
Abstract
The incidence of type 2 diabetic osteoporosis (T2DOP), which seriously threatens elderly people’s health, is rapidly increasing in recent years. However, the specific mechanism of the T2DOP is still unclear. Studies have shown the relationship between iron overload and T2DOP. Mitochondrial ferritin (FtMt) is a protein that stores iron ions and intercepts toxic ferrous ions in cells mitochondria. Ferroptosis, an iron-dependent cell injured way, may be related to the pathogenesis of T2DOP. In this study, we intend to elucidate the effect of FtMt on ferroptosis in osteoblasts and explain the possible mechanism. We first detected the occurrence of ferroptosis in bone tissue and the expression of FtMt after inducing T2DOP rat model. Then we used hFOB1.19 cells to study the influence of high glucose on FtMt, ferroptosis, and osteogenic function of osteoblasts. Then we observed the effect of FtMt on ferroptosis and osteoblast function by lentiviral silencing and overexpression of FtMt. We found ferroptosis in T2DOP rats bone. Overexpression of FtMt reduced osteoblastic ferroptosis under high glucose condition while silent FtMt induced mitophagy through ROS / PINK1/Parkin pathway. Then we found increased ferroptosis in osteoblasts after activating mitophagy by carbonyl cyanide-m-chlorophenyl-hydrazine (CCCP, a mitophagy agonist). Our study demonstrated that FtMt inhibited the occurrence of ferroptosis in osteoblasts by reducing oxidative stress caused by excess ferrous ions, and FtMt deficiency induced mitophagy in the pathogenesis of T2DOP. This study suggested that FtMt might serve as a potential target for T2DOP therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
With the aging of the population, various chronic diseases affect our health. The impact of diabetes on the elderly is particularly serious, while type 2 diabetes accounts for approximately 90% of all diabetes cases [1]. However, about 425,000 new patients appeared every year in the past few decades [2]. Osteoporosis has a great impact on the living quality of the elderly among the complications of diabetes, causing a serious economic burden. Compared with type 1 diabetes, type 2 diabetic osteoporosis (T2DOP) has not decreased bone density, but increased bone fragility [3]. Therefore, we have not studied its complex pathological mechanism.
Iron disorder is a representative ion disorder in diabetic patients [4], and the imbalance of iron homeostasis is a momentous factors causing osteoporosis [5]. Studies showed that increased divalent metal transporter 1 (DMT1) expression under high glucose caused iron overload in osteoblasts and oxidative stress damage, which led to decreased osteogenic function [6, 7]. Ferroptosis, a cell damage mode closely related to iron metabolism, was newly proposed and named by Dixon in 2012 [8]. In recent years, many studies have been carried out in cancer, neurodegenerative disease, and ischemia-reperfusion injury [9,10,11]. Ge et al. found ferroptosis in diabetic cardiomyopathy [12]. The main manifestation of ferroptosis was the formation of abundant reactive oxygen species (ROS) through Fenton reaction, which caused the lipid peroxides accumulation and cell damage [13]. Therefore, the imbalance of iron homeostasis and the ferroptosis of osteoblasts may be the mechanism of T2DOP.
Mitochondrial ferritin (FtMt) stored iron in mitochondria. Its structure was similar to ferritin heavy chain which also had ferrous oxidase activity, and could intercept excessive iron ions in mitochondria to reduce the production of oxygen-free radicals [14]. Many studies showed that FtMt reduced oxidative stress and maintained the stability of intracellular iron [15,16,17]. Wang et al. suggested that FtMt inhibited the ferroptosis in SH-SY5Y cells induced by erastin [18]. Therefore, FtMt may be played a considerable role in T2DOP.
Mitophagy decomposes damaged mitochondria and reduces cell damage, which belong to a selective autophagy in cells [19]. Studies have shown that mitophagy is widely involved in apoptosis, necrosis, and other terminal regulatory processes [20, 21]. Wang and Wei et al. found that ROS induced mitophagy via PINK1/Parkin pathway [22, 23]. Therefore, mitophagy was considered to reduce the production of mitochondrial ROS [24]. However, increased mitochondrial ROS inhibited mitophagy in muscle tissue [25]. Meanwhile, the increased ROS in melanoma cells induced mitophagy, which caused cell death due to more ROS produced [26]. Therefore, the relationship between mitophagy and ROS needs further study. FtMt may be involved in the occurrence of mitophagy because of regulating ROS.
