
Abstract
Glutathione peroxidase 4 (Phospholipid hydroperoxide glutathione peroxidase, PHGPx) can directly reduce phospholipid hydroperoxide. Depletion of GPx4 induces lipid peroxidation-dependent cell death in embryo, testis, brain, liver, heart, and photoreceptor cells of mice. Administration of vitamin E in tissue specific GPx4 KO mice restored tissue damage in testis, liver, and heart. These results indicate that suppression of phospholipid peroxidation is essential for cell survival in normal tissues in mice. Ferroptosis is an iron-dependent non-apoptotic cell death that can elicited by pharmacological inhibiting the cystine/glutamate antiporter, system Xc− (type I) or directly binding and loss of activity of GPx4 (Type II) in cancer cells with high level RAS-RAF-MEK pathway activity or p53 expression, but not in normal cells. Ferroptosis by Erastin (Type I) and RSL3 (RAS-selective lethal 3, Type II) treatment was suppressed by an iron chelator, vitamin E and Ferrostatin-1, antioxidant compound. GPx4 can regulate ferroptosis by suppression of phospholipid peroxidation in erastin and RSL3-induced ferroptosis. Recent works have identified several regulatory factors of erastin and RSL3-induced ferroptosis. In our established GPx4-deficient MEF cells, depletion of GPx4 induce iron and 15LOX-independent lipid peroxidation at 26 h and caspase-independent cell death at 72 h, whereas erastin and RSL3 treatment resulted in iron-dependent ferroptosis by 12 h. These results indicated the possibility that the mechanism of GPx4-depleted cell death might be different from that of ferroptosis induced by erastin and RSL3.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Phospholipid Hydroperoxide
- Mitochondrial Death Pathway
- Phospholipid Peroxidation
- Phospholipid Hydroperoxide Glutathione Peroxidase
- Acute Kidney Failure
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Ferroptosis is an iron- and lipid peroxidation-dependent and caspase-independent novel form of regulated cell death (RCD), which was recently named in 2012 by Dr. Brent R. Stockwell (Dixon et al. 2012). Ferroptosis is distinct from other types of cell death such as apoptosis, autophagic cell death, and necroptosis (Dixon et al. 2012; Yang and Stockwell 2008). Ferroptosis inducer, Ras-selective lethal small molecule (RSL) including erastin and RSL-3 were found by high-throughput small molecule-screening that selectively killed the mutant Ras oncogene transformed human foreskin fibroblasts (BJeJR), but not their isogenic primary counterparts and normal cells in a non-apoptotic manner (Yang and Stockwell 2008; Yagoda et al. 2007). Multiple inhibitors of apoptosis, necrosis, and autophagy (e.g., Z-VAD-FMK, Boc-D-FMK, wortmannin, and necrostatin-1) cannot rescue ferroptosis by erastin and RSL3 treatment. In contrast, antioxidants [e.g., vitamin E and butylated hydroxytoluene (BHT)] and iron chelator (deferoxamine mesylate) inhibit RSLs induced cell death. These results indicated that ferroptosis refers to an iron-dependent, non-apoptotic cell death.
Glutathione peroxidase 4 (GPx4, Phospholipid hydroperoