
Abstract
Copper (Cu) is one of the most significant trace elements in the body, but it is also a widespread environmental toxicant health. Ferroptosis is a newly identified programmed cell death, which involves various heavy metal–induced organ toxicity. Nevertheless, the role of ferroptosis in Cu-induced hepatotoxicity remains poorly understood. In this study, we found that 330 mg/kg Cu could disrupt the liver structure and cause characteristic morphological changes in mitochondria associated with ferroptosis. Additionally, Cu treatment increased MDA (malondialdehyde) and LPO (lipid peroxide) production while reducing GSH (reduced glutathione) content and GCL (glutamate cysteine ligase) activity. However, it is noticeable that there were no appreciable differences in liver iron content and key indicators of iron metabolism. Meanwhile, our further investigation found that 330 mg/kg Cu-exposure changed multiple ferroptosis-related indicators in chicken livers, including inhibition of the expression of SLC7A11, GPX4, FSP1, and COQ10B, whereas enhances the levels of ACLS4, LPCAT3, and LOXHD1. Furthermore, the changes in the expression of NCOA4, TXNIP, and Nrf2/Keap1 signaling pathway–related genes and proteins also further confirmed 330 mg/kg Cu exposure-induced ferroptosis. In conclusion, our results indicated that ferroptosis may play essential roles in Cu overload–induced liver damage, which offered new insights into the pathogenesis of Cu-induced hepatotoxicity.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Copper (Cu) is a necessary microelement, which also has an irreplaceable function in promoting the activity of essential enzymes in the energy metabolism and many cellular processes [1]. To date, formula feeds containing Cu are still widely used in the livestock industry as growth-promoting stimulants [2]. Noticeably, Cu is also a dangerous toxicant and environmental pollutant [3]. Recently, multiple studies have proposed that Cu pollution in rivers, lakes, soils, and even air around the world is becoming more and more serious due to the long-term abuse of Cu-based agricultural and animal husbandry products (pesticides, fungicides, and feed additives) and the discharge of industrial wastewater [4, 5]. What is more worrying is that high concentrations of Cu in the environment can accumulate in organisms at all trophic levels through the food chain and pose a serious threat to human health [4, 6]. The liver is an important organ that performs several biological functions such as metabolism, detoxification, immune defense, and the storage of trace elements [7, 8]. A continuous intake of a high Cu diet can lead to excessive accumulation of Cu in the liver, which leads to an imbalance of Cu homeostasis in the liver and deposition into other organs and tissues through the circulatory system [2, 9]. In addition, the hepatotoxicity of Cu to a variety of animal species has been confirmed in multiple studies [10]. And the main toxicity mechanism is that excessive hepatic Cu accumulation may lead to oxidative damage by overproduction of ROS, which can induce various types of cell death including autophagy, apoptosis, and necrosis [11,12,13]. Nevertheless, the current research data on whether other types of cell death patterns are associated with Cu-induced hepatotoxicity remain relatively limited. Thus, the underlying mechanism of Cu-induced hepatotoxicity is a concern that deserves sustained attention.
Ferroptosis is a new form of cell death mode that unlike other programmed cell death [14]. The characteristics of ferroptosis primarily involve inhibition of systemic Xc and GPX4 activity, GSH depletion, disturbances in iron metabolism, and abnormal lipid metabolism [15,16,17]. In addition, the morphological characteristics of ferroptosis are mainly manifested by as increased mitochondrial membrane density, mitochondrial contraction, and mitochondrial crista damage [15, 17]. It is noteworthy that some researchers have shown that ferroptosis has become a pivotal mechanism for heavy metal exposure to induce cell death. Wei et al. indicated that mitochondrial dysfunction and ferroptosis are important mechanisms for NiCl2 exposure–induced liver injury [18]. Additionally, the findings of research by Meng et al. have revealed that arsenate could induce iron accumulation and mitochondrial crista breakage and activates ferroptosis-related pathways in the testes of mice [19]. It was also found that Cu can inhibit GPX4 expression by inducing autophagic degradation, which leads to ferroptotic cell death [20]. Nevertheless, the role and detailed mechanism of ferroptosis in Cu-induced hepatotoxicity in chickens remain unknown. Notably, several of our recent works have found that intake of excess Cu causes mitochondrial dysfunction, oxidative damage, and lipid peroxidation in chicken livers [12, 21]. Thus, we speculate that exposure to Cu-induced hepatotoxicity may be associated with activation of the ferroptosis-related signaling pathway.