Previous studies showed that FtMt stabilized iron ions in mitochondria and inhibited oxidative stress, which indicated that FtMt was related to the occurrence of mitophagy. Ferroptosis, which was closely related to intracellular oxidative stress, might also be related to mitophagy. These results suggested that FtMt could regulate the occurrence of ferroptosis in osteoblasts through mitophagy, and further affect the osteogenic function in T2DOP.
Material and Methods
Experimental Animals
Forty-five SD rats (3 months old, 200 ± 20 g) were obtained from the Department of Experimental Animals in China Medical University (Animal Certificate Number: SCXK (Liaoning) 2008-0005). Fifteen rats grew as control while other thirty rats were established T2DOP model. The model rats were given a high-fat feed and 12 h/day water for 2 months. Then streptozotocin was intraperitoneally injected at 30 mg/kg. Seventy-two hours later, the model was successfully established when insulin sensitivity index decreased and fasting plasma glucose exceeded 7.8 mmol/L [27]. Then all rats continue grew 3 months to cause osteoporosis. Thirty model rats were divided into two groups. One was fifteen T2DOP rats only, and other was fifteen T2DOP rats with deferoxamine (DFO) treatment (60 mg/kg/day, intraperitoneally inject, last for the last 1 month). All the rats grew under standard laboratory conditions and were killed at the 9th month by cervical dislocation. The tibias were aseptically removed immediately, stored in 4% phosphate-buffered formalin solution at 4 °C.
Cell Line
The human osteoblast cell line hFOB 1.19 was bought from Cell Bank of Chinese Academy of Sciences (GNHu14, Shanghai, China). Cell line had been tested for mycoplasma and short tandem repeat (STR). Cells were developed in the mixture of Dulbecco’s Modified Eagle Medium and Ham’s F12 Medium (D-MEM/F-12) (SH30023.01; Hyclone; GE Healthcare Life Sciences, Logan, UT, USA) which contains 10% fetal bovine serum (SH30070.03; Hyclone; GE Healthcare Life Sciences, Logan, UT, USA) and 0.3 mg/ml G418 (A1720; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) under the condition of humidified 5% CO2 and 33.5 °C. Cells were disposed by trypsin/EDTA (SH30042.01; Hyclone; GE Healthcare Life Sciences, Logan, UT, USA) for subcultivation. The medium was replaced every other day. Besides, cells grew in high concentration of glucose (HG, 35 mmol/l) for 72 h [28]. Carbonyl cyanide-m-chlorophenyl-hydrazine (C2759; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was used to activate mitophagy in CCCP group, and ferrostatin-1 (Fer-1) (SML0583; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was used to inhibit ferroptosis.
Lentiviral Transfection
Small interfering RNA sequences targeting human FtMt (ccTGGAAACCTACTACCTGAA) was utilized. LV-FtMt-RNAi (73112-1; NM_177478) and flag tag were used for transfection to knock down the expression of FtMt. The siRNA sequence (TTCTCCGAACGTGTCACGT) was utilized as a negative control (LV-Ctrl). LV-FtMt (44045-3; NM_177478) and flag tag were used for FtMt overexpression, and the same lentiviral vector containing flag only was served as negative control. The lentiviral was produced by GeneChem Corporation (Shanghai, China).
Mitochondrion Isolation
Mitochondria were separated using cell mitochondria isolation kit (C3601; Beyotime Institute of Biotechnology, Shanghai, China). Osteoblasts were collected and washed by cold phosphate buffer saline (PBS). We added mitochondrion isolation reagents to cells on ice and waited 15 min. Then the cells were homogenized in a ground glass homogenizer. Solution was centrifuged at 1000×g at 4 °C for 10 min. The supernatant was centrifuged again at 3500×g at 4 °C for 10 min. Sediment was the mitochondria. After adding mitochondrial lysis buffer, we collected mitochondrial protein.
Analysis of Intracellular ROS
An oxidation-sensitive fluorescent probe (DCFH-DA) was used to detect intracellular ROS. At first, partial osteoblasts grew in different conditions for 72 h. We collected and washed cells twice using PBS afterward. Osteoblasts were incubated using 10 μmol/l DCFH-DA at 37 °C for 20 min. Then DCF fluorescence of 10,000 osteoblasts was detected at an excitation wavelength of 488 nm and emission wavelength of 535 nm by flowcytometry (BD Biosciences, Franklin Lakes, NJ, USA).
Western Blot
Approximately 5 × 106 osteoblasts were washed using PBS and lysed for 30 min by RIPA buffer. Then we scraped to collect the protein and centrifuged at 12,000×g at 4 °C for 30 min. We measured and unified the protein concentration of various samples by the bicinchoninic acid (BCA) method and denatured proteins. Then the 30 μg total protein was separated by SDS-PAGE under 80 V and transferred to PVDF membrane under 110 mA for 60 min. We dissolved 5% skimmed milk in TBST and soaked the membranes for 1 h. The membranes were incubated for 12 h at 4 °C with the antibody. We washed the membranes three times using TBST. Then they were incubated in peroxidase-conjugated secondary antibody for 2 h. The membrane was detected by the EC3 Imaging System (UVP; Analytik Jena AG, Jena, Germany). The software Image J (National Institutes of Health, Bethesda, MD, USA) was utilized to measure the optical density of each membrane.
Lipid Peroxidation Detection
According to the reaction between malondialdehyde (MDA) and thiobarbituric acid (TBA), we used a lipid peroxidation MDA assay kit (S0131; Beyotime Institute of Biotechnology, Shanghai, China) to measure the change of lipid peroxidation. Supernatant was collected after cells were lysed and centrifuged which procedure is similar to western blot. Then 200 μl MDA working solution was added to 100 μl supernatant. The mixture was heated to 100 °C for 15 min. After cooled to room temperature, centrifuged at 1000×g for 10 min, and 200 μl supernatant was detected at 532 nm by a microplate reader.
Alizarin Red S Staining
The bone nodule formation was detected by Alizarin Red S Staining after osteogenic differentiation. After washing three times by PBS, we fixed cells with 95% ethanol for 10 min. Then the cells were stained with 40 mmol/l alizarin red S solution for 40 min. We used phase-contrast microscopy (Nikon, Japan) to take photos of the cells and bone nodule. The density of staining was assessed by Image J.
Alkaline Phosphatase Detection
The floating cells were collected after digested and centrifugation. Using lysis buffer to lyse the cells after washing by PBS twice, we measured the ALP activity by alkaline phosphatase activity assay kit (Beyotime, China). Absorbance was showed using 96-well plate reader at 405 nm.
Transmission Electron Microscopy
The floating cells were collected after digested and centrifugation. Cold PBS was utilized to wash the cells twice and the 5% glutaraldehyde was used to fix the cells. Then cells were dehydrated, embedded, sectioned, and stained conventionally. The morphology of mitochondria was observed by transmission electron microscopy. The number of injured mitochondria was counted in every 10 fields.
Immunohistochemistry (IHC)
Bone tissues were fixed with paraformaldehyde (4%, 15 min, room temperature) first, and then permeabilized with Triton X-100 (0.2%, 5 min, room temperature). Using a 5% BSA blocking buffer (30 min, room temperature) and then incubating the sections with anti-GPX4 or anti-FtMt antibodies (4 °C, overnight) were followed by secondary antibodies incubation for 120 min. Then sections were incubated with DAPI (0.1%, 5 min) and washed with PBS followed by transferring cover slips onto the glass slides. A fluorescent microscope (Olympus, Japan) was used to obtain the images, and a imaging software (Image J) was utilized to perform the semi-quantitative analysis.
Statistical Analysis
The statistical analysis results were presented by GraphPad Prism 7 software (GraphPad Software, Inc., La Jolla, CA, USA). We used Student’s t test to evaluate the differences of every two groups and one-way analysis of variance to analyze multiple groups. It was considered to be statistically significant when p < 0.05.
Results
There Was Ferroptosis in T2DOP Rats
We induced T2DOP model rats by high-fat feeding and STZ. Then we evaluated the model through bone mineral density (BMD), fasting blood glucose (FBG), fasting insulin (FINS), and insulin sensitivity index (ISI), which confirmed that the model was successfully established (Fig. 1c–f) [6]. DFO was used to treat T2DOP rats. The results of western blot and immunohistochemistry suggested that the content of glutathione peroxidase 4 (GPX4, ferroptosis marker protein) in T2DOP group was significantly decreased. After treatment with DFO, the content of GPX4 in T2DOP rats recovered (Fig. 1a, b). The results suggested that ferroptosis existed in T2DOP rat.

There was ferroptosis and increased FtMt in T2DOP. a IHC detected GPX4 and FtMt in Ctrl, T2DOP, T2DOP + DFO 3 group rats. b Western blot showed GPX4 and FtMt expression level. c BMD decreased in T2DOP group rats. d FBG increased in T2DOP and T2DOP + DFO group rats. e FINS increased in T2DOP and T2DOP + DFO group rats. f ISI decreased in T2DOP and T2DOP + DFO group rats. n = 15 per group. IHC scale = 20 μm. Data are means ± SD. *p < 0.05 vs Ctrl, #p < 0.05 vs T2DOP
High Glucose Led to Ferroptosis and Decreased Osteogenic Function
After 72 h of treatment with high glucose, we found the decreased expression of GPX4 (Fig. 2b), the increased level of ROS (Fig. 5b), and the accumulation of lipid peroxide (Fig. 2c). Electron microscopy showed that the mitochondrial morphological was consistent with the change of ferroptosis (Fig. 4a). These results showed that high glucose promoted ferroptosis of osteoblasts. At the same time, we detected the related indicators of osteogenic function, and found a decreased expression of osteoprotegerin (OPG) and osteocalcin (OCN) (Fig. 2b), a decreased activity of alkaline phosphatase (ALP) (Fig. 2d), and decreased formation of mineralized nodules (Fig. 4b). The results indicated that high glucose decreased the osteogenic function of osteoblasts.

Overexpressed FtMt reduced osteoblastic ferroptosis and increased osteogenic function under high glucose. a Western blot showed mitochondrial DMT1 expression level increased in high-glucose (HG) group. b Western blot showed upregulated FtMt, increased GPX4, OPG, and OCN under high glucose. c Lipid peroxidation was detected according to the reaction between malondialdehyde (MDA) and thiobarbituric acid (TBA). d ALP activity increased when FtMt upregulated. The means ± SD of three independent experiments were utilized to present all the values. *p < 0.05 vs Ctrl, #p < 0.05 vs HG, &p < 0.05 vs LV-Ctrl
FtMt Increased Under High Glucose Condition or in T2DOP Rats
Immunohistochemistry and western blot showed that FtMt increased in T2DOP rats, but decreased after DFO treatment (Fig. 1a, b). At the same time, we found that the expression of FtMt in osteoblasts was still increased under high glucose condition (Fig. 2b). Studies have shown that high glucose caused iron overload in osteoblasts by upregulating the expression of DMT1. Wolff et al. have found that DMT1 was also expressed in mitochondria [29]. Therefore, we extracted the osteoblast mitochondria. The results showed that the expression of mitochondrial DMT1 increased under high glucose condition (Fig. 2a). These results suggested that high glucose caused mitochondrial iron overload.
Effects of Overexpression of FtMt on Ferroptosis and Function of Osteoblasts
We observed the effect of FtMt on osteoblasts by using FtMt lentivirus to upregulate the expression of FtMt in osteoblasts. The results showed that overexpressed FtMt reversed the changes of osteoblasts induced by high glucose (GPX4 decreased, ROS level increased, lipid peroxide increased, OPG and OCN decreased, ALP activity decreased, mineralized nodules decreased) (Figs. 2b–d, 4b, and 5b). These results suggested that upregulated FtMt reduced intracellular ROS level and inhibited ferroptosis, then improved osteoblast function under high glucose condition. At the same time, it also indirectly indicated that the increased amount of FtMt was still not enough to protect osteoblasts from ROS damage, so ferroptosis stiil occurred in T2DOP model.
Downregulated FtMt Induced Mitophagy Via ROS/PINK1/Parkin Pathway
The effect of downregulated FtMt on osteoblasts was similar to that of high glucose treatment. Ferroptosis and decreased osteogenic function was observed. However, these results were changed after using ferroptosis inhibitor (Figs. 3a–d, 4b, and 5b). At the same time, we found that the expression of PINK1, parkin, and LC3 increased, which indicated that FtMt silencing might lead to mitophagy (Fig. 3b). After antioxidant NAC inhibiting ROS, the expression of mitophagy-related proteins decreased (Fig. 3b), which indicated that downregulated FtMt induced mitophagy via ROS/PINK1/parkin pathway.

