
Abstract
Identifying prosurvival mechanisms in stressed neuronal cells would provide protective strategies to hinder neurodegeneration. Recent evidence shows that vascular endothelial growth factor (VEGF), a well-established mitogen in endothelial cells, can mediate neuroprotection against damaging insults through the activation of its cognate receptor VEGFR2. In addition, growth factor receptor signaling pathways have been shown to crosstalk with cAMP-dependent Protein Kinase A (PKA) to protect neuronal cells from harmful stimuli. Whether a relationship exists between VEGFR2 and PKA in mediating neuroprotection under stressful conditions is unknown. Using SK-N-SH neuronal cells as a model system, we show that serum deprivation induces an upregulation in VEGF and VEGFR2 that concomitantly serves as a prosurvival signaling pathway. Inhibitor studies revealed that PKA functioned concurrently with VEGFR2 pathway to signal the activation of the extracellular signal-regulated protein kinases (ERK1/2) as protection against caspase-3/7 activation and a subsequent cell death. The loss in cell viability induced by VEGFR2 and PKA inhibition was prevented by caspase inhibition or overexpression of ERK1. Overexpression of the antiapoptotic protein Bcl-xL also promoted survival when VEGFR2 function was blocked. However, the protection elicited by all three treatments were prevented by the inclusion of a selective inhibitor of mitogen-activated protein kinase kinase (MEK), the upstream kinase that activates ERK1/2. Taken together, these findings suggested that PKA and VEGFR2 converge at the MEK/ERK1/2 pathway to protect serum starved neuronal cells from a caspase-dependent cell death.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Identifying survival mechanisms that respond to stressful stimuli would provide an insight on neuroprotective processes that function under pathological conditions. Vascular endothelial growth factor (VEGF) is a potent angiogenic factor that has been shown to protect neurons from the cell death induced by harmful insults [1, 2]. Evidence that VEGF exhibits neurotrophic properties was demonstrated by its ability to stimulate axonal outgrowth, increase the survival of rat mesencephalic neurons, and rescue hippocampal cells from death induced by serum withdrawal, hypoxia, ischemia, and glutamate-induced toxicity [3–8]. More recently, VEGF was shown to delay neurodegeneration in vivo in axotomized retinal ganglion cells [9] and a mouse model of amyotrophic lateral sclerosis [10]. While the downstream pathways targeted by VEGF are well characterized in endothelial cells, the signaling mechanisms induced by VEGF in neuronal cells require further clarification. A delineation of these pathways is complicated by the fact that VEGF can mediate biological effects through two cell surface receptors, VEGFR1 or VEGFR2, either alone or in concert with the neuropilin family of class 3 semaphorin cell surface receptors, Neuropilin 1 (NRP1) and 2 (NRP2) [11]. In endothelial cells, VEGFR2 interactions signal cell survival, proliferation, migration, differentiation, and angiogenesis, while VEGFR1 is presumed to function as a decoy receptor that sequesters VEGF from activating VEGFR2 [11]. NRP1 binds both VEGF and class 3 semaphorins and plays a functional role in axonal pathfinding, retraction and collapse during development and neuronal death [12, 13]. The demonstration that VEGFR2 is activated by VEGF to protect against harmful insults both in vitro and in vivo implicates this pathway as a critical mediator of neuronal cell survival under stressful conditions [6–8, 14]. VEGF was shown to signal neuroprotection through the activation of downstream pathways for phosphatidylinositol 3′-kinase/Protein Kinase B (PI3K/Akt) [6, 14, 15], the mitogen-activated protein kinase kinase/extracellular signal-regulated protein kinases (MEK/ERK1/2) [7] or PI3K/Akt and MEK/ERK1/2 in combination [8, 9]. In addition, VEGF can promote survival by suppressing caspase-3 activation [16].
The cAMP-inducible PKA is a well-established survival pathway in neuronal cells that modulates the ERK1/2 signaling cascade either directly, or via stimulatory or inhibitory interactions with growth factor-mediated mechanisms [17]. In this context, cAMP-dependent PKA activation was shown to inhibit ERK1/2 stimulation by platelet-derived growth factor (PGDF) and epidermal growth factor (EGF) in different cell types by modulating growth factor-mediated signaling through the Ras/Raf/MEK/ERK1/2 pathway [18]. In endothelial cells, cAMP-dependent PKA was shown to inhibit VEGF-mediated ERK1/2 activation through a phosphorylation-dependent inactivation of the MEK kinase Raf-1 [19]. In neuronal cells, nerve growth factor (NGF) and cAMP analogues were shown to stimulate ERK1/2 activation to induce differentiation [20] while ERK1/2 activation by cAMP alone was shown to protect against the damaging effects of exocitotoxicity [21] and oxidative stress [22]. More recently, the neuroprotective properties of cAMP-dependent PKA were shown to resemble that elicited by VEGF in that PKA can also activate ERK1/2 and Akt and suppress caspase-3 to promote survival [23, 24]. Therefore, we investigated whether the PKA pathway may function concurrently with the VEGF signaling pathway in stressed neuronal cells to promote survival through similar downstream targets. The existence of crosstalk between the PKA and VEGF-mediated survival pathways has not been explored in neuronal cells.
Herein, we show that the PKA and VEGFR2 signaling pathways activate ERK1/2 to protect against caspase activation and cell death in serum deprived SK-N-SH neuronal cells. Caspase inhibition or overexpression of ERK1 or Bcl-xL prevented cell death, but inhibition of MEK, the upstream activator if ERK1/2, eliminated these protective effects. These findings implicate the MEK/ERK1/2 signaling cascade as a critical downstream effector of VEGFR2 and PKA-mediated neuroprotection against a caspase-dependent cell death.
Material and methods
Materials
Recombinant human vascular endothelial growth factor 165 (VEGF165) and the anti-human VEGF neutralizing antibody were obtained from PeproTech Inc (Rocky Hill, NJ). SU1498, LY294002, H89 and PKI 14–22 Amide were obtained from EMD Biosciences Inc (San Diego, CA), U0126 was obtained from Promega Corporation (Madison, WI) and z-VAD-fmk and 8-(4-chlorophenylthio)-adenosine-3′,5′-cyclic monophosphate (8-CPT-cAMP) were obtained from Biomol International (Plymouth Meeting, PA).
Cell culture
Human neuroblastoma SK-N-SH cells were obtained from the American Type Culture Collection (Rockville, MD) and maintained at 37°C in DMEM-F10 (1:1) supplemented with 5% fetal bovine serum (FBS) (Invitrogen Corporation, Carlsbad, CA). For each experiment, cells were grown to 80% confluency followed by 48 h of serum starvation without and with the treatments as indicated.
Transient transfection
SK-N-SH cells were transfected with 0.05 μg of pcDNA3-Bcl-xL (gift of Dr. Shigemi Matsuyama, Case Comprehensive Cancer Center, Cleveland, OH) and pCEP4-Erk1 (gift of Dr. Melanie H. Cobb, University of Texas Southwestern Medical Center, TX) using siIMPORTER transfection reagent (Upstate Cell Signaling Solutions, Charlottesville, Virginia), in 96-well plates according to the manufacturer’s instructions. An empty pcDNA3 vector served as the control. For each transfection, cells were incubated for 4 h in the transfection medium at 37°C followed by 48 h treatments as indicated under serumfree conditions.
RNA interference
Cells were transfected with 20 μM of KDR/Flk-1/VEGFR2 SMARTpool® siRNA duplexes (Dharmacon RNA Technologies, Lafayette, CO) according to the manufacture’s directions. Cells were then treated as indicated and incubated for 48 h under serumfree conditions. A mixture of non-specific siRNA duplexes served as a negative control.
Reverse transcriptase PCR
Total RNA was purified from cells treated as indicated using the RNeasy RNA isolation Kit (QIAGEN, Valencia, CA) using the manufacturer’s protocol. Total RNA (2 μg) was reverse transcribed at 50°C for 30 min with gene specific primers followed by an amplification of the resultant cDNA for 35 cycles as instructed by the QIAGEN OneStep RT-PCR Kit. The gene specific primers were as follows: VEGF165, forward 5′-AGCCTTGCCTTGCTGCTCTA-3′; reverse 5′-GTGCTGGCCTTGGTGAGG-3′; VEGFR2, forward 5′-GCATCTCATCTGTTACAGC-3′; reverse 5′-CTTCATCAATCTTTACCCC-3′; VEGFR1, forward 5′-GACGTCTAGAGTTTGACACGAAGC-3′; reverse 5′-GCATGCAACACTGAGTAACATGAC-3′; NRP1, forward 5′-AAAAGCCCACGGTCATAG-3′; reverse 5′-TGTCATCCACAGCAATCC-3′; NRP2, forward 5′-CAAGTTGCTGTGGGTCATC-3′; reverse 5′-AATTGCTCCAGTCCACCTC-3′; and actin, forward 5′-TGACGGGGTCACCCACACTGTGCCCATCTA-3′; reverse 5′-CTAGAAGCATTTGCGGTGGACGATGGAGGG-3′ PCR products were visualized by electrophoresis on 2% agarose gels.
Cell viability
Cells were plated in 96-well microtiter plates and treated as indicated under serumfree conditions for 48 h at 37°C. Cell viability was determined using a colorimetric MTS assay (Promega Corp, Madison, WI) and quantified according to manufacturer’s instruction. Survival measurements are expressed as the percent of the untreated control.
Caspase-3/7 activities
Caspase activity was measured in cells plated in a 96-well format using the fluorescence cell-based Apo-ONE® Homogeneous Caspase-3/7 Assay (Promega) according to manufacturer’s directions. Caspase activity was quantified using the Molecular Dynamics TyphoonTM 9410 Imaging System with ImageQuant software (Amersham Pharmacia Biotech, Piscataway, NJ). Data were normalized as fluorescent units/μg protein.
Nuclei staining
Cells were pretreated with 20 μM z-VAD-fmk for 1 h followed by treatments as indicated for 48 h at 37°C. Cells were then washed in PBS, fixed with 3.7% paraformaldehyde for 20 min at 37°C, and stained with 1 μg/ml of the chromatin-specific dye Hoechst 33342 (Sigma) for 20 min. Stained nuclei were assessed for chromatin condensation and images were captured at 60X magnification using a Nikon OPTIPHOT-2 fluorescent microscope.
Protein extraction and western blotting
Total cell lysates were obtained from cells treated for 48 h at 37°C under serum deprivation and harvested in a lysis buffer, as described previously [25]. Protein concentrations were determined with a bicinchoninic acid assay (BCA) according to manufacturer’s instructions (Pierce, Rockford, IL). Equal amounts of protein (25 μg) from each lysate were resolved by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Blots were then blocked and incubated overnight at 4°C with the following primary antibodies: rabbit antibodies that recognized both phosphorylated and total forms of ERK1/2 and Akt (1:1000; Cell Signaling Technology, Beverly, MA) and mouse antibodies against Bcl-xL (1:500; BD Biosciences, San Jose CA) and β-actin (1:1000; Sigma-Aldrich) which was used as a loading control. Immunoreactive bands were detected with anti-mouse or anti-rabbit secondary antibodies conjugated to horseradish peroxidase and visualized with the SuperSignal West Pico Chemiluminescent Substrate (Pierce Endogen, Rockford, IL).
Protein immunoprecipitation
VEGFR2 was selectively immunoprecipitated from equal concentrations (500 μg) of total protein lysate using a rabbit antibody raised against VEGFR2 (1:200; Upstate Cell Signaling Solutions). Antibody-antigen complexes were selectively removed by binding to protein A Sepharose beads (Sigma-Aldrich) in a final volume of 200 μl overnight at 4°C. Beads were then collected by centrifugation, washed, resuspended in 25 μl of 2x SDS loading buffer and subjected to Western blotting analysis. In order to detect protein expression of VEGFR2, blots were probed with a rabbit antibody raised against VEGFR2 (1:1000; Santa Cruz Biotechnology, Inc, Santa Cruz CA). In order to detect activated VEGFR2, blots were probed with a mouse antibody that specifically recognizes phosphotyrosine (1:1000; Santa Cruz Biotechnology, Inc, Santa Cruz CA) and immunoreactive bands were detected as described in the protocol for Western Blotting.
