
Abstract
Autosomal dominant gain of function mutations in the gene encoding PI3K p110δ were recently associated with a novel combined immune deficiency characterized by recurrent sinopulmonary infections, CD4 lymphopenia, reduced class-switched memory B cells, lymphadenopathy, CMV and/or EBV viremia and EBV-related lymphoma. A subset of affected patients also had elevated serum IgM. Here we describe three patients in two families who were diagnosed with HIGM at a young age and were recently found to carry heterozygous mutations in PIK3CD. These patients had an abnormal circulating B cell distribution featuring a preponderance of early transitional (T1) B cells and plasmablasts. When stimulated in vitro, PIK3CD mutated B cells were able to secrete class-switched immunoglobulins. This finding implies that the patients’ elevated serum IgM levels were unlikely a product of an intrinsic B cell functional inability to class switch. All three patients developed malignant lymphoproliferative syndromes that were not associated with EBV. Thus, we identified a novel subset of patients with PIK3CD mutations associated with HIGM, despite indications of preserved in vitro B cell class switch recombination, as well as susceptibility to non-EBV-associated malignancies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The findings of elevated serum IgM with low IgG, IgA, or IgE in the setting of immune deficiency leads most immunologists toward a diagnosis of HIGM caused by X-linked (XL) or autosomal recessive (AR) defects in the CD40 ligand/CD40 signaling pathway or AR disorders involving AID or UNG [1, 2]. Angulo et. al. and Lucas et. al. recently described a novel immune deficiency resulting from a gain of function mutation in the gene encoding the PI(3)K subunit p110δ [3, 4]. Patients presented with sinopulmonary infections, lymphadenopathy, CMV and/or EBV viremia, sometimes with EBV-related lymphoma. They had reduced numbers of naïve CD4+ T cells and class-switched memory B cells, and a subset also had elevated serum IgM levels. Here we describe 3 patients from 2 families who are heterozygous for PIK3CD mutations, were diagnosed with a classic hyper IgM phenotype during childhood and as young adults developed hematologic malignancies that were not EBV driven.
Case Reports
F1P1 is a Caucasian female born to non-consanguineous parents 21 years ago (Fig. 1a). She suffered from recurrent bacterial infections (otitis media, sinopulmonary) from 18 months onward and initiated IVIG at age 7. Diffuse lymphadenopathy and splenomegaly were first noted at age 3, and a lymph node biopsy done at age 8 was reported as “lymphoid hyperplasia.” EBV and CMV were repeatedly negative by PCR. Her immunologic phenotype was characterized by high IgM (700–800 mg/dL), low IgA (40 mg/dL) with normal total IgG but low IgG2 (<12 mg/dL) and IgG4 (<9 mg/dL). Her response to rubella was normal (81.9 kIU/L), but reportedly her response to Pneumovax was suboptimal. She was CD3 lymphopenic (463/uL) with an absolute CD4+ T cell count of 209/uL; CD8+ T cell, NK cell and B cell numbers were normal, as were T cell responses to mitogens.

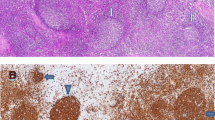
Family trees and characteristic pathology: a Family 1 and b Family 2 – gray color indicates an affected individual. c Lymph node biopsy on patient F1P1 showing effacement of the nodal architecture due to a diffuse proliferation of large atypical lymphoid cells with abundant cytoplasm, large nuclei with prominent central nucleolus (H&E 400×); also negative for EBV by in situ hybridization using EBER probe (inset). d Colon biopsy on patient F2P3 showing diffuse proliferation of small to medium atypical lymphoid cells with evidence of lymphoepithelial lesions involving the lamina propria of the colon (H&E 200×); in situ hybridization for EBV using EBER probe (inset) was negative
At age 19, she presented with diffuse, bulky adenopathy and her biopsy and staging were consistent with Stage IVA, EBV-negative, diffuse large B cell lymphoma. After 6 cycles of R-CHOP chemotherapy, she achieved a complete remission. She had immune thrombocytopenia on two occasions. Sanger sequencing identified a mutation in PIK3CD p110δ subunit (E1021K) previously described to be deleterious [3, 4].
F2P2 is a 36-year-old Caucasian female born to healthy parents after an uneventful pregnancy (Fig. 1b). Because her half sister had elevated serum IgM, IgM was measured in her cord blood and found to be elevated (104 mg/dL). During childhood she developed high serum IgM (peak 4500 mg/dL), and low IgG and IgA (nadir 34 mg/dL and <7 mg/d, respectively). Despite IgG supplementation, she experienced recurrent, severe sinopulmonary infections and otitis media, lymphadenitis, rectal abscesses, cellulitis, and pneumococcal sepsis, resulting in >40 lifetime hospitalizations. Her CD4+ T cell count was low (133/uL) with naïve CD4 cells being most significantly reduced (2/uL). T cell responses to mitogens and antigens were normal, but humoral responses to protein and polysaccharide vaccines were poor. In addition to immune deficiency, the patient had massive hypertrophy of lymphoid tissue in her gastrointestinal tract, mediastinum, spleen and peripheral nodes. At age 1, the lymphoproliferation resulted in an intestinal obstruction requiring surgical resection of a benign ileocecal mass. Pathology demonstrated florid follicular hyperplasia, but not massive GCs as reported in HIGM-AID. At age 12, another bulky abdominal mass responded to corticosteroid therapy. She developed severe bronchiectasis after multiple episodes of post obstructive pneumonia. The patient was diagnosed with EBV (−) non-Hodgkin’s lymphoma at age 16, achieved a complete remission with chemotherapy (CHOP) but relapsed 3 years later. She was successfully treated again, though her high IgM recurred post chemotherapy. Of note, she was persistently EBV and CMV negative by PCR. A second malignancy, basal cell carcinoma, was removed from her forehead. In addition, the patient had eczematous dermatitis with S. aureus and HSV super-infection, dysmorphic facial features, osteoporosis complicated by vertebral collapse, nephro- and cholelithiasis, and delayed sexual maturation.
F2P3 is a 24-year-old Caucasian male (Fig. 1b) with elevated IgM, recurrent sinopulmonary infections, lymphadenopathy, warts, poor responses to Pneumovax and H. influenza vaccines and a low naïve CD4 cell count. At one point he had a low positive EBV titer (1950 copies/mL) but then developed an EBV (−) MALT lymphoma affecting his colon. A recent lymph node biopsy showed hyperplastic GCs and contained B cells expressing IgG and IgM in equal amounts with a normal kappa and lambda distribution. As shown in Fig. 1b, F2P3’s mother was the maternal half sister to F2P2. She was also diagnosed with HIGM during childhood, presented with recurrent pyogenic infections, had Hodgkin lymphoma at age 21, and died at age 22 from AML (the possibility that the AML was therapy-related could not be ruled out). No genetic material was available from this patient for diagnosis.
Flow cytometry on peripheral blood from F2P2 and F2P3 demonstrated elevated proportions of T1 transitional B cells and plasmablasts (Fig. 2a). Upon in vitro stimulation, B cells from both patients were capable of producing both IgG and IgA as measured by ELISPOT (Fig. 2b). At least a portion of IgG secretion was likely induced by in vitro class switching, given the lack of surface IgG expression on pre-culture B cells and evidence that in vitro expression of IgG was associated with proliferation (not shown). Results from F2M, F2P2’s mother and F2P3’s grandmother, a healthy 70-year-old woman, were not different from other healthy controls. Proliferation-dependent IgG and IgA expression of peripheral blood mononuclear cells (PBMCs) from F2M and F2P2, using conditions previously described [5], were also qualitatively similar, although lower in frequency for F2P2. Finally, reconstitution of CD19-depleted PBMCs with naïve or naïve/transitional B cells of healthy donors also demonstrated that these in vitro conditions enabled class switching to occur (Fig. 2b). F2P2 and F2P3 were found to have a previously unreported gain of function mutation of PIK3CD (c.1246T>C, p. C416R), resulting in AKT hyperphosphorylation (not shown). F2M, who ought to be an obligate carrier, was found to be PIK3CD wild type on sequencing of a genomic DNA sample extracted from peripheral blood. Mutations in CD40, CD40L, AICDA, and UNG were sought and ruled out in F2P3.