Downregulated FtMt increased osteoblastic ferroptosis, decreased osteogenic function, and induced mitophagy. a Western blot showed GPX4, OCN, and OPG expression level which decreased when FtMt was downregulated. b The deficiency of FtMt caused PINK1, Parkin, and LC3 increase. c MDA increased after FtMt was downregulated. d ALP activity decreased when FtMt was downregulated. The means ± SD of three independent experiments were utilized to present all the values. *p < 0.05 vs Ctrl, #p < 0.05 vs LV-FtMt-RNAi

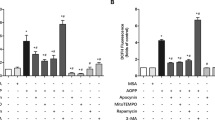
Osteoblasts were observed by electron microscopy and ARS staining. a Electron microscopy showed increased ferroptosis in HG and CCCP groups. b ARS staining showed the mineralized extracellular matrix of osteoblast cells under different conditions. The means ± SD of three independent experiments were utilized to present all the values. *p < 0.05 vs Ctrl, #p < 0.05 vs HG, &p < 0.05 vs LV-FtMt-RNAi, &#p < 0.05 vs CCCP

CCCP promoted ferroptosis and ROS variation after cells had been treated. a Western blot showed GPX4, OCN, and OPG expression levels decreased after adding CCCP. b Flowcytometry analysis of ROS variation using fluorescent probe (DCFH-DA). c ALP activity decreased after adding CCCP. d MDA increased after adding CCCP. The means ± SD of three independent experiments were utilized to present all the values. *p < 0.05 vs Ctrl, #p < 0.05 vs HG, &p < 0.05 vs LV-FtMt-RNAi, &#p < 0.05 vs CCCP
Activation of Mitophagy Promoted Ferroptosis in Osteoblasts
To investigate the relationship between mitophagy and ferroptosis, we used a mitophagy agonist (Carbonyl cyanide-m-chlorophenyl-hydrazine, CCCP). The results of electron microscopy indicated that the ferroptosis of osteocytes in CCCP group significantly increased (Fig. 4a). Other results such as the decreased expression of GPX4, OPG, and OCN (Fig. 5a), increased ROS level (Fig. 5b), increased lipid peroxide (Fig. 5d), decreased ALP activity (Fig. 5c), and decreased mineralized nodules (Fig. 4b) were observed at the same time. However, the ferroptosis was reversed after adding ferroptosis inhibitor in CCCP group. The results showed that mitophagy promoted the occurrence of ferroptosis in osteoblasts.
Discussion
Compared with other types of osteoporosis, T2DOP has a complex pathological mechanism, which is still unclear. Li et al. found that iron overload had a great influence on the function of bone tissue [30]. Another study showed that iron overload aggravated diabetic nerve injury [31]. Iron overload was considered as one dangerous factor of ferroptosis [32]. Therefore, it is of great significance to study the osteoblastic ferroptosis caused by high glucose for explaining the pathogenesis of T2DOP.
Mitochondrion is the place to synthesize intracellular heme and Fe/S protein complexes, which requires a substantial amount of iron ions [33]. Mitochondrion also generates ROS during the process of producing ATP [34], which may bring about ferroptosis. The structure of FtMt is similar to the heavy chain structure of intracellular ferritin. Excess ferrous ions can produce ROS through the fenton reaction, while FtMt has ferrous oxidase activity that can reduce the amount of free ferrous ions in the mitochondrion, thereby decreasing the content of ROS [35]. Wang et al. also found that FtMt had a certain inhibitory effect on erastin-induced ferroptosis [18]. However, the effect of FtMt on ferroptosis of osteoblasts induced by T2DOP and high glucose has not been studied. Our study demonstrated that there was ferroptosis and increased FtMt in T2DOP rats. After treatment of high glucose, the expression of osteoblasts mitochondrial DMT1 and FtMt both increased, which suggested high glucose caused mitochondrial iron overload. Moreover, high glucose caused the reduction of GPX4 while ROS and lipid peroxidation increased, leading to ferroptosis and low osteogenic function. GPX4 was one reductase that reduced lipid peroxidation and ROS. The deficiency of GPX4 and the increase of lipid peroxidation and ROS are considered to be the occurrences of ferroptosis [36]. However, the results were opposite when FtMt overexpressed. We deemed that overexpressed FtMt decreased the degree of iron overload and ROS generated by fendon reaction. Therefore, FtMt could inhibit the occurrence of osteoblastic ferroptosis caused by high glucose and recover osteogenic function.