Nuclear and cytoplasmic protein extraction
In order to delineate the subcellular localization of ERK1/2, cells were serum starved for 48 h followed by a replacement with fresh media containing 10 ng/ml of exogenous VEGF and incubated for 10, 30 and 60 min. A 0 time control represents cells incubated without VEGF. For each time frame, cells were collected in a dissociation buffer (0.3 mM EDTA/5% glycerol), centrifuged at 4°C and washed twice with ice-cold PBS. Nuclear and cytoplasmic extracts were then prepared using the NE-PER® Nuclear and Cytoplasmic Extraction protocol (Pierce Biotechnology) according to manufacturers’ instructions. Each extract was subjected to Western blot analyses for the detection of phosphorylated and total ERK1/2 or actin as described above.
Statistical analyses
Data are expressed as the mean ± SEM of experiments that were replicated at least three times. Statistical significance was assessed by a one-way ANOVA followed by pairwise contrasts (Bonferroni analysis). A difference resulting in a P < 0.05 is considered significant.
Results
The mRNA expression levels of VEGF and its receptors, VEGFR1, VEGFR2, and NRP1 are upregulated in serum deprived neuronal cells
Increasing evidence suggests that VEGF confers neuroprotection under stress conditions through interactions with cognate receptors that are expressed on neuronal cells [1]. We found that 48 h of serum deprivation in the human neuroblastoma cell line, SK-N-SH, upregulated the mRNA expression levels of VEGF, VEGFR1, VEGFR2 and NRP1 and downregulated NRP2 (Fig. 1A). Since VEGF signals most biological functions through VEGFR2, we examined whether the upregulation in VEGFR2 gene expression coincided with increased levels of protein. Immunoprecipitation experiments showed that VEGFR2 protein was only detected in serum starved cells at similar levels in the absence and presence of exogenous VEGF (Fig. 1B). Consistent with these results, VEGFR2 phosphorylation was only detected in immunoprecipitates from serum starved cells (Fig. 1C, lane 2) and densitrometry scans revealed that this activation was augmented 2-fold by exogenous VEGF (lane 3) and reduced by a selective inhibitor of VEGFR2 activation, SU1498, to 25% of that observed in the absence (compare lanes 2 and 4) and presence (compare lanes 3 and 5) of VEGF. In order to establish whether VEGF or VEGFR2 mediate neuronal cell survival, cell viability was measured in SK-N-SH cells following incubations for 48 h in the presence (Fig. 2A) and absence (Fig. 2B) of serum with either exogenous VEGF, a neutralizing antibody against VEGF or SU1498. Whereas no changes were observed in the viability of serum treated cells, VEGF increased survival by 20% in serum deprived cells while inhibition of either VEGF or VEGFR2 incubated without and with VEGF elicited a significant (P < 0.001) loss in viability. The stimulation in survival by exogenous VEGF in serum starved cells was observed at concentrations ranging from 20 to 100 ng/ml of the growth factor (data not shown). The specificity of the effects of the pharmacological inhibitor SU1498 was confirmed in cell viability assays with serum starved SK-N-SH cells that were transfected with siRNA that specifically silenced VEGFR2 gene expression (Fig. 2C, supplementary data Fig. 2A). These findings suggested that VEGF signals protection in serum deprived neuronal cells through an autocrine or paracrine activation of VEGFR2. However, we cannot rule out the possibility that NRP1 may heterodimerize with VEGFR2 as a co-receptor to signal survival under these conditions (11, 12).

VEGF and its receptors are upregulated by serum deprivation in neuronal cells and VEGF signals VEGFR2 activation. (A) RT-PCR analyses for VEGF and receptor expression were performed with SK-N-SH cells cultured for 48 h at 37°C under serum and serumfree conditions as described in “Materials and Methods.” (B) VEGFR2 protein was immunoprecipitated from total lysates of cells grown as described in A with the addition of serum deprived cells treated with 10 ng/ml of exogenous VEGF. (C) VEGFR2 protein was immunoprecipitated from total lysates of cells grown as described in B with the addition of serum deprived cells treated with 10 μM of the VEGFR2 inhibitor SU1498 in the absence and presence of 10 ng/ml of exogenous VEGF. Immunoprecipitated VEGFR2 was analyzed by Western blotting for total protein in B and phosphorylated receptor in C using an anti-VEGFR2 or anti-phosphotyrosine antibody (1:1000), respectively

Inhibition of VEGF or VEGFR2 decreases cell viability only in serum starved neuronal cells. Cells were incubated with (A) and without (B) serum in the absence and presence of VEGF (VE) or 0.5 μg/ml of neutralizing antibody against human VEGF (anti-VE), or 10 μM SU1498 (SU) or SU1498 in combination with VEGF (SU/VE). After 48 h at 37°C, cells were assayed for viability using a MTS assay as described in “Materials and Methods.” (C) Serum starved cells were also transfected with 20 μM of VEGFR2 siRNA duplexes (siRNA) or non-specific siRNA duplexes as a control (siC) or treated with SU1498 (SU) and assayed for viability. Results represent the ± S.E.M. of the percent cell viability relative to untreated (A) or serum serumfree (B, C) control cells (100%) from at least three independent experiments. (***P < 0.001) indicates significance between VE or siC and inhibitor treated cells
PKA contributes to survival and ERK1/2 activation in response to serum deprivation
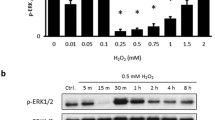
The cAMP-dependent PKA pathway plays a prosurvival role in neuronal cells by mediating protection through ERK1/2 activation [20–22, 24]. In order to test whether PKA activation also contributed to survival, SK-N-SH cells were assayed for cell viability following 48 h of incubation with a selective inhibitor of PKA, H89 in the presence and absence of serum. Consistent with the results in Figs. 2A and 2B, H89 induced a significant decrease in cell viability only in serum deprived SK-N-SH cells (Fig. 3A). In order to insure that the inhibitory effect of H89 was specific for PKA, cell viability measurements were confirmed in comparative assays with a highly selective peptide inhibitor PKI 14–22 Amide (supplementary data, Fig. 2B). However, treatments with H89 potentiated the cell death induced by VEGFR2 siRNA (Fig. 3B), suggesting that VEGFR2 and PKA functioned in serum deprived cells as distinct prosurvival mechanisms. In order to examine whether both pathways activate similar prosurvival mechanisms, we compared the effects of VEGF and the cAMP analogue 8-(4-chlorophenylthio)-adenosine-3′,5′-cyclic monophosphate (8-CPT-cAMP), a PKA activator, on ERK1/2 phosphorylation. Whereas 8-CPT-cAMP increased the endogenous levels of ERK phosphorylation in serum starved SK-N-SH cells (Fig. 3C, compare lanes 1 and 3), maximal induction was achieved with VEGF independent of 8-CPT-cAMP (Fig. 3C, lanes 2 and 4).

The VEGFR2 and PKA pathways signal survival in serum deprived neuronal cells. (A) Cells were incubated with and without serum for 48 h in the absence and presence of 10 μM H89 and assayed for viability as described in “Materials and Methods.” (B) Cell viability was measured in cells transfected with 20 μM of control non-specific siRNA duplexes (siC), 20 µM VEGFR2 siRNA dupexes (siRNA), 10 µM H89 alone (siC/H89) and VEGFR2 siRNA and H89 in combination (siRNA/H89) for 48 h under serumfree conditions. (C) Lysates were prepared from cells treated in the absence and presence of 10 ng/ml VEGF and 250 μM 8-cAMP-CPT (CPT) alone or in combination and then analyzed by Western blotting as described in “Materials and Methods” for phosphorylated and total forms of ERK1/2. Actin serves as a protein loading control. Results represent the ± S.E.M. of the percent cell viability of H89 treated cells relative to serum and serumfree controls (100%) in A or serumfree cells transfected with siC (100%) in B from at least three independent experiments. (***P < 0.001 and **P < 0.01) indicate significance between serum and serumfree cells treated with H89 in A or between VEGFR2 siRNA and siC or siC/H89 and siRNA/H89 in B
Since VEGF and PKA can also signal activation of the prosurvival kinase Akt [8, 9, 23] lysates from serum starved cells treated with SU1498 and H89 in the absence and presence of VEGF were analyzed by Western blotting analyses for phosphorylation of Akt and ERK1/2. While H89 reduced ERK1/2 activation irrespective of exogenous VEGF, the inhibitor had little effect on the phosphorylation of Akt (Fig. 4A compare lanes 1 with 5 and 2 with 6). Conversely, VEGFR2 inhibition abrogated ERK1/2 and Akt phosphorylation alone or in combination with H89 (Fig. 4A lanes 3, 4, 7, and 8) independent of exogenous VEGF. These findings suggested that both the VEGF/VEGFR2 and PKA pathways differentially signal the phosphorylation of ERK1/2 while VEGFR2 is the primary regulator of Akt activation. The specificity of VEGF/VEGFR2 signaling was assured in experiments showing that treatments with VEGFR2 siRNA mimicked SU1498 in preventing ERK1/2 and Akt activation by endogenous VEGF in serum starved cells (supplementary data, Fig. 3). The partial activation of ERK1/2 in H89 treated cells and its loss by VEGFR2 inhibition in Fig. 4A also suggested that PKA contributes to ERK1/2 activation downstream from VEGFR2.

VEGR2 and PKA differentially activate ERK1/2 and Akt in serum deprived SK-N-SH neuronal cells. (A) Lysates were prepared from serum starved cells that were either untreated or treated with 10 μM SU1498 (SU) or 10 μM H89 in the presence and absence of exogenous VEGF (VE) and subjected to Western blot analyses for ERK1/2 and Akt activation. Total ERK1/2, Akt and actin serve as protein loading controls. (B) Nuclear and cytoplasmic extracts were prepared from serum starved cells incubated in the absence (0 min) and presence of 10 ng/ml VEGF for 10, 30, and 60 min as described in “Materials and Methods.” Immunoblot analyses were then performed to detect the total and phosphorylated forms of ERK1/2. Actin serves as a protein loading control. (C) Lysates were prepared from serum starved cells that were treated with and without 10 μM U0126 (U) each in the absence and presence of 10 ng/ml VEGF and analyzed for activated and total ERK1/2
In order to assess the subcellular activation of ERK1/2 by VEGF, cytoplasmic and nuclear extracts were prepared from serum starved cells treated with VEGF for 10, 30 and 60 min and analyzed by Western blotting. VEGF stimulated a rapid phosphorylation of ERK1/2 in the nucleus that peaked at 10 min (Fig. 4B, compare lanes 6 and 8) and an activation of ERK1/2 in the cytoplasm that was only evident following 30 min of stimulation (Fig. 4B, lanes 3 and 4). No change occurred in the total levels of ERK1/2 in the cytoplasm and nucleus. In order to assess a role for MEK in VEGFR2-mediated activation of ERK1/2, serum starved cells were treated with the MEK selective inhibitor U1026 in the absence and presence of VEGF. As shown in Fig. 4C (lanes 3 and 4) MEK inhibition abolished ERK1/2 activation, suggesting that VEGFR2 mediates survival through the MEK/ERK1/2 pathway in serum deprived neuronal cells.