B cell Phenotype: a Peripheral blood B cell subtypes in affected patients (F2P2 and F2P3) and unaffected mother (F2M) versus healthy donor (HD) controls as measured using flow cytometry of isolated peripheral blood mononuclear cells (PBMCs). Numbers in parenthesis represent the percentage of CD19+ B cells out of total PBMCs. B cell subsets were defined using cell surface markers as specified by Moir, et. al. (2013) [10]. b Number of antibody-secreting cells measured by ELISPOT (spots per 106 CD19+ B cells). PBMCs from Patients (F2P2 and F2P3) and unaffected mother (F2M) were stimulated in vitro with S. aureus Cowan strain I (SAC), CpG, IL-21 and pokeweed mitogen (PWM) for 4 days. CD19-depleted PBMCs from a healthy donor (HD) reconstituted with naïve (N) or naïve and transitional (N + Tr) B cells (CD27/IgG-negative fraction of CD10-depleted or CD10-containing B cells respectively of a donor whose CD10+ transitional B cells represented 20 % of total B cells) were included. Results for healthy donor cells were representative of three independent replicates
Discussion
HIGM encompasses multiple immune defects with the common laboratory findings of low serum IgG, IgA and/or IgE with elevated to normal IgM [1]. Underlying defects alter B cell maturation pathways, class switch recombination (CSR) and somatic hypermutation (SHM). They include abnormalities in cell-cell interaction (CD40/CD40L; 70 % of cases; predisposing to opportunistic and sinopulmonary infections, autoimmunity, neuroendocrine and biliary malignancy; lymph nodes are missing GCs and class-switched B cells), B cell intrinsic defects (AID/UNG; predisposing to sinopulmonary infections and autoimmunity; lymph nodes have giant GCs; B cells demonstrate defective CSR and SHM) and other diseases like NEMO, Ataxia-Telangiectasia, and PMS2 mutations/Lynch syndrome that have been also associated with HIGM phenotypes [1]. There are several features that clinically distinguish our patients from the classical HIGM phenotype. None of our patients experienced opportunistic infections, and their lymph nodes had GCs, both of which argue against a CD40L/CD40 defect. On the other hand, those GCs were not giant and, by all indications, their B cells were capable of CSR when stimulated in vitro, both of which argue against AID/UNG mutations.
PIK3CD mutations were recently associated with primary immunodeficiency [3, 4]. Patients presented with a diverse array of infectious, lymphoproliferative, and malignant processes. Elevated serum IgM, bronchiectasis and poor T cell responses to mitogens/antigens were also seen in some individuals. Malignancies reported were mostly associated with EBV, and in one series, persistent EBV and/or CMV infection were a key component of their immune deficiency syndrome. Our patients shared some clinical and laboratory features with the patients in these other cohorts, but they also fulfilled the diagnostic criteria for HIGM and, during their 2nd to 3rd decades of life, developed different forms of EBV-independent hematologic malignancies. Non-hematopoietic malignancies, such as basal cell carcinoma in F2P2, might also be associated with PIK3CD mutations [6]. Cancer susceptibility associated with PIK3CD mutations is not that surprising a finding: Zhang et al. have recently identified and characterized PIK3CD as an oncogene associated with diffuse large B-cell lymphomas [7]. Moreover, Xing et al. demonstrated a synergistic cytotoxic effect against human AML progenitors using a combination of PI3K p110δ and mitogen-extracellular activated protein kinase (MEK) inhibitors [8].
Regarding HIGM associated with PIK3CD mutations, in our cohort, patients presented with high proportions of T1 transitional B cells and plasmablasts along with indications of intact CSR in vitro, suggesting that their elevated serum IgM levels were not primarily related to a B cell-intrinsic defect. However, it should be noted that intact or normal CSR cannot be formally ascertained given direct comparison of B-cell subsets was not possible due to limitations in cell numbers and the extremely skewed nature of B-cell subsets within the PBMC compartment of these patients. Lymph node histopathology demonstrated hyperplastic GCs, which differed from HIGM associated with defects in either CD40/CD40L in which GCs are absent, or AID/UNG in which GCs are present but giant. Studies to further characterize both B and T lymphocytes and their interactions in these patients are underway.
While in some HIGMs as CD40L deficiency, hematopoietic stem cell transplantation (HSCT) is a definitive therapy, its role in patients with PI3CKD mutations has yet to be elucidated. In the cohort described by Angulo et al. [3] a single pediatric patient was transplanted, showing dramatic improvement at 1 year of follow up. While encouraging, it is too early to determine whether HSCT completely reverses the phenotype: our patients did not develop malignancies until their second and third decades of life, unlike other patients with PIK3CD their lymphomas were not EBV positive, and the mechanism (or spectrum) of their cancer susceptibility is not yet clearly determined.
Another intriguing feature detected in F2 was the PIK3CD mutation segregation pattern. PIK3CD associated disease is inherited as an autosomal dominant trait and de novo mutations have been previously reported [3, 4]. Although we cannot formally exclude that two independent, identical, de novo mutational events have occurred in F2 (one in F2P2 and the other on F2P3’s mother), the possibility of germinal mosaicism in F2M appears the more plausible explanation for disease segregation in this family [9].
The clinical implications of this new variant of HIGM are profound, because unlike other HIGM phenotypes, PIK3CD is associated with aggressive hematologic malignancy on the background of chronic benign lymphoproliferation. Frequent and rigorous screening for progression to malignant disease, including maintaining a low threshold for biopsy of suspicious lesions, is crucial to early initiation of successful, potentially life-saving therapy. For these reasons, we believe it is important for physicians caring for patients with HIGM, or families with increased cancer susceptibility, to include PIK3CD in their differential diagnosis and perform genetic testing in the setting of the appropriate clinical phenotype.
Abbreviations
- AID:
-
Activated cytidine deaminase
- AML:
-
Acute myeloid leukemia
- HIGM:
-
Hyper IgM syndrome
- IVIG:
-
Intravenous immunoglobulin
- GC:
-
Germinal center
- MALT:
-
Mucosal associated lymphoid tissue
- PI(3)K:
-
Phosphoinositide 3 kinase
- PIK3CD :
-
Phosphatidylinositol-4,5-Bisphosphonate 3-Kinase, Catalytic subunit delta
- R-CHOP:
-
Rituximab, Cyclophosphamide, Doxorubicin, Vincristine, Vrednisone
- UNG:
-
uracyl-N-glycosylase
References
Davies EG, Thrasher AJ. Update on the hyper immunoglobulin M syndromes. Br J Haematol. 2010;149(2):167–80.
Notarangelo LD, Duse M, Ugazio AG. Immunodeficiency with hyper-IgM (HIM). Immunodefic Rev. 1992;3(2):101–21.
Angulo I, Vadas O, Garcon F, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science. 2013;342(6160):866–71.
Lucas CL, Kuehn HS, Zhao F, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat Immunol. 2013;15(1):88–97.
Kotlarz D, Zietara N, Uzel G, et al. Loss-of-function mutations in the IL-21 receptor gene cause a primary immunodeficiency syndrome. J Exp Med. 2013;210(3):433–43.
Zhong M, Bian Z, Wu Z. miR-30a suppresses cell migration and invasion through downregulation of PIK3CD in colorectal carcinoma. Cell Physiol Biochem. 2013;31(2–3):209–18.
Zhang J, Grubor V, Love CL, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013;110(4):1398–403.
Xing Y, Hogge DE. Combined inhibition of the phosphoinosityl-3-kinase (PI3Kinase) P110delta subunit and mitogen-extracellular activated protein kinase (MEKinase) shows synergistic cytotoxicity against human acute myeloid leukemia progenitors. Leuk Res. 2013;37(6):697–704.
Zlotogora J. Germ line mosaicism. Hum Genet. 1998;102(4):381–6.
Moir S, Fauci AS. Insights into B cells and HIV-specific B-cell responses in HIV-infected individuals. Immunol Rev. 2013;254(1):207–24.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH. The content of this publication does not necessarily reflect the views or policies of the DHHS, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Author information
Authors and Affiliations
Corresponding author
Additional information
M. C. Crank and J. K. Grossman contributed equally to this work.
Rights and permissions
About this article
Cite this article
Crank, M.C., Grossman, J.K., Moir, S. et al. Mutations in PIK3CD Can Cause Hyper IgM Syndrome (HIGM) Associated with Increased Cancer Susceptibility. J Clin Immunol 34, 272–276 (2014). https://doi.org/10.1007/s10875-014-0012-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-014-0012-9