Upregulated FtMt could reduce iron overload and ROS to decrease cell damage [17] while FtMt deficiency caused opposite results [37]. However, increased ROS damaged mitochondrial membrane and induced mitophagy via PINK1/Parkin pathway [38]. Interestingly, Basit et al. found that mitochondrial complex I inhibition promoted mitophagy and ROS production [26]. However, study showed that mitophagy decreased ROS level [24, 39]. Zhang et al. explored the role of autophagy in vascular smooth muscle cells and concluded that autophagy could promote or inhibit cell death through different pathways [40]. Accordingly, we guessed mitophagy might play different roles through various pathways in different cells. We have demonstrated that downregulated FtMt enhanced mitophagy in osteoblasts via ROS/PINK1/Parkin pathway. Through adding mitophagy agonist CCCP to osteoblasts, we found the increase of ROS and lipid peroxidation while GPX4 decreased. Mitophagy might remove damaged mitochondria but produced more ROS to increase cell oxidative stress, which indicated that mitophagy promoted ferroptosis. We guess the possible reason was that mitophagy decomposed injured mitochondria causing excessive free iron ions, which produced more ROS through fenton reaction and induced ferroptosis. After adding Fer-1 to CCCP group, the opposite results appeared. However, the specific mechanism of mitophagy-inducing ferroptosis needs more studies. At the same time, high glucose led to osteoblastic ferroptosis even though the expression of FtMt increased. We thought the possible explanation was that the iron overload increased expression of FtMt, but actually free iron was still excessive, so it also induced ferroptosis. Moreover, there are many ways to induce ferroptosis in cells, including the increase of free ferrous ions caused by ferritinophagy, and the decrease of GPX4 caused by system Xc- [41,42,43]. Therefore, the roles of FtMt and mitophagy in ferroptosis remain to be further studied.
Conclusion
Iron is essential for maintaining cell function. The discovery of ferroptosis offers a new direction for studies of various diseases with iron homeostasis imbalance. Our study suggests that FtMt can reduce the content of ROS and decrease the occurrence of ferroptosis while FtMt deficiency do the opposite through mitophagy, playing a vital role in intracellular iron homeostasis regulation. Therefore, FtMt may be a valid target for T2DOP therapy.
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Zheng Y, Ley SH, Hu FB (2018) Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol 14:88–98. https://doi.org/10.1038/nrendo.2017.151
Jaacks LM, Siegel KR, Gujral UP, Narayan KMV (2016) Type 2 diabetes: a 21st century epidemic. Best Pract Res Clin Endocrinol Metab 30:331–343. https://doi.org/10.1016/j.beem.2016.05.003
Anagnostis P, Paschou SA, Gkekas NN, Artzouchaltzi AM, Christou K, Stogiannou D, Vryonidou A, Potoupnis M, Goulis DG (2018) Efficacy of anti-osteoporotic medications in patients with type 1 and 2 diabetes mellitus: a systematic review. Endocrine 60:373–383. https://doi.org/10.1007/s12020-018-1548-x
Altamura S, Kopf S, Schmidt J, Müdder K, da Silva AR, Nawroth P, Muckenthaler MU (2017) Uncoupled iron homeostasis in type 2 diabetes mellitus. J Mol Med 95:1387–1398. https://doi.org/10.1007/s00109-017-1596-3
Che J, Yang J, Zhao B, Zhang G, Wang L, Peng S, Shang P (2020) The effect of abnormal iron metabolism on osteoporosis. Biol Trace Elem Res 195:353–365. https://doi.org/10.1007/s12011-019-01867-4
Zhang W-L, Meng H-Z, Yang M-W (2015) Regulation of DMT1 on bone microstructure in type 2 diabetes. Int J Med Sci 12:441–449. https://doi.org/10.7150/ijms.11986
Liu F, Zhang W-L, Meng H-Z, Cai ZY, Yang MW (2017) Regulation of DMT1 on autophagy and apoptosis in osteoblast. Int J Med Sci 14:275–283. https://doi.org/10.7150/ijms.17860
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B III, Stockwell BR (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Liang C, Zhang X, Yang M, Dong X (2019) Recent progress in ferroptosis inducers for cancer therapy. Adv Mater 31:e1904197. https://doi.org/10.1002/adma.201904197
