pivotal role in controlling proliferation and survival of tumor cells. In particular, class I PI3K plays a crucial role in oncogenesis [34]. In addition, (chronic) viral infections may contribute to this preexisting tumor risk. Our result showed that about half of the reported APDS patients (48.1%) had at least one episode of viral infection and that the risk to develop lymphomas was significantly higher in patients with a previous history of viral infections especially EBV. EBV has a particular affinity to B cells and that these act as reservoir for the virus [35, 36]. Moreover, underlying immune dysregulation prevents effective viral elimination and creates a hostile inflammatory environment further stimulating cell survival and proliferation [37,38,39]. This dysregulation in APDS1 patients involves exhaustion, senescence, and diminished cytotoxicity of CD8+ T and NK cells induced by PIK3CD mutation which may increase herpesviruses susceptibility and tumor surveillance [9].
Unfortunately, the PI3K/AKT/mTOR activation is an unfavorable prognostic factor for DLBCL [40] and in our study, 66.6% of patients who died of lymphoma received a diagnosis of DLBCL. Therefore, periodic EBV screening and considering the risk of lymphoma in those with a history of chronic EBV infection may be beneficial for APDS patients. Nevertheless, further cohort studies are needed to investigate to what extent EBV infections predict risk of developing malignancy in the future.
In APDS, gain of function (GOF) mutations in PI3Kδ either in the leukocyte-restricted p110δ catalytic (PIK3CD) subunit or in the ubiquitously expressed p85α regulatory (PIK3R1) subunit leads to APDS1 and APDS2, respectively. In spite of heterozygous loss-of-function (LOF) mutations in PIK3R1, due to the net outcome of increased PI3Kδ activity, these are also considered GOF mutations [41]. Homozygous loss-of-function mutations either in PIK3R1 [42, 43] or in PIK3CD [44] result in different PID entities with PI3Kδ deficiency and autosomal recessive inheritance. Since the early publications, 11 activating missense mutations in the PIK3CD gene, one splice site, and one missense mutation in PIK3R1 gene have been reported (Table 4). These mutations augment PI3Kδ activity through enhancing membrane association and kinase activity (E1021K [1, 45], E1025G, and R929C [19, 46, 47]), increasing kinase activity through conformational changing (E81K and G124D [48]), interrupting inhibitory interaction between p110δ helical domain and the p85α nSH2 domain (E525K, E525A, and Y524N [2, 18, 49]) and between p85α iSH2 domain and p110δ C2 domain (N334K, R405C, and C416R [2, 50]). Accordingly, two mutations in iSH2 domain of p85α (434–475 deletion and N564K) interrupt inhibitory function of this protein [47, 50] and also diminish proteins stability [12]. PIK3CD and PIK3R1 GOF variants display genetic pleiotropy and their mutations also have oncogenic potential [51, 52]. Mutations in p110δ are found in analogous locations to oncogenic mutations in p110α [48]. P85α acts as a tumor suppressor; hence, mutations in this protein may drive cancer development through p110α hyperactivation [53, 54]. Several mutations in p85α have been reported in the context of endometrial cancer [55], glioma [56], and colon cancer [57]. Interestingly, the p.N564K mutation (that reported in APDS2) is an oncotic mutation in endometrial [58] and breast [59] cancers, while p.434–475del has not been reported in cancerous samples [60]. Considering the different impacts of p85α mutations on p110δ and p110α activity [45], it is maybe not surprising that these oncogenic mutations are restricted in lymphoma.
The therapeutic approach in APDS patients includes prophylactic antibiotics, immunoglobulin replacement therapy, use of immunosuppressive agents, and HSCT. Different immunosuppressive agents have been used to control autoimmune and inflammatory complications. About one-third of the reported APDS patients had autoimmune complications and received at least one course of immunosuppressive agents including steroids, rapamycin, rituximab, mycophenolate mofetil, tacrolimus, azathioprine, and cyclosporine. Prednisolone in combination with mesalazine improved the inflammatory bowel disease [61] in one patient, and rituximab reduced non-neoplastic lymphoproliferation in five APDS cases [7]. Among the immunosuppressive agents, rapamycin was shown to ameliorate the severity of benign lymphoproliferative manifestations and improve immunologic markers in APDS [25, 62, 63] while APDS-related cytopenia and GI inflammation responded less well [63]. Rapamycin has also shown to restore the NK cell function, which is likely relevant in the context of the known susceptibility to viral infections [64]. However, long-term side effects of rapamycin need to be monitored as one patient developed a lymphoma under rapamycin therapy [63]. By getting more familiar with the underlying mechanism of APDS, several targeted therapies have been proposed; however, because of limited experiences, it remains to be seen to what extent these therapies will lead to a significant clinical benefit. Selective PI3Kδ inhibitors have been suggested to provide greater efficacy and fewer side effects. Rao et al. [65] investigated the therapeutic effect of Leniolisib (CDZ173) in APDS individuals and observed normalization of circulating transitional and naïve B cells, reduction in senescent T cells, decreases in elevated serum IgM levels, and cytokine/chemokine modulation. Furthermore, after 3 months of therapy, lymph node and spleen sizes reduced and an increase of well-being was reported in all patients [64]. The patients received treatment for over 9 months, and during this period, no significant side effects have been detected [65, 66]. Another PI3Kδ inhibitor Idelalisib (GS-1101) has shown to reduce hyperactivity of the mutant PI3Kδ, but the in vivo function remains to be surveyed in future studies [45]. Nemiralisib as a PI3Kδ inhibitor has been shown to improve the survival of mice with early Streptococcus pneumonia infection and reduce IL-10 levels in the lung [67]. PI3Kδ hyperactivation promotes the development of CD19+B220− cells that are functionally considered equivalent to the expanded transitional B cells in APDS patients. Both cell types produce IL-10 leading to an increased susceptibility to S. pneumonia infections. Therefore, it is postulated that nemiralisib may also prevent S. pneumonia infection in APDS patients. Inhaled nemiralisib is currently being investigated in APDS patients and has been proposed to benefit patients primarily affected by airway infection and bronchiectasis. However, this ongoing clinical trial has not reported any results yet [67]. Recently, it has been shown that APDS mouse model exhibited aberrant autoantibody production against commensal microbiome which considerably controlled with antibiotics [68]. This may imply modification of gut microbiota as a treatment option for autoimmune manifestations in APDS patients.
HSCT in APDS patients has been specifically evaluated in two unrelated studies [69, 70]. Age at transplant ranged from 4 to 18 years. HLA-matched donors were used in 45.5% (71) and 80% (70) of individuals. Most cases, especially younger patients, improved their clinical symptoms in particular infection susceptibility, lymphoproliferation, hypogammaglobinemia, and enteropathy. Most of the patients (71.4% (70) and 100% (71)) were weaned of IVIG by day 100. However, adverse complications (90% (70) and 90.9% (71)) and engraftment failure (0% (70) and 36.4% (71)) were frequent, and graft versus host disease occurred in five and nine patients, respectively. Furthermore, HSCT carried a mortality risk of 10–20% and two patients in each study died from sepsis, multiorgan failure, CMV/adenovirus pneumonitis, and idiopathic pneumonitis [69, 70]. This indicates that HSCT should likely be restricted to those APDS patients with severe and complicated clinical manifestations with no or insufficient response to the currently available conventional therapies.
References
Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR et al (2013) Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science. 342(6160):866–871
Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U et al (2014) Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol 15(1):88–97
Deau MC, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P et al (2015) Erratum: a human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest 125(4):1764–1765
Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, McElwee J et al (2014) Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med 211(13):2537–2547
Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W et al (2018) The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol 38(1):129–143
Michalovich D, Nejentsev S (2018) Activated PI3 kinase delta syndrome: from genetics to therapy. Front Immunol 9:369
Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N et al (2017) Clinical spectrum and features of activated phosphoinositide 3-kinase delta syndrome: a large patient cohort study. J Allergy Clin Immunol 139(2):597–606 e4
Elkaim E, Neven B, Bruneau J, Mitsui-Sekinaka K, Stanislas A, Heurtier L et al (2016) Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase delta syndrome 2: a cohort study. J Allergy Clin Immunol 138(1):210–218 e9
Edwards ESJ, Bier J, Cole TS, Wong M, Hsu P, Berglund LJ et al (2018) Activating PIK3CD mutations impair human cytotoxic lymphocyte differentiation and function and EBV immunity. J Allergy Clin Immunol 143(1):276–291 e6
Lawrence MG, Uzel G (2015) 6-Year-old boy with recurrent sinopulmonary infections and lymphadenopathy. J Allergy Clin Immunol Pract 3(3):461–463 e1
Crank MC, Grossman JK, Moir S, Pittaluga S, Buckner CM, Kardava L et al (2014) Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. J Clin Immunol 34(3):272–276
Deau MC, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P et al (2014) A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest 124(9):3923–3928
Kannan JA, Davila-Saldana BJ, Zhang KJ, Filipovich AH, Kucuk ZY (2015) Activated phosphoinositide 3-kinase delta syndrome in a patient with a former diagnosis of common variable immune deficiency, bronchiectasis, and lymphoproliferative disease. Ann Allergy Asthma Immunol 115(5):452–454
Heurtier L, Lamrini H, Chentout L, Deau MC, Bouafia A, Rosain J et al (2017) Mutations in the adaptor-binding domain and associated linker region of p110delta cause activated PI3K-delta syndrome 1 (APDS1). Haematologica. 102(7):e278–ee81
Mettman D, Thiffault I, Dinakar C, Saunders C (2017) Immunodeficiency-associated lymphoid hyperplasia as a cause of intussusception in a case of activated PI3K-delta syndrome. Front Pediatr 5:71
Baleydier F, Ranza E, Schappi M, Rougemont AL, Merlini L, Ansari M, et al. (2018). Activated phosphoinositide 3 kinase delta syndrome (APDS): a primary immunodeficiency mimicking lymphoma. J Pediatr Hematol Oncol
Hong CR, Lee S, Hong KT, Choi JY, Shin HY, Choi M et al (2018) Successful haploidentical transplantation with post-transplant cyclophosphamide for activated phosphoinositide 3-kinase delta syndrome. J Allergy Clin Immunol Pract 7(3):1034–1037 e1
Luo Y, Xia Y, Wang W, Li Z, Jin Y, Gong Y et al (2018) Identification of a novel de novo gain-of-function mutation of PIK3CD in a patient with activated phosphoinositide 3-kinase delta syndrome. Clin Immunol 197:60–67
Wang X, Qi X, Yang B, Chen S, Wang J (2018) Autophagy benefits the replication of egg drop syndrome virus in duck embryo fibroblasts. Front Microbiol 9:1091
Lougaris V, Faletra F, Lanzi G, Vozzi D, Marcuzzi A, Valencic E et al (2015) Altered germinal center reaction and abnormal B cell peripheral maturation in PI3KR1-mutated patients presenting with HIGM-like phenotype. Clin Immunol 159(1):33–36
Kuhlen M, Hoenscheid A, Loizou L, Nabhani S, Fischer U, Stepensky P et al (2016) De novo PIK3R1 gain-of-function with recurrent sinopulmonary infections, long-lasting chronic CMV-lymphadenitis and microcephaly. Clin Immunol 162:27–30
Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova JL et al (2016) Genetic diagnosis using whole exomesequencing in common variable immunodeficiency. Front Immunol 7(JUN):220
Martinez-Saavedra MT, Garcia-Gomez S, Acosta AD, Quintana JJM, Paez JP, Garcia-Reino EJ et al (2016) Gain-of-function mutation in PIK3R1 in a patient with a narrow clinical phenotype of respiratory infections. Clin Immunol 173:117–120
Asano T, Okada S, Tsumura M, Yeh TW, Mitsui-Sekinaka K, Tsujita Y et al (2018) Enchanced AKT phosphorylation of circulating B cells in patients with activated PI3K delta syndrome. Front Immunol 9:568
Olbrich P, Lorenz M, Cura Daball P, Lucena JM, Rensing-Ehl A, Sanchez B et al (2016) Activated PI3Kdelta syndrome type 2: two patients, a novel mutation, and review of the literature. Pediatr Allergy Immunol: official publication of the European Society of Pediatric Allergy and Immunology 27(6):640–644
Ge Q, Moir LM, Trian T, Niimi K, Poniris M, Shepherd PR et al (2012) The phosphoinositide 3′-kinase p110delta modulates contractile protein production and IL-6 release in human airway smooth muscle. J Cell Physiol 227(8):3044–3052
Moir LM, Trian T, Ge Q, Shepherd PR, Burgess JK, Oliver BG et al (2011) Phosphatidylinositol 3-kinase isoform-specific effects in airway mesenchymal cell function. J Pharmacol Exp Ther 337(2):557–566
Petrovski S, Parrott RE, Roberts JL, Huang H, Yang J, Gorentla B et al (2016) Dominant splice site mutations in PIK3R1 cause hyper IgM syndrome, lymphadenopathy and Short stature. J Clin Immunol 36(5):462–471
Garcia-Morato MB, Garcia-Minaur S, Garicano JM, Simarro FS, Molina LDP, Lopez-Granados E et al (2017) Mutations in PIK3R1 can lead to APDS2, SHORT syndrome or a combination of the two. Clin Immunol 179:77–80
Goncalves MD, Hopkins BD, Cantley LC (2018) Phosphatidylinositol 3-kinase, growth disorders, and cancer. N Engl J Med 379(21):2052–2062
Ueki K, Fruman DA, Brachmann SM, Tseng YH, Cantley LC, Kahn CR (2002) Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol Cell Biol 22(3):965–977
Lanning NJ, Carter-Su C (2006) Recent advances in growth hormone signaling. Rev Endocr Metab Disord 7(4):225–235
Kracker S, Curtis J, Ibrahim MAA, Sediva A, Salisbury J, Campr V et al (2014) Occurrence of B-cell lymphomas in patients with activated phosphoinositide 3-kinase δ syndrome. J Allergy Clin Immunol 134(1):233–236 e3
Majchrzak A, Witkowska M, Smolewski P (2014) Inhibition of the PI3K/Akt/mTOR signaling pathway in diffuse large B-cell lymphoma: current knowledge and clinical significance. Molecules (Basel, Switzerland) 19(9):14304–14315
Thorley-Lawson DA, Hawkins JB, Tracy SI, Shapiro M (2013) The pathogenesis of Epstein-Barr virus persistent infection. Curr Opin Virol 3(3):227–232
Faulkner GC, Burrows SR, Khanna R, Moss DJ, Bird AG, Crawford DH (1999) X-linked agammaglobulinemia patients are not infected with Epstein-Barr virus: implications for the biology of the virus. J Virol 73(2):1555–1564