As the main protein source of the human diets, chickens are more susceptible to toxic effects caused by dietary intake of copper than other poultry. Thus, this study is designed to reveal the role of ferroptosis in Cu-caused hepatotoxicity by using a model of Cu poisoning in chickens and to explore its potential molecular mechanisms. Our results will provide novel insights and useful information for Cu-induced hepatotoxicity.
Materials and Methods
Animals and Treatment
In the present study, a total of two hundred and forty (240) broiler chicks (specific pathogen-free) were purchased from a local hatchery. The chicks were reared in suitable controlled environments and they were free to acquire the dietary and water. All chicks were randomized into four groups: control group (11 mg Cu/kg diet), low Cu group (110 Cu/kg diet), middle Cu group (220 Cu/kg diet), and high Cu group (330 Cu/kg diet). Copper sulfate (CuSO4) was added to the basic diet as a source of Cu, and the dosages of Cu were selected based on our previous investigation [3]. After all treatments lasted 49 days, chickens were euthanized and the livers were excised rapidly. All animal procedures were approved and implemented by the Ethics Committee of South China Agricultural University (2017A087).
Histopathological Studies and Ultrastructural Observation
At the end of the experiment, liver samples used for histopathological studies were extracted and embedded in paraffin by following standard histopathologic techniques. Next, sections were cut into 5 μm for H&E staining [22]. Histopathological examination was performed under an optical microscope (Leica, Germany).
Ultrastructural observation by transmission electron microscope (TEM, Thermo Fisher, USA) was preformed to evaluate the influence of Cu on liver tissues. Briefly, after liver tissues fixing with 2.5% glutaraldehyde solution, the liver’s ultra-thin sections were prepared using the previously described method [23].
Liver Iron Content Assay
The total-iron content in the livers was evaluated using a commercial tissue iron assay kit (Dojindo, Tokyo, Japan). Moreover, liver tissue sections were prepared per the method of H&E staining, and then divalent iron were measured by the Lillie staining solution (Solarbio, China). In brief, sections were dye with Lillie staining solution for 60 min, and then re-dye with nuclear fast red for 3 min, and finally conventionally dehydrate, transparent, and sealed with neutral resin.
Oxidative Stress Parameter Assays
The levels of lipid peroxidation (LPO, Nanjing-Jiancheng, China), malondialdehyde (MDA, Beyotime, China), reduced glutathione, and oxidized glutathione disulfide (GSH and GSSG, Beyotime, China) as well as the activity of γ-glutamyl cysteine ligase (GCL, Solarbio, China) in liver tissues were measured following the instructions for the respective kit.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Analysis
All experimental steps for qRT-PCR analysis referred to the previous methods [23]. The sequences of target gene primers are provided in Table 1 (Supplementary Materials).
Western Blot Analysis
Sample preparation and western blot analysis were performed as previously described [23]. The primary antibodies used in the present study include β-actin/GPX4/NCOA4 (Bioss, Beijing, China), Nrf2/Keap1/TF/TFR1/FTL/FSP1/SLC7A11 (Proteintech, Wuhan, China), FTH1/ACLS4 (Affinity, Jiangsu, China), and HO-1/SOD1/TXNIP/TRX (Abcam, MA, USA). The protein band intensity was quantified using Image J software and normalized by β-actin.
Immunohistochemistry and Immunofluorescence Staining Analyses
This study further analyzed the situ expression levels of SLC7A11 and GPX4, where the methods used by immunohistochemical and immunofluorescence staining techniques were performed in accordance with previous methods [23]. SLC7A11 antibody was purchased from Proteintech (NO. 26864-1-AP, 1:500, Wuhan, China), GPX4 was purchased from Proteintech (NO. 67763-1-Ig, 1:800, Wuhan, China).
Statistical Analysis
Data analysis was conducted using GraphPad Prism 8.0 (GraphPad Inc., USA). The results are shown in the form of mean ± standard error of means (SEM). Differences among groups were analyzed using one-way analysis of variance (ANOVA). A P value < 0.05 was deemed statistically significant difference.
Results
Liver Pathological Analysis and Ultrastructural Observation
As presented in Fig. 1A, the histological observation revealed that the structure and morphology of liver in control group were normal. Furthermore, compared to control group, no obvious abnormalities were found after treatment with 110 and 220 mg/kg Cu. However, in the 330 mg/kg Cu treatment group, the hepatocyte vacuolar degeneration (black arrow) and hepatic sinusoid widened were observed. In addition, we further evaluated the ultrastructure changes of the liver by using TEM, and observed characteristic morphological characteristics associated with ferroptosis in 330 mg/kg Cu group, such as mitochondrial membrane rupture (red arrow), mitochondrial cristae disappeared (yellow arrow), and uniform round-shaped mitochondria (Fig. 1B).

Effects of Cu on the histopathological and ultrastructure in chicken liver. A Cu-induced histopathological changes in chicken liver (× 400; scale bar, 50 μm), hepatocyte vacuolar degeneration (black arrow). B Ultrastructural observations in the liver tissues; N, nucleus; Mito, mitochondria; mitochondrial membrane rupture (red arrow); mitochondrial cristae disappeared (yellow arrow). The pseudo-color in the figure represents mitochondria
Effects of Cu Exposure on Oxidative Stress Indicators in Chicken Livers
Cellular lipid peroxidation and glutathione depletion are considered to be the important features of ferroptosis. Therefore, we assessed the effects of Cu exposure on LPO, MDA, GSH, GSSG, and GCL changes in livers. As shown in Fig. 2A, B, 330 mg/kg Cu treatment potently increased the contents of LPO, while markedly decreasing the activities of GCL compared to control. Meanwhile, the MDA content displayed an increasing trend after Cu-treated, and a significant difference was observed in the 220 and 330 mg/kg Cu-treated groups (Fig. 2C, P < 0.05). Furthermore, the contents of GSH and GSSG showed a similar trend in the livers, which were dramatically decreased in the 220 and 330 mg/kg Cu-treatment groups than in the control group (Fig. 2D, E, P < 0.05). Additionally, the ratios of GSH/GSSG in 330 mg/kg Cu-treatment group were also decreased (Fig. 2F, P < 0.05).

Cu exposure induced oxidative stress in chicken liver. A LPO content. B GCL activity. C MDA content. D GSH content. E GSSG content. F GSH/GSSG. All data were expressed as mean ± SEM; n = 6 for each group. “*” indicates statistically significant difference with the control group (*P < 0.05, **P < 0.01, and ***P < 0.001)
Effects of Cu Exposure on the Nrf2 Signaling Pathway in Chicken Livers
Considering the important role of Nrf2 in ferroptosis [24], the levels of Nrf2 and its associated indicators (SOD1, HO-1, TRX, and TXNIP) were assessed. As illustrated in Fig. 3A–C, the mRNA expression levels of Nrf2 and SOD-1 were dramatically downregulated, while Keap1 expression was upregulated in 330 mg/kg Cu-treatment group compared with control group (P < 0.05). Furthermore, the levels of Nrf2 signaling pathway-related proteins were consistent in their corresponding gene expression levels. We found that 330 mg/kg Cu treatment significantly reduced protein levels of Nrf2 (nuclei), SOD1, and TRX (Fig. 3E, F, J, K, P < 0.05). Meanwhile, treatment with 330 mg/kg Cu increased the protein expression of Keap1 and TXNIP (Fig. 3G, I, P < 0.05). Intriguingly, both mRNA and protein levels of HO-1 followed a decreasing trend in the 330 mg/kg Cu treatment group, but were not significantly different (Fig. 3D, H, P > 0.05).

Cu exposure inhibited the Nrf2 signaling pathway in chicken liver. A–D Effects of Cu on the relative mRNA expressions of Nrf2, Keap1, SOD1, and HO-1. E–K Effects of Cu on the relative protein expressions of Nrf2, Keap1, SOD1, HO-1, TXNIP, TRX. All data were expressed as mean ± SEM; n = 6 for each group. “*” indicates statistically significant difference with the control group (*P < 0.05, **P < 0.01, and ***P < 0.001)
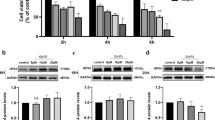
Effects of Cu Exposure on Iron Metabolism in Chicken Livers
To validate whether Cu exposure instigates ferroptosis in hepatocytes by inducing iron disturbances, we measured iron content and the expression of iron metabolism-related factors. The results revealed that exposure to Cu had no obvious effect on the iron content of the liver, and the concentration of hepatic iron presented a trend of first rising and then decreasing in the three Cu treatment groups (Fig. 4A, P > 0.05). Meanwhile, iron accumulation in the liver tissues was not observed by Lillie divalent iron staining results (Fig. 4B). Moreover, the mRNA levels of TF, TFR1, STEAP3, and FTH1 have no significant difference in all groups (Fig. 4C–F, P > 0.05). Similarly, there was no significant change in the protein expression levels of TF, TFR1, and FTH1 between the Cu-exposed group and the control group (Fig. 4I–L, P > 0.05). Nevertheless, the mRNA expression levels of DMT1 in the 330 mg/kg Cu treatment group and FTL in all Cu treatment groups were remarkably increased compared to control (Fig. 4G, H, P < 0.05). Additionally, the protein of FTL was remarkably increased in 330 mg/kg Cu-treated group compared to control (Fig. 4M, P < 0.05).

Effects of Cu exposure on iron metabolism in chicken liver. A Total iron content. B Lillie staining. C–H Effects of Cu on the relative mRNA expressions of TF, TFR1, STEAP3, FTH1, DMT1, and FTL. I–M Effects of Cu on the relative protein expressions of TF, TFR1, FTH1, and FTL. All data were expressed as mean ± SEM; n = 6 for each group. “*” indicates statistically significant difference with the control group (*P < 0.05, **P < 0.01, and ***P < 0.001)
Effects of Cu Exposure on Ferroptosis Pathway in Chicken Livers
Next, we further examined the protein and mRNA levels of ferroptosis correlative indicators. As illustrated in Fig. 5A, compared to control, the mRNA levels of GPX4, SLC7A11, FSP1, and COQ10B in the 330 mg/kg Cu-treatment group were obviously downregulated (P < 0.05), whereas LPCAT3 and LOXHD1 were significantly upregulated. Moreover, the NCOA4 and ACSL4 mRNA transcriptional levels were obviously upregulated in the 220 and 330 mg/kg Cu-treatment groups compared to control group (P < 0.05). Notably, there was no significant change of the mRNA expression levels of SLC3A2 between the Cu exposed group and the control group (P > 0.05). Heatmap visually presented the mRNA expression levels of ferroptosis-related genes. In addition, coincident with the changes in mRNA levels, 330 mg/kg Cu treatment significantly decreased the protein levels of GPX4, SLC7A11, FSP1 and increased the levels of ACLS4 and NCOA4 (Fig. 5B a–f, P < 0.05). Meanwhile, the result of immunofluorescence and immunohistochemistry showed that the positive staining of GPX4 and SLC7A11 in liver was obviously decreased in the 330 mg/kg Cu treatment group (Fig. 5B g–j, P < 0.05).

Cu exposure activated ferroptosis-related signaling pathways in chicken liver. A, a–j Effects of Cu on the relative mRNA expressions of GPX4, SLC7A11, FSP1, NCOA4, COQ10B, ACSL4, LPCAT3, LOXHD1, and SLC3A2. A, a–f Effects of Cu on the relative protein expressions of FSP1, NCOA4, ACSL4, SLC7A11, and GPX4. B, g Immunofluorescent staining analysis the expression of GPX4 protein in chicken liver. B, h Immunohistochemistry staining analysis of the expression of SLC7A11 protein in chicken liver. All data were expressed as mean ± SEM; n = 6 for each group. “*” indicates statistically significant difference with the control group (*P < 0.05, **P < 0.01, and ***P < 0.001)
Discussion
Cu is a widely distributed toxic heavy-metal pollutant, and Cu exposure seriously threatens human health [6, 25]. Thus, revealing the toxicological mechanisms of Cu-induced hepatotoxicity may contribute to the prevention and treatment of Cu poisoning. Our previous studies have demonstrated that the liver Cu content was accumulated remarkably with the increased intake of Cu, and exposure to Cu induces chicken hepatotoxicity by inducing apoptosis and inhibiting mitophagy [3]. Our findings showed that Cu exposure could cause liver damage including steatosis and ultrastructural changes in liver tissues. One of the interesting findings is that we observed characteristic morphological characteristics associated with ferroptosis in the 330 mg/kg Cu-treated group, such as mitochondrial membrane rupture, mitochondrial cristae disappeared, and uniform round-shaped mitochondria. Furthermore, in agreement with previous research, Cu exposure increases liver GSH depletion and lipid peroxidation [21]. These results suggested that ferroptosis might be another crucial contributing mechanism by which Cu induced hepatotoxicity. Therefore, we further explored the underlying molecular mechanisms of this possibility.
The increase in the intracellular labile iron pool (LIP) and the promotion of lipid peroxidation by catalyzing the formation of free radicals through the Fenton reaction are considered to be the critical factor to ferroptosis [26, 27]. Under normal circumstances, extracellular Fe3+ can be introduced into the endosome by binding to transferrin (TF) on the cell membrane to form TF-Fe3+ and forming a complex with the membrane protein transferrin receptor 1 (TFR1) [27]. Fe3+ is then reduced to Fe2+ by the six-transmembrane epithelial antigen of the prostate 3 (STEAP3) and transported via the divalent metal transporter 1 (DMT1) to LIP in the cytoplasm [15, 27]. Additionally, excess iron is stored in ferritin consisting of ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1), or Fe2+ is oxidized to Fe3+ by ferroportin1 (FPN1, SLC40A1) and transported to the extracellular and circulatory systems [27, 28]. Previous studies demonstrated that polystyrene microplastic-induced ferroptosis in the mice liver was featured by inhibited the expression of FTH1 and TFR1 [29]. In addition, Xiao et al. have reported that arsenite perturbs the iron-homeostasis by altering the levels of TfR1, DMT1, and FPN1, thereby increasing the ferroptosis sensitivity of hippocampal tissues and PC-12 cells [30]. Nevertheless, the effect of copper ions accumulated in the organism on iron ion transport and iron homeostasis remains an outstanding question. Studies have shown that intake of 60 ppm dietary copper has no significant effect on iron concentrations in pigs’ livers, while intake of 120 and 240 ppm dietary Cu reduces liver iron concentrations by 50% and 60%, respectively [31]. Furthermore, adding 1000 mg/kg Cu to the diet of laying hens leads to an increase in iron concentrations in their livers [32]. However, after 9 weeks of treating sheep with large amounts of Cu (20 mg of CuSO4/kg body wt. per day), there was no significant change in the concentration of iron in the liver [33]. These preliminary studies suggested that the effect of Cu on iron transport and iron homeostasis is complex and varies with the change of receptor and Cu concentration. In the present study, our findings demonstrated that the iron content and the levels of TF, TFR1, STEAP3, and FTH1 in the liver were not significantly affected by Cu exposure. This is also consistent with previous research which found that Cu supplementation did not affect the expression of TF and TFR1 in HepG2 cells [34]. Interestingly, we observed that the expressions of DMT1 and FTL were remarkably upregulated following Cu treatment. A possible explanation for the DMT1 increase is that the DMT1 also transports Cu in some cells and under certain circumstances [35]. In addition, elevated FTL expression levels indicate that the storage of iron may be in an activated state [36]. Therefore, we speculate that Cu-induced ferroptosis is independent of iron metabolism in the liver of chickens.
To further determine whether Cu induces ferroptosis in vivo, we investigated the effects of Cu exposure on ferroptosis-related pathways, including lipid metabolism, amino acid, and glutathione metabolism pathways. System Xc- (xCT) is an amino acid antiporter composed of the two subunits SLC7A11 and SLC3A2. xCT that is localized on the cell membrane can exchange cystine and glutamate in and out of the cell at a 1:1 ratio, and the cystine that enters the cell is reduced to cysteine under the catalytic action of cystine reductase [15, 37]. Under the enzymatic activity of GCL and GSS, cysteine, glutamic acid, and glycine together can synthesize the endogenous antioxidant glutathione (GSH) [37]. In addition to being an essential part of cell membranes or organelle membranes, lipids regulate a variety of biological functions [26]. But different types of lipids from varied sources regulate various biological processes. Free polyunsaturated fatty acids (PUFAs) can be reacted with phosphatidylethanolamine (PE) to synthesize PUFA-containing phospholipids (PUFAs-PE) under the action of ACSL4 and LPCAT3 [26, 27, 38]. The formed PUFAs-PE can be oxidized to produce a toxic lipid peroxide (L-ROS) by lipoxygenase (LOX)-mediated enzymatically manner, or by a non-enzymatic manner involved in a Fenton reaction mediated by free redox active iron [15, 27]. It is worth noting that both Cu ion and iron ion can exist as divalent metal cations in the body, and Cu (II) ions can also directly catalyze the formation of ROS through Fenton and Haber-Weiss reactions [39]. GPX4 is the only enzyme in the body that converts GSH into GSSG and reduces toxic lipid peroxides to the non-toxic lipid alcohols [16, 40]. Therefore, GPX4 has important implications for the removal of lipid peroxides and the prevention of ferroptosis [40]. In present study, the mRNA and protein expressions of GPX4 and SLC7A11 were significantly reduced in the Cu-treated group, which is consistent with the law of ferroptosis. Moreover, Cu exposure significantly depleted the contents of GSH and inhibited the enzymatic activity of GCL. Meanwhile, the increased expression levels of ACSL4, LPCAT3, and LOXHD1 also substantiated the induction of ferroptosis after Cu treatment. These results were in agreement with those published previously [41, 42]. Moreover, the levels of FSP1 and COQ10B in the Cu treatment group were obviously reduced compared with the control group, while the mRNA and protein expression levels of NCOA4 were significantly increased. Ferroptosis suppressor protein 1 (FSP1) catalyzes the regeneration of non-mitochondrial CoQ10 by NAD(P)H, which can inhibit ferroptosis by directly scavenging lipid peroxyl radicals [27]. Nuclear receptor co-activator 4 (NCOA4) is a selective cargo receptor for the selective autophagy of ferritin in ferroptosis, and its upregulation is correlated to ferroptosis [43]. These data together suggested that GSH metabolism disorders caused by inhibition of SLC7A11 and GPX4 expression and lipid peroxidation are an important factor in Cu overload-induced ferroptosis in chicken livers.
The Nrf2/Keap1 is a critical signaling pathway for cellular antioxidant stress responses [44]. Under oxidative stress conditions, Nrf2 is dissociated from the Nrf2-Keap1 complex and translocated into the nucleus to regulate various antioxidant genes (e.g., HO-1, SOD1) [44, 45]. Furthermore, many recent studies have identified that Nrf2 is involved in regulating the expression of multiple ferroptosis-related genes, such as SLC7A11, GPX4, SLC40A1, and FTH1 [24, 43]. Both HO-1 and SOD1 are powerful antioxidant enzymes in cells, while the function of HO-1 in ferroptosis is controversial because an increase in HO-1 might also increase intracellular concentrations of labile iron [26, 46]. In addition, thioredoxin-interacting/inhibiting protein (TXNIP) and thioredoxin (TRX) have also been shown to be regulators of ferroptosis because of their capacity to maintain oxidative stress balance [47]. Thus, inactivation of the Nrf2/Keap1 signaling could contribute to the promotion of ferroptosis [48]. Consistent with this notion, we found a dramatic increase in the expression levels of keap1 and TXNIP in chicken livers after Cu treatment, and accompanied by a decrease in the levels of Nrf2, HO-1, TRX, and SOD1. These results corroborate that the mechanism of Cu-induced ferroptosis in chicken livers also involves the inhibition of the Nrf2/Keap1 signaling pathway.
Conclusions
Taken together, our findings revealed for the first time that ferroptosis might be a newly characterized form of Cu exposure-induced liver damage. And the potential mechanism involves the disturbances in GSH metabolism, lipid peroxidation, ferritinophagy, and inhibition of FSP1-CoQ10 axis in chicken livers. Our study provides a new cognize for the mechanisms of Cu-induced hepatotoxicity.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Tsang T, Davis CI, Brady DC (2021) Copper biology. Curr Biol 31:R421–R427
Gao Y, Yang W, Che D, Adams S, Yang L (2020) Advances in the mechanism of high copper diets in restraining pigs growth. J Anim Physiol An N 104:667–678
Yang F, Liao J, Yu W, Qiao N, Guo J, Han Q, Li Y, Hu L, Pan J, Tang Z (2021) Exposure to copper induces mitochondria-mediated apoptosis by inhibiting mitophagy and the PINK1/parkin pathway in chicken (Gallus gallus) livers. J Hazard Mater 408:124888
Fagnano M, Agrelli D, Pascale A, Adamo P, Fiorentino N, Rocco C, Pepe O, Ventorino V (2020) Copper accumulation in agricultural soils: risks for the food chain and soil microbial populations. Sci Total Environ 734:139434
Li X, Zhang J, Gong Y, Liu Q, Yang S, Ma J, Zhao L, Hou H (2020) Status of copper accumulation in agricultural soils across China (1985-2016). Chemosphere 244:125516
Zhong G, He Y, Wan F, Wu S, Jiang X, Tang Z, Hu L (2021) Effects of long-term exposure to copper on the Keap1/Nrf2 signaling pathway and Msr-related redox status in the kidneys of rats. Biol Trace Elem Res 199:4205–4217
Cao P, Nie G, Luo J, Hu R, Li G, Hu G, Zhang C (2022) Cadmium and molybdenum co-induce pyroptosis and apoptosis via the PTEN/PI3K/AKT axis in the livers of Shaoxing ducks (Anas platyrhynchos). Food Funct 13:2142–2154
Michalopoulos GK, Bhushan B (2021) Liver regeneration: biological and pathological mechanisms and implications. Nat Rev Gastro Hepat 18:40–55
Linder MC (2020) Copper homeostasis in mammals, with emphasis on secretion and excretion. a review. Int J Mol Sci 21
Johncilla M, Mitchell KA (2011) Pathology of the liver in copper overload. Semin Liver Dis 31:239–244
Wang X, Wang H, Li J, Yang Z, Zhang J, Qin Z, Wang L, Kong X (2014) Evaluation of bioaccumulation and toxic effects of copper on hepatocellular structure in mice. Biol Trace Elem Res 159:312–319
Yang F, Pei R, Zhang Z, Liao J, Yu W, Qiao N, Han Q, Li Y, Hu L, Guo J, Pan J, Tang Z (2019) Copper induces oxidative stress and apoptosis through mitochondria-mediated pathway in chicken hepatocytes. Toxicol In Vitro 54:310–316