DeGregorio-Rocasolano N, Martí-Sistac O, Gasull T (2019) Deciphering the iron side of stroke: neurodegeneration at the crossroads between iron dyshomeostasis, excitotoxicity, and ferroptosis. Front Neurosci 13:85. https://doi.org/10.3389/fnins.2019.00085
Huang L-L, Liao X-H, Sun H, Jiang X, Liu Q, Zhang L (2019) Augmenter of liver regeneration protects the kidney from ischaemia-reperfusion injury in ferroptosis. J Cell Mol Med 23:4153–4164. https://doi.org/10.1111/jcmm.14302
Ge Z-D, Lian Q, Mao X, Xia Z (2019) Current status and challenges of NRF2 as a potential therapeutic target for diabetic cardiomyopathy. Int Heart J 60:512–520. https://doi.org/10.1536/ihj.18-476
Forcina GC, Dixon SJ (2019) GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics 19:e1800311. https://doi.org/10.1002/pmic.201800311
Drysdale J, Arosio P, Invernizzi R, Cazzola M, Volz A, Corsi B, Biasiotto G, Levi S (2002) Mitochondrial ferritin: a new player in iron metabolism. Blood Cells Mol Dis 29:376–383. https://doi.org/10.1006/bcmd.2002.0577
Wu W, Chang S, Wu Q, Xu Z, Wang P, Li Y, Yu P, Gao G, Shi Z, Duan X, Chang YZ (2016) Mitochondrial ferritin protects the murine myocardium from acute exhaustive exercise injury. Cell Death Dis 7:e2475. https://doi.org/10.1038/cddis.2016.372
Guan H, Yang H, Yang M, Yanagisawa D, Bellier JP, Mori M, Takahata S, Nonaka T, Zhao S, Tooyama I (2017) Mitochondrial ferritin protects SH-SY5Y cells against H2O2-induced oxidative stress and modulates α-synuclein expression. Exp Neurol 291:51–61. https://doi.org/10.1016/j.expneurol.2017.02.001
Gao G, Zhang N, Wang Y-Q, Wu Q, Yu P, Shi ZH, Duan XL, Zhao BL, Wu WS, Chang YZ (2017) Mitochondrial ferritin protects hydrogen peroxide-induced neuronal cell damage. Aging Dis 8:458–470. https://doi.org/10.14336/AD.2016.1108
Wang Y-Q, Chang S-Y, Wu Q, Gou YJ, Jia L, Cui YM, Yu P, Shi ZH, Wu WS, Gao G, Chang YZ (2016) The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front Aging Neurosci 8:308. https://doi.org/10.3389/fnagi.2016.00308
Green DR, Galluzzi L, Kroemer G (2011) Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333:1109–1112. https://doi.org/10.1126/science.1201940
Liang X, Wang S, Wang L, Ceylan AF, Ren J, Zhang Y (2020) Mitophagy inhibitor liensinine suppresses doxorubicin-induced cardiotoxicity through inhibition of Drp1-mediated maladaptive mitochondrial fission. Pharmacol Res 157:104846. https://doi.org/10.1016/j.phrs.2020.104846
Wang H, Ni H-M, Chao X, Ma X, Rodriguez YA, Chavan H, Wang S, Krishnamurthy P, Dobrowsky R, Xu DX, Jaeschke H, Ding WX (2019) Double deletion of PINK1 and Parkin impairs hepatic mitophagy and exacerbates acetaminophen-induced liver injury in mice. Redox Biol 22:101148. https://doi.org/10.1016/j.redox.2019.101148
Wang Y, Nartiss Y, Steipe B, McQuibban GA, Kim PK (2012) ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 8:1462–1476. https://doi.org/10.4161/auto.21211
Wei X, Qi Y, Zhang X, Qiu Q, Gu X, Tao C, Huang D, Zhang Y (2014) Cadmium induces mitophagy through ROS-mediated PINK1/Parkin pathway. Toxicol Mech Methods 24:504–511. https://doi.org/10.3109/15376516.2014.943444
Liang D, Zhuo Y, Guo Z, He L, Wang X, He Y, Li L, Dai H (2019) SIRT1/PGC-1 pathway activation triggers autophagy/mitophagy and attenuates oxidative damage in intestinal epithelial cells. Biochimie 170:10–20. https://doi.org/10.1016/j.biochi.2019.12.001
Sakellariou GK, Pearson T, Lightfoot AP, Nye GA, Wells N, Giakoumaki II, Vasilaki A, Griffiths RD, Jackson MJ, McArdle A (2016) Mitochondrial ROS regulate oxidative damage and mitophagy but not age-related muscle fiber atrophy. Sci Rep 6:33944. https://doi.org/10.1038/srep33944
Basit F, van Oppen LM, Schöckel L et al (2017) Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis 8:e2716. https://doi.org/10.1038/cddis.2017.133
Li B, Wang Y, Liu Y, Ma J, Li Y (2013) Altered gene expression involved in insulin signaling pathway in type II diabetic osteoporosis rats model. Endocrine 43:136–146. https://doi.org/10.1007/s12020-012-9757-1
Zhao W, Zhang W, Ma H, Yang M (2020) NIPA2 regulates osteoblast function by modulating mitophagy in type 2 diabetes osteoporosis. Sci Rep 10:3078. https://doi.org/10.1038/s41598-020-59743-4