Rezaei N, Hedayat M, Aghamohammadi A, Nichols KE (2011) Primary immunodeficiency diseases associated with increased susceptibility to viral infections and malignancies. J Allergy Clin Immunol 127(6):1329–1341.e2; quiz 42-3
Rezk SA, Weiss LM (2007) Epstein-Barr virus-associated lymphoproliferative disorders. Hum Pathol 38(9):1293–1304
Philip M, Rowley DA, Schreiber H (2004) Inflammation as a tumor promoter in cancer induction. Semin Cancer Biol 14(6):433–439
Xu ZZ, Xia ZG, Wang AH, Wang WF, Liu ZY, Chen LY et al (2013) Activation of the PI3K/AKT/mTOR pathway in diffuse large B cell lymphoma: clinical significance and inhibitory effect of rituximab. Ann Hematol 92(10):1351–1358
Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K (2016) PI3Kdelta and primary immunodeficiencies. Nat Rev Immunol 16(11):702–714
Conley ME, Dobbs AK, Quintana AM, Bosompem A, Wang YD, Coustan-Smith E et al (2012) Agammaglobulinemia and absent B lineage cells in a patient lacking the p85α subunit of PI3K. J Exp Med 209(3):463–470
Tang P, Upton JEM, Barton-Forbes MA, Salvadori MI, Clynick MP, Price AK et al (2018) Autosomal recessive agammaglobulinemia due to a homozygous mutation in PIK3R1. J Clin Immunol 38(1):88–95
Sogkas G, Fedchenko M, Dhingra A, Jablonka A, Schmidt RE, Atschekzei F (2018) Primary immunodeficiency disorder caused by phosphoinositide 3-kinase delta deficiency. J Allergy Clin Immunol 142(5):1650–3 e2
Dornan GL, Siempelkamp BD, Jenkins ML, Vadas O, Lucas CL, Burke JE (2017) Conformational disruption of PI3Kdelta regulation by immunodeficiency mutations in PIK3CD and PIK3R1. Proc Natl Acad Sci U S A 114(8):1982–1987
Dulau Florea AE, Braylan RC, Schafernak KT, Williams KW, Daub J, Goyal RK et al (2017) Abnormal B-cell maturation in the bone marrow of patients with germline mutations in PIK3CD. J Allergy Clin Immunol 139(3):1032–1035 e6
Wentink M, Dalm V, Lankester AC, van Schouwenburg PA, Scholvinck L, Kalina T et al (2017) Genetic defects in PI3Kdelta affect B-cell differentiation and maturation leading to hypogammaglobulineamia and recurrent infections. Clin Immunol 176:77–86
Burke JE, Perisic O, Masson GR, Vadas O, Williams RL (2012) Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110alpha (PIK3CA). Proc Natl Acad Sci U S A 109(38):15259–15264
Tsujita Y, Mitsui-Sekinaka K, Imai K, Yeh TW, Mitsuiki N, Asano T et al (2016) Phosphatase and tensin homolog (PTEN) mutation can cause activated phosphatidylinositol 3-kinase delta syndrome-like immunodeficiency. J Allergy Clin Immunol 138(6):1672–1680 e10
Burke JE, Williams RL (2015) Synergy in activating class I PI3Ks. Trends Biochem Sci 40(2):88–100
Rae W, Gao Y, Ward D, Mattocks CJ, Eren E, Williams AP (2017) A novel germline gain-of-function variant in PIK3CD. Clin Immunol 181:29–31
Dominguez-Pinilla N, Allende LM, Rosain J, Gallego MDC, Chaves F, Deswarte C, et al. (2018) Disseminated abscesses due to mycoplasma faucium in a patient with activated PI3Kdelta syndrome type 2. J Allergy Clin Immunol Pract
Thorpe LM, Spangle JM, Ohlson CE, Cheng H, Roberts TM, Cantley LC et al (2017) PI3K-p110alpha mediates the oncogenic activity induced by loss of the novel tumor suppressor PI3K-p85alpha. Proc Natl Acad Sci U S A 114(27):7095–7100
Sun M, Hillmann P, Hofmann BT, Hart JR, Vogt PK (2010) Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proc Natl Acad Sci U S A 107(35):15547–15552
Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B et al (2011) High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov 1(2):170–185
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR et al (2013) The somatic genomic landscape of glioblastoma. Cell. 155(2):462–477
Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB et al (2012) Recurrent R-spondin fusions in colon cancer. Nature. 488(7413):660–664
Urick ME, Rudd ML, Godwin AK, Sgroi D, Merino M, Bell DW (2011) PIK3R1 (p85alpha) is somatically mutated at high frequency in primary endometrial cancer. Cancer Res 71(12):4061–4067
Chen L, Yang L, Yao L, Kuang XY, Zuo WJ, Li S et al (2018) Characterization of PIK3CA and PIK3R1 somatic mutations in Chinese breast cancer patients. Nat Commun 9(1):1357
Burke JE (2018) Structural basis for regulation of phosphoinositide kinases and their involvement in human disease. Mol Cell 71(5):653–673
Elgizouli M, Lowe DM, Speckmann C, Schubert D, Hulsdunker J, Eskandarian Z et al (2016) Activating PI3Kdelta mutations in a cohort of 669 patients with primary immunodeficiency. Clin Exp Immunol 183(2):221–229
Rae W, Ramakrishnan KA, Gao Y, Ashton-Key M, Pengelly RJ, Patel SV et al (2016) Precision treatment with sirolimus in a case of activated phosphoinositide 3-kinase delta syndrome. Clin Immunol 171:38–40
Maccari ME, Abolhassani H, Aghamohammadi A, Aiuti A, Aleinikova O, Bangs C et al (2018) Disease evolution and response to rapamycin in activated phosphoinositide 3-kinase delta syndrome: the European Society for Immunodeficiencies-Activated Phosphoinositide 3-Kinase delta Syndrome Registry. Front Immunol 9:543
Ruiz-García R, Vargas-Hernández A, Chinn IK, Angelo LS, Cao TN, Coban-Akdemir Z et al (2018) Mutations in PI3K110δ cause impaired natural killer cell function partially rescued by rapamycin treatment. J Allergy Clin Immunol 142(2):605–617 e7
Rao VK, Webster S, Dalm V, Sediva A, van Hagen PM, Holland S et al (2017) Effective “activated PI3Kdelta syndrome”-targeted therapy with the PI3Kdelta inhibitor leniolisib. Blood. 130(21):2307–2316
Coulter TI, Cant AJ (2018) The treatment of activated PI3Kdelta syndrome. Front Immunol 9:2043
Stark AK, Chandra A, Chakraborty K, Alam R, Carbonaro V, Clark J et al (2018) PI3Kdelta hyper-activation promotes development of B cells that exacerbate Streptococcus pneumoniae infection in an antibody-independent manner. Nat Commun 9(1):3174
Preite S, Cannons JL, Radtke AJ, Vujkovic-Cvijin I, Gomez-Rodriguez J, Volpi S et al (2018) Hyperactivated PI3Kdelta promotes self and commensal reactivity at the expense of optimal humoral immunity. Nat Immunol 19(9):986–1000
Nademi Z, Slatter MA, Dvorak CC, Neven B, Fischer A, Suarez F et al (2017) Hematopoietic stem cell transplant in patients with activated PI3K delta syndrome. J Allergy Clin Immunol 139(3):1046–1049
Okano T, Imai K, Tsujita Y, Mitsuiki N, Yoshida K, Kamae C et al (2018) Hematopoietic stem cell transplantation for progressive combined immunodeficiency and lymphoproliferation in patients with activated phosphatidylinositol-3-OH kinase delta syndrome type 1. J Allergy Clin Immunol 143(1):266–275
Funding
This work was supported by vice chancellor for research, Alborz University of Medical Sciences, under Grant No. IR.ABZUMS.REC.1397.051.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jamee, M., Moniri, S., Zaki-Dizaji, M. et al. Clinical, Immunological, and Genetic Features in Patients with Activated PI3Kδ Syndrome (APDS): a Systematic Review. Clinic Rev Allerg Immunol 59, 323–333 (2020). https://doi.org/10.1007/s12016-019-08738-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-019-08738-9