Yu W, Liao J, Yang F, Zhang H, Chang X, Yang Y, Bilal RM, Wei G, Liang W, Guo J, Tang Z (2021) Chronic tribasic copper chloride exposure induces rat liver damage by disrupting the mitophagy and apoptosis pathways. Ecotox Environ Safe 212:111968
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison BR, Stockwell BR (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–1072
Cepelak I, Dodig S, Dodig DC (2020) Ferroptosis: regulated cell death. Arh Hig Rada Toksiko 71:99–109
Tang D, Chen X, Kang R, Kroemer G (2021) Ferroptosis: molecular mechanisms and health implications. Cell Res 31:107–125
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D (2016) Ferroptosis: process and function. Cell Death Differ 23:369–379
Wei L, Zuo Z, Yang Z, Yin H, Yang Y, Fang J, Cui H, Du Z, Ouyang P, Chen X, Chen J, Geng Y, Zhu Y, Chen Z, Huang C, Wang F, Guo H (2022) Mitochondria damage and ferroptosis involved in Ni-induced hepatotoxicity in mice. Toxicology 466:153068
Meng P, Zhang S, Jiang X, Cheng S, Zhang J, Cao X, Qin X, Zou Z, Chen C (2020) Arsenite induces testicular oxidative stress in vivo and in vitro leading to ferroptosis. Ecotox Environ Safe 194:110360
Xue Q, Yan D, Chen X, Li X, Kang R, Klionsky DJ, Kroemer G, Chen X, Tang D, Liu J (2023) Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy 19:1982–1996
Yang F, Liao J, Pei R, Yu W, Han Q, Li Y, Guo J, Hu L, Pan J, Tang Z (2018) Autophagy attenuates copper-induced mitochondrial dysfunction by regulating oxidative stress in chicken hepatocytes. Chemosphere 204:36–43
Zhang C, Lin T, Nie G, Hu R, Pi S, Wei Z, Wang C, Li G, Hu G (2021) In vivo assessment of molybdenum and cadmium co-induce nephrotoxicity via causing calcium homeostasis disorder and autophagy in ducks (Anas platyrhyncha). Ecotox Environ Safe 230:113099
Wan F, Zhong G, Ning Z, Liao J, Yu W, Wang C, Han Q, Li Y, Pan J, Tang Z, Huang R, Hu L (2020) Long-term exposure to copper induces autophagy and apoptosis through oxidative stress in rat kidneys. Ecotox Environ Safe 190:110158
Dodson M, Castro-Portuguez R, Zhang DD (2019) NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol 23:101107
Liao J, Li Q, Hu Z, Yu W, Zhang K, Ma F, Han Q, Zhang H, Guo J, Hu L, Pan J, Li Y, Tang Z (2022) Mitochondrial miR-1285 regulates copper-induced mitochondrial dysfunction and mitophagy by impairing IDH2 in pig jejunal epithelial cells. J Hazard Mater 422:126899
Liu J, Kang R, Tang D (2021) Signaling pathways and defense mechanisms of ferroptosis. Febs J 289(22):7038–7050
Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, Lei P (2021) Ferroptosis: mechanisms and links with diseases. Signal Transduct Tar 6:49
Chen X, Li J, Kang R, Klionsky DJ, Tang D (2021) Ferroptosis: machinery and regulation. Autophagy 17:2054–2081
Mu Y, Sun J, Li Z, Zhang W, Liu Z, Li C, Peng C, Cui G, Shao H, Du Z (2022) Activation of pyroptosis and ferroptosis is involved in the hepatotoxicity induced by polystyrene microplastics in mice. Chemosphere 291:132944
Xiao J, Zhang S, Tu B, Jiang X, Cheng S, Tang Q, Zhang J, Qin X, Wang B, Zou Z, Chen C (2021) Arsenite induces ferroptosis in the neuronal cells via activation of ferritinophagy. Food Chem Toxicol 151:112114
Bradley BD, Graber G, Condon RJ, Frobish LT (1983) Effects of graded levels of dietary copper on copper and iron concentrations in swine tissues. J Anim Sci 56:625–630
Stevenson MH, Jackson N (1980) Effects of level of dietary copper sulphate and period of feeding on the laying, domestic fowl, with special reference to tissue mineral content. Brit J Nutr 43:205–215
Theil EC, Calvert KT (1978) The effect of copper excess on iron metabolism in sheep. Biochem J 170:137–143