PKA or VEGFR2 inhibition induces a caspase-dependent cell death that is prevented by treatments with z-VAD-fmk
VEGF and PKA can protect neuronal cells from stress-induced cell death by suppressing caspase-3 activation [16, 24]. In our neuronal model, serum deprivation induced over a 2-fold increase in caspase-3/7 activation (Fig. 5A) that was reduced significantly by treatments with VEGF or 8-CPT-cAMP (Fig. 5B) and increased even 2-fold higher by VEGFR2 or PKA inhibition either alone or in combination (Fig. 5C). Treatments with exogenous VEGF had little effect on caspase-activation when VEGFR2 was inhibited by SU1498 (Fig. 5C) or VEGFR2 siRNA (supplementary data, Fig. 4). However, pretreatments with the pan caspase inhibitor z-VAD-fmk decreased caspase activity (Fig. 6A) and promoted survival (Fig. 6B) when serum starved cells were incubated with SU1498 or H89 alone but not in combination. Similarly, Hoechst staining of nuclei in serum starved cells treated with SU1498, H89 or the selective inhibitor of PKA, PKI 14–22 Amide showed that the condensation and chromatin fragmentation associated with apoptosis were prevented by z-VAD-fmk (Fig. 6C; compare arrows in left panel with right panel). Together, these data suggest that the VEGFR2 or PKA pathway signals protection against a caspase-dependent cell death induced by serum deprivation. However, the finding that z-VAD-fmk failed to rescue cells when VEGFR2 and PKA were inhibited simultaneously suggests that survival requires either pathway to function.

Serum deprivation induces caspase activation which is suppressed by the VEGFR2 and PKA signaling pathways. (A) Caspase-3/7 was measured as described in “Materials and Methods” in cells incubated in the presence and absence of serum and (B) in serum starved cells treated with 10 ng/ml VEGF (VE) or 250 μM of 8-CPT-cAMP (CPT) or (C) with serum starved cells that were untreated or treated with VEGF (VE) alone or 10 μM SU1498 in the absence (SU) and presence of exogenous VEGF (SU/VE) or 10 μM H89 (H89) alone or in combination with SU1498 (SU/H89). Results represent the ± S.E.M. of the caspase activity (units/μg protein) relative to serumfree control (100%) from at least three independent experiments. (*P < 0.05; ***P < 0.001) indicates significance between VEGF or CPT and serumfree control in B and VEGF or serumfree control and SU, SU/VE, H89 and SU/H89 in C

Caspase inhibition prevents the cell death induced by a blockade of VEGFR2 or PKA function. (A) Caspase-3/7 activation and (B) cell viability were measured in serum starved cells pretreated without and with 20 μM of the caspase inhibitor z-VAD-fmk (z-VAD) followed by incubations with 10 μM SU1498 (SU) and H89 either alone or in combination. Results represent the ± S.E.M. of the percent cell viability or caspase activity (units/μg protein) relative to serumfree control (100%) from at least three independent experiments. (***P < 0.001) indicates significance between cells pretreated without and with z-VAD alone or in combination with SU or H89 or SU/H89 in A and SU and H89 in B. NS, not significant. (C) Nuclear morphology of cells treated with SU, H89 and the PKA inhibitor PKI 14–22 Amide (PKI) without and with z-VAD were analyzed for chromatin condensation (arrows) by staining with Hoechst 33342 as described in “Materials and Methods.” Scale bar is 20 μm in length
VEGFR2 inhibition induces Bcl-xL depletion
In many paradigms of cellular stress, death can result from a mitochondrial dysfunction involving decreased levels of the antiapoptotic protein Bcl-xL (26). Bcl-xL functions by preventing the mitochondrial membrane permeabilization that leads to cytochrome c release, caspase activation and cell death [26]. Since activated ERK1/2 pathway can prevent mitochondrial dysfunction by modulating Bcl-xL expression levels [26, 27], we examined whether the differential loss in ERK1/2 phosphorylation by VEGFR2 or PKA inhibition (Fig. 4A) was paralleled by similar changes in the protein levels of Bcl-xL. As shown in Fig. 7A (lanes 3 and 4), Bcl-xL protein levels were depleted by SU1498 or VEGFR2 siRNA irrespective of VEGF and reduced by H89 alone (lane 5) but abrogated when H89 was in combination with VEGFR2 siRNA (Fig. 7A, lane 6). These results implicated VEGFR2 in regulating Bcl-xL expression levels possibly through activated ERK1/2. In order to address a role for Bcl-xL in neuronal cell survival, serum starved cells were transfected with a construct that overexpresses Bcl-xL and treated with SU1498. Indeed, Bcl-xL overexpression decreased caspase activation (Fig. 7B) and promoted survival (Fig. 7C), indicating that the protection provided by VEGFR2 is also Bcl-xL-dependent. However, Bcl-xL overexpression had no effect on H89-induced cell death (data not shown).

Overexpression of Bcl-xL prevents the cell death induced by VEGFR2 inhibition. (A) Lysates from serum starved cells treated without and with 10 ng/ml VEGF alone or in combination with 20 μM VEGFR2 siRNA, 10 μM SU1498 (SU) or H89 alone or in combination with VEGFR2 siRNA were analyzed by Western blotting for detection of Bcl-xL with actin as a loading control. (B) Capase activation and (C) cell viability were measured in cells transfected with a construct that overexpresses Bcl-xL and then incubated without and with SU1498 (SU). Cells transfected with a pcDNA empty vector served as a control. Results represent the ± S.E.M. of the percent cell viability or caspase activity (units/μg protein) relative to serumfree controls (100%) from at least three independent experiments. (***P < 0.001) denote significance between SU and SU/Bcl-xL in B and C
ERK1 overexpression protects against a caspase-dependent cell death
Since our results suggested that VEGFR2 and PKA mediate survival through ERK1/2, we examined whether ERK1 activation alone would protect against the cell death induced by VEGFR2 and PKA inhibition. Overexpression of ERK1 in SU1498 or H89 treated cells diminished caspase-3/7 activation (Fig. 8A) and increased cell viability (Fig. 8B) to levels that resembled that observed with z-VAD-fmk (Fig. 6A and 6B) and Bcl-xL overexpression (Figs. 7B and 7C). However, ERK1 overexpression diminished caspase-3/7 activity in cells cotreated with SU1498 and H89 but failed to prevent cell death. The inclusion of the MEK inhibitor U0126 abrogated the protection elicited by z-VAD-fmk and ERK1 and Bcl-xL overexpression in SU1498 treated cells (Fig. 8C), indicating that VEGFR2 signals MEK activation of ERK1/2 to promote survival. Similarly, MEK inhibition also prevented the protection induced by caspase inhibition and ERK1 overexpression in H89 treated cells (data not shown). Collectively, these findings indicate that the protection mediated through the VEGFR2 or PKA signaling pathways in serum starved neuronal cells is dependent upon activation of the MEK/ERK1/2 signaling cascade. A model of this neuroprotective mechanism is given in Fig. 9.