Wolff NA, Garrick LM, Zhao L, Garrick MD, Thévenod F (2014) Mitochondria represent another locale for the divalent metal transporter 1 (DMT1). Channels (Austin) 8:458–466. https://doi.org/10.4161/19336950.2014.956564
Li Y, Bai B, Zhang Y (2018) Expression of iron-regulators in the bone tissue of rats with and without iron overload. Biometals 31:749–757. https://doi.org/10.1007/s10534-018-0133-3
Zhao S, Zhang L, Xu Z, Chen W (2013) Neurotoxic effects of iron overload under high glucose concentration. Neural Regen Res 8:3423–3433. https://doi.org/10.3969/j.issn.1673-5374.2013.36.008
Yamada N, Karasawa T, Wakiya T, Sadatomo A, Ito H, Kamata R, Watanabe S, Komada T, Kimura H, Sanada Y, Sakuma Y, Mizuta K, Ohno N, Sata N, Takahashi M (2020) Iron overload as a risk factor for hepatic ischemia-reperfusion injury in liver transplantation: Potential role of ferroptosis. Am J Transplant 20:1606–1618. https://doi.org/10.1111/ajt.15773
Braymer JJ, Stümpfig M, Thelen S, Mühlenhoff U, Lill R (2019) Depletion of thiol reducing capacity impairs cytosolic but not mitochondrial iron-sulfur protein assembly machineries. Biochim Biophys Acta, Mol Cell Res 1866:240–251. https://doi.org/10.1016/j.bbamcr.2018.11.003
Walker O’LS, Ragos R, Wong MK, Adam M, Cheung A, Raha S (2020) Reactive oxygen species from mitochondria impacts trophoblast fusion and the production of endocrine hormones by syncytiotrophoblasts. PLoS One 15:e0229332. https://doi.org/10.1371/journal.pone.0229332
Levi S, Corsi B, Bosisio M, Invernizzi R, Volz A, Sanford D, Arosio P, Drysdale J (2001) A human mitochondrial ferritin encoded by an intronless gene. J Biol Chem 276:24437–24440. https://doi.org/10.1074/jbc.C100141200
Imai H, Matsuoka M, Kumagai T et al (2017) Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol 403:143–170. https://doi.org/10.1007/82_2016_508
Wang L, Wang L, Dai Z, Wu P, Shi H, Zhao S (2017) Lack of mitochondrial ferritin aggravated neurological deficits via enhancing oxidative stress in a traumatic brain injury murine model. Biosci Rep 37. https://doi.org/10.1042/BSR20170942
Zhang Y, Xi X, Mei Y, Zhao X, Zhou L, Ma M, Liu S, Zha X, Yang Y (2019) High-glucose induces retinal pigment epithelium mitochondrial pathways of apoptosis and inhibits mitophagy by regulating ROS/PINK1/Parkin signal pathway. Biomed Pharmacother 111:1315–1325. https://doi.org/10.1016/j.biopha.2019.01.034
Li S, Lin Q, Shao X, Zhu X, Wu J, Wu B, Zhang M, Zhou W, Zhou Y, Jin H, Zhang Z, Qi C, Shen J, Mou S, Gu L, Ni Z (2019) Drp1-regulated PARK2-dependent mitophagy protects against renal fibrosis in unilateral ureteral obstruction. Free Radic Biol Med 152:632–649. https://doi.org/10.1016/j.freeradbiomed.2019.12.005
Zhang Y-Y, Shi Y-N, Zhu N, Wang W, Deng CF, Xie XJ, Liao DF, Qin L (2019) Autophagy: a killer or guardian of vascular smooth muscle cells. J Drug Target 28:1–22. https://doi.org/10.1080/1061186X.2019.1705312
Zhang Z, Guo M, Li Y, Shen M, Kong D, Shao J, Ding H, Tan S, Chen A, Zhang F, Zheng S (2019) RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy 16:1–24. https://doi.org/10.1080/15548627.2019.1687985
Sui S, Zhang J, Xu S, Wang Q, Wang P, Pang D (2019) Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis 10:331. https://doi.org/10.1038/s41419-019-1564-7
Wang L, Liu Y, Du T et al (2019) ATF3 promotes erastin-induced ferroptosis by suppressing system Xc. Cell Death Differ 27:662–675. https://doi.org/10.1038/s41418-019-0380-z
Acknowledgements
We would like to thank all the people and institution to help and support this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
All the animals experiments were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Review Board of the First Hospital of China Medical University.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, X., Ma, H., Sun, J. et al. Mitochondrial Ferritin Deficiency Promotes Osteoblastic Ferroptosis Via Mitophagy in Type 2 Diabetic Osteoporosis. Biol Trace Elem Res 200, 298–307 (2022). https://doi.org/10.1007/s12011-021-02627-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-021-02627-z