Fosset C, Danzeisen R, Gambling L, McGaw BA, McArdle HJ (2009) Cu loading alters expression of non-IRE regulated, but not IRE regulated, Fe dependent proteins in HepG2 cells. J Inorg Biochem 103:709–716
Jiang L, Garrick MD, Garrick LM, Zhao L, Collins JF (2013) Divalent metal transporter 1 (Dmt1) mediates copper transport in the duodenum of iron-deficient rats and when overexpressed in iron-deprived HEK-293 cells. J Nutr 143:1927–1933
Lane DJ, Merlot AM, Huang ML, Bae DH, Jansson PJ, Sahni S, Kalinowski DS, Richardson DR (2015) Cellular iron uptake, trafficking and metabolism: key molecules and mechanisms and their roles in disease. Biochim Biophys Acta 1853:1130–1144
Stockwell BR et al (2017) Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171:273–285
Qiu Y, Cao Y, Cao W, Jia Y, Lu N (2020) The application of ferroptosis in diseases. Pharmacol Res 159:104919
Sachdev PK, Freeland-Graves J, Beretvas SN, Sanjeevi N (2018) Zinc, copper, and iron in oral submucous fibrosis: a meta-analysis. Int J Dent 2018:3472087
Ursini F, Maiorino M (2020) Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radical Bio Med 152:175–185
Liu Z, Lv X, Yang B, Qin Q, Song E, Song Y (2021) Tetrachlorobenzoquinone exposure triggers ferroptosis contributing to its neurotoxicity. Chemosphere 264:128413
Zhao L, Feng Y, Xu ZJ, Zhang NY, Zhang WP, Zuo G, Khalil MM, Sun LH (2021) Selenium mitigated aflatoxin B1-induced cardiotoxicity with potential regulation of 4 selenoproteins and ferroptosis signaling in chicks. Food Chem Toxicol 154:112320
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, Sun B, Wang G (2020) Ferroptosis: past, present and future. Cell Death Dis 11:88
Bellezza I, Giambanco I, Minelli A, Donato R (2018) Nrf2-Keap1 signaling in oxidative and reductive stress. Bba-Mol Cell Res 1865:721–733
Fang Y, Xing C, Wang X, Cao H, Zhang C, Guo X, Zhuang Y, Hu R, Hu G, Yang F (2021) Activation of the ROS/HO-1/NQO1 signaling pathway contributes to the copper-induced oxidative stress and autophagy in duck renal tubular epithelial cells. Sci Total Environ 757:143753
Kerins MJ, Ooi A (2018) The roles of NRF2 in modulating cellular iron homeostasis. Antioxid Redox Sign 29:1756–1773
Yumnamcha T, Devi TS, Singh LP (2019) Auranofin mediates mitochondrial dysregulation and inflammatory cell death in human retinal pigment epithelial cells: implications of retinal neurodegenerative diseases. Front Neurosci-Switz 13:1065
Lu J, Zhao Y, Liu M, Lu J, Guan S (2021) Toward improved human health: Nrf2 plays a critical role in regulating ferroptosis. Food Funct 12:9583–9606
Funding
This work was supported by the National Natural Science Foundation of China (No. 32072930 and 31572585).
Author information
Authors and Affiliations
Contributions
Conceptualization: Gaolong Zhong; methodology: Gaolong Zhong, Jianzhao Liao, and Feiyang Ma; formal analysis and investigation: Gaolong Zhong, Yuanxu Li, Feiyang Ma, and Yihui Huo; writing—original draft preparation: Gaolong Zhong; writing—review and editing: Qingyue Han and Lianmei Hu; funding acquisition: Zhaoxin Tang; resources: Zhaoxin Tang; supervision: Zhaoxin Tang.
Corresponding author
Ethics declarations
Ethics Approval
The procedures of animal experiments were authorized and performed by the Ethics Committee of South China Agricultural University (Permit Number: 2017A087).
Consent for Publication
All the authors have consented for publication of this research.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhong, G., Li, Y., Ma, F. et al. Copper Exposure Induced Chicken Hepatotoxicity: Involvement of Ferroptosis Mediated by Lipid Peroxidation, Ferritinophagy, and Inhibition of FSP1-CoQ10 and Nrf2/SLC7A11/GPX4 Axis. Biol Trace Elem Res 202, 1711–1721 (2024). https://doi.org/10.1007/s12011-023-03773-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-023-03773-2