The protection provided by caspase inhibition and overexpression of ERK1 and Bcl-xL in SU1498 treated cells is MEK-dependent. (A) Caspase activity and (B) cell viability were measured in serum starved cells transfected with a construct that overexpresses ERK1 and incubated with 10 μM SU1498 (SU) or 10 μM H89 alone or in combination. Results represent the ±S.E.M. of the percent cell viability or caspase activity (units/μg protein) relative to serumfree controls (100%) from at least three independent experiments. (***P < 0.001) denotes significance between ERK1 and ERK1/SU/H89. (C) Cell viability was measured in cells treated with 20 μM z-VAD-fmk (z-VAD) or transfected with the Bcl-xL or ERK1 construct and incubated with 10 μM SU1498 (SU) in the absence and presence of U0126 (U). Results represent the ± S.E.M. of the percent cell viability or caspase activity (units/μg protein) relative to serumfree control (100%) from at least three independent experiments. (***P < 0.001) denotes significance between SU/Bcl-xL or SU/ERK1 or SU/z-VAD without and with U in combination

Model for protective mechanism by VEGF and PKA in neuronal cells after serum deprivation. Serum deprivation induces increased levels of VEGF and VEGFR2 (upward arrows) which together with PKA signal protection through the MEK/ERK1/2 pathway. VEGFR2 also activates the PI3K/Akt pathway. Activated ERK1/2 (or ERK1 overexpression) mediates a decrease in caspase-3/7 activation and cell death (downward arrows) to provide neuroprotection perhaps through transcriptional-dependent and -independent mechanisms. VEGFR2 also regulates Bcl-xL protein expression levels possibly through ERK1/2 (hatched arrow) to protect against caspase activation
Discussion
Herein, we show that the PKA and the VEGF/VEGFR2 pathways converge at the MEK/ERK1/2 signaling cascade to promote survival in serum starved neuronal cells. Unlike the demonstration that PKA inhibits VEGF-directed activation of ERK1/2 in endothelial cells [19], our findings are consistent with evidence that PKA stimulates ERK1/2 signaling in neuronal cells to mediate growth factor-induced cellular processes [20, 28]. Our demonstration that ERK1/2 activation by VEGF and 8-cAMP-CPT (Fig. 3C) is associated with a parallel decrease in caspase-3/7 activity (Fig. 5B) is also in agreement with the notion that the PKA and VEGF signaling pathways provide neuroprotection by suppressing caspase-3 activation [16, 24].
One important aspect of these studies is that prolonged serum deprivation in neuronal cells upregulates the gene expression of VEGF and its cognate receptors VEGFR1, VEGFR2 and NRP1. While all three receptors may function under these conditions, the demonstrations that exogenous VEGF increased the phosphorylation levels of VEGFR2 (Fig. 1C) and a blockade of this event results in cell death (Figs. 2B and 2C) is consistent with previous evidence that VEGF signals through VEGFR2 to promote neuronal cell survival [7]. Our results indicate that VEGFR2 and PKA differentially activate ERK1/2 to promote survival in response to serum deprivation. In this regard, densitometry scans of ERK1/2 phosphorylation by both pathways (Fig. 3C) showed that activation by VEGF was 2-fold greater than that induced by the PKA activator, 8-cAMP-CPT. The observation that the maximal levels of activated ERK1/2 induced by VEGF (Fig. 3C lane 2; Fig. 4A lane 2) are abrogated only by the inhibition of VEGFR2 but not PKA (Fig. 4A, lanes 4 and 6) suggests that VEGFR2 resides upstream from PKA in signaling the activation of the MEK/ERK1/2 pathway. In accordance with this finding, VEGFR2 inhibition also diminshed the protein levels of Bcl-xL while PKA inhibition only reduced this event by approximately 30% (Fig. 7A). However, the fact that VEGF fails to restore ERK1/2 phosphorylation to peak levels and promote survival in H89 treated cells (Fig. 4A, lane 6; data not shown) also suggests that VEGFR2 and PKA may depend upon common regulatory points to fully activate the MEK/ERK1/2 cascade. One possible regulatory mechanism is through the small GTPase binding proteins, such as Ras, Rac and Rap1 which together with the Raf kinases, A-Raf, B-Raf, and Raf-1 are key elements in regulating the MEK/ERK1/2 pathway by receptor tyrosine kinases and PKA [29]. In this context, VEGF-mediated activation of VEGFR2 is known to signal ERK1/2 phosphorylation through the sequential activations of Ras/Raf-1 [30] while Rap-1-mediated activation of B-Raf links PKA activation to MEK1 stimulation in neuronal cells [28]. However, PKA activation may also require Ras as well as protein kinase C (PKC) which is also a downstream effector of VEGFR2-directed activation of ERK1/2 [11, 17, 31, 32]. While further experimentation is needed, it is tempting to speculate that VEGFR2 and PKA signaling share Ras or PKC as activators of the MEK/ERK1/2 pathway in response to serum deprivation. However, the observation that a simultaneous blockade of VEGFR2 and PKA (Fig. 3B) induces an increased cell death that cannot be alleviated by caspase inhibition (Fig. 6B) or ERK1 overexpression (Fig. 8B) also suggests that both pathways are distinct in mediating survival in serum deprived neuronal cells.
Our finding that VEGFR2 modulates Akt phosphorylation (Fig. 4A) is in agreement with increasing evidence that VEGF-mediated activation of the PI3K/Akt and MEK/ERK1/2 pathways serve as effectors of neuroprotection against damaging insults [8, 9]. Interestingly, we found that only MEK inhibition induced a significant increase in caspase activation (supplementary data, Fig. 5A). Cell viability was diminished only when PI3K/Akt and MEK/ERK1/2 were blocked simultaneously (supplementary data, Fig. 5B), suggesting that both may serve as compensatory prosurvival mechanisms. Nevertheless, our observation that MEK alone is required to signal protection (Fig. 8C) when VEGFR2-directed activation of ERK1/2 and Akt is blocked (Fig. 4A) suggests that the MEK/ERK1/2 signaling cascade is necessary and sufficient to suppress apoptosis in our paradigm of neuronal cell death. In agreement with these findings, brain-derived neurotrophic factor (BDNF) was shown to protect neurons from the damaging effects of hypoxia through ERK1/2 but not PI3K [33]. Similarly, blocking VEGF-mediated signaling of MEK but not PI3K in cortical neurons sensitized neuronal cells toward an apoptotic cell death [7], suggesting that ERK1/2 plays a more direct role in preventing the activation of cell death pathways. Furthermore, survival in prostate cancer cells was attributed to a coordinated activation of ERK1/2 by the PKA, epidermal growth factor (EGF) and insulin-like growth factor I (IGF-I) signaling pathways [34]. Collectively, these data implicate ERK1/2 as a downstream effector of VEGFR2 and PKA-mediated protection against a caspase-dependent cell death in serum deprived neuronal cells.
The observation that VEGF stimulates a cytoplasmic and nuclear phosphorylation of ERK1/2 (Fig. 4B) raises the possibility that ERK1/2 may target mechanisms in both cellular compartments to promote survival. This hypothesis is based on emerging evidence that ERK1/2 can prevent mitochondrial dysfunction in neuronal cells through transcriptional-dependent and -independent mechanisms [26, 27]. The integrity of the mitochondria is tightly regulated by the Bcl-2 family of proteins where a balance in the ratio of antiapoptotic (e.g., Bcl-2 and Bcl-xL) and proapoptotic (e.g., Bax and Bak) members protect against outer membrane permeabilization that leads to cytochrome c release and caspase activation [35]. In our studies, the observation that the cell death induced by VEGFR2 inhibition was manifested by coincident losses in activated ERK1/2 and Bcl-xL protein levels and prevented by Bcl-xL or ERK1 overexpression suggests that VEGF may modulate mitochondrial function through an ERK1/2-mediated regulation of Bcl-xL protein expression. Activated ERK1/2 was shown to prevent apoptosis in various cancer cell lines by regulating Bcl-xL expression levels through an ERK1/2-mediated transcriptional activation of cAMP-responsive element binding protein (CREB) [36–38]. In this regard, we have observed that VEGF stimulates an ERK1/2-dependent phosphorylation of CREB in serum deprived SK-N-SH cells that localizes in the nucleus (unpublished data). It is conceivable that activated PKA may also stimulate the nuclear translocation of activated ERK1/2 in neuronal cells to mediate CREB-dependent transcription of genes that promote survival [39]. Alternatively, activated ERK1/2 in the cytoplasm would prevent neuronal cell death by directly preventing mitochondrial membrane depolarization and cytochrome c release [40] or by inactivating the proapoptotic protein BAD by phosphorylation [41]. Indeed, the phosphorylation of ERK1/2 by VEGF in serum-deprived nonneuronal cells was shown to suppress apoptosis by modulating the Bax/Bcl-2 ratio and by preventing cytochrome c release and caspase-3 activation [42].
In conclusion, the present data demonstrate that the VEGF/VEGFR2 and PKA signaling pathways crosstalk in neuronal cells to protect against a caspase-dependent cell death induced by serum starvation. Both pathways promote neuronal cell survival through differential stimulation of the MEK/ERK1/2 pathway with maximal activation requiring VEGF-mediated signaling through VEGFR2. These findings implicate the MEK/ERK1/2 cascade as a critical effector of VEGF and PKA-mediated neuroprotection in response to stressful stimuli.
References
Zachary I (2005) Neuroprotective role of vascular endothelial growth factor: signalling mechanisms, biological function, and therapeutic potential. Neurosignals 14:207–221
Gora-Kupilas K, Josko J (2005) The neuroprotective function of vascular endothelial growth factor (VEGF). Folia Neuropathol 43:31–39
Sondell M, Sundler F, Kanje M (2000) Vascular endothelial growth factor is a neurotrophic factor which stimulates axonal outgrowth through the flk-1 receptor. Eur J Neurosci 12:4243–4254
Silverman WF, Krum JM, Mani N et al (1999) Vascular, glial and neuronal effects of vascular endothelial growth factor in mesencephalic explant cultures. Neuroscience 90:1529–1541
Jin KL, Mao XO, Greenberg DA (2000) Vascular endothelial growth factor rescues HN33 neural cells from death induced by serum withdrawal. J Mol Neurosci 14:197–203
Jin KL, Mao XO, Nagayama T et al (2000) Induction of vascular endothelial growth factor receptors and phosphatidylinositol 3-Kinase/Akt signaling by global cerebral ischemia in the rat. Neuroscience 100:713–717
Ogunshola OO, Antic A, Donoghue MJ et al (2002) Paracrine and autocrine functions of neuronal vascular endothelial growth factor (VEGF) in the central nervous system. J Biol Chem 277:11410–11415
Matsuzaki H, Tamatani M, Yamaguchi A et al (2001) Vascular endothelial growth factor rescues hippocampal neurons from glutamate-induced toxicity: signal transduction cascades FASEB J 15:1218–1220
Kilic I, Kilic E, Ja¨rve A et al (2006) Human vascular endothelial growth factor protects axotomized retinal ganglion cells in vivo by activating ERK1/2 and Akt Pathways. J Neurosci 26:12439–12446
Wang Y, Mao XO, Xie L et al (2007) Vascular endothelial growth factor overexpression delays neurodegeneration and prolongs survival in amyotrophic lateral sclerosis mice. J Neurosci 27:304–307
Olsson AK, Dimberg A, Kreuger J et al (2006) VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol 7:359–371
Neufeld G, Cohen T, Shraga N et al (2002) The neuropilins: multifunctional semaphorin and VEGF receptors that modulate axon guidance and angiogenesis. Trends Cardiovasc Med 12:13–19
Tamagnone L, Comoglio PM (2000) Signalling by semaphorin receptors: cell guidance and beyond. Trends Cell Biol 10:377–383
Kilic E, Kilic U, Wang Y et al (2006) The phosphatidylinositol-3 kinase/Akt pathway mediates VEGF’s neuroprotective activity and induces blood brain barrier permeability after focal cerebral ischemia. FASEB J 20:1185–1187
Wick A, Wick W, Waltenberger J et al (2002) Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J Neurosci 15:6401–6407
Jin K, Mao XO, Batteur SP et al (2001) Caspase-3 and the regulation of hypoxic neuronal death by vascular endothelial growth factor. Neuroscience 108:351–358
Stork PJ, Schmitt JM (2002) Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol 12:258–266
Dumaz N, Marais R (2005) Integrating signals between cAMP and the RAS/RAF/MEK/ERK signalling pathways. FEBS J 272:3491–3504
D’Angelo G, Lee H, Weiner RI (1997) cAMP-dependent protein kinase inhibits the mitogenic action of vascular endothelial growth factor and fibroblast growth factor in capillary endothelial cells by blocking Raf activation. J Cell Biochem 67:353–366
Yao H, York RD, Misra-Press A et al (1998) The cyclic adenosine monophosphate–dependent protein kinase (PKA) is required for the sustained activation of mitogen activated kinases and gene expression by nerve growth factor. J Biol Chem 273:8240–8247
Park K, Luo JM, Hisheh S et al (2004) Cellular mechanisms associated with spontaneous and ciliary neurotrophic factor-cAMP-induced survival and axonal regeneration of adult retinal ganglion cells. J Neurosci 24:10806–10815
Troadec JD, Marien M, Mourlevat S et al (2002) Activation of the mitogen-activated protein kinase (ERK(1/2)) signaling pathway by cyclic AMP potentiates the neuroprotective effect of the neurotransmitter noradrenaline on dopaminergic neurons. Mol Pharmacol 62:1043–1052
Filippa N, Sable C L, Filloux C et al (1999) Mechanism of Protein Kinase B Activation by Cyclic AMP-Dependent Protein. Kinase Mol Cell Biol 19:4989–5000
Wang G, Qi C, Fan GH et al (2005) PACAP protects neuronal differentiated PC12 cells against the neurotoxicity induced by a mitochondrial complex I inhibitor, rotenone. FEBS Lett 579:4005–4011
Rockwell P, Martinez J, Papa L et al (2004) Redox regulates COX-2 upregulation and cell death in the neuronal response to cadmium. Cell Signal 16:343–353
Horbinski C, Chu CT (2005) Kinase signaling cascades in the mitochondrion: a matter of life or death. Free Radic Biol Med 38:2–11
Hetman M, Gozdz A (2004) Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. Eur J Biochem 271:2050–2055
Vossler MR, Yao H, York RD et al (1997) cAMP activates MAP Kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell 89:73–82
Chong H, Vikis HG, Guan KL (2003) Mechanisms of regulating the Raf kinase family. Cell Signal 15:463–469
Olsson AK, Dimberg A, Kreuger J et al (2006) VEGF receptor signalling — in control of vascular function. Nat Rev Mol Cell Biol 7:359–371
Bouschet T, Perez V, Fernandez C et al (2003) Stimulation of the ERK pathway by GTP-loaded Rap1 requires the concomitant activation of Ras, Protein Kinase C, and Protein Kinase A in neuronal cells. J Biol Chem 278:4778–4785
Iida N, Namikawa K, Kiyama H et al (2001) Requirement of Ras for the activation of Mitogen-Activated Protein Kinase by calcium influx, cAMP, and neurotrophin in hippocampal neurons. J Neurosci 21:6459–6466
Han BH, Holtzman DM (2000) BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J Neurosci 20:5775–5781
Putz T, Culig Z, Eder IE et al (1999) Epidermal growth factor (EGF) receptor blockade inhibits the action of EGF, insulin-like growth factor I, and a protein kinase A activator on the mitogen-activated protein kinase pathway in prostate cancer cell lines. Cancer Res 59:227–233
Cory S, Adams JM (2002) The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2:647–656
Eliseev RA, VanWinkle B, Rosier RN et al (2004) Diazoxide-mediated preconditioning against apoptosis involves activation of cAMP-response element-binding Protein (CREB) and NFκB. J Biol Chem 279:46748–46754
Boucher MJ, Morisset J, Vachon PH et al (2000) MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-xL, and Mcl-1 and promotes survival of human pancreatic cancer cells. J Cell Biochem 79:355–369
Mori M, Uchida M, Watanabe T et al (2003) Activation of extracellular signal-regulated kinases ERK1 and ERK2 induces Bcl-xL up-regulation via inhibition of caspase activities in erythropoietin. J Cell Physiol 195:290–297
Impey S, Obrietan K, Wong ST et al (1998) Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron 21:869–883
Lee HJ, Bach JH, Chae HS et al (2004) Mitogen-activated protein kinase/extracellular signal-regulated kinase attenuates 3-hydroxykynurenine-induced neuronal cell death. J Neurochem 88:647–656
Jin K, Mao XO, Zhu Y et al (2002) MEK and ERK protect hypoxic cortical neurons via phosphorylation of Bad. J Neurochem 80:119–125
Baek JH, Jang JE, Kang CM et al (2000) Hypoxia-induced VEGF enhances tumor survivability via suppression of serum deprivation-induced apoptosis. Oncogene 19:4621–4631
Acknowledgments
This project was supported by the National Institutes of Health Grant (NIGMS SCORE to P.R.) and Grant Number RR03037 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material
Rights and permissions
About this article
Cite this article
Gomes, E., Papa, L., Hao, T. et al. The VEGFR2 and PKA pathways converge at MEK/ERK1/2 to promote survival in serum deprived neuronal cells. Mol Cell Biochem 305, 179–190 (2007). https://doi.org/10.1007/s11010-007-9542-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-007-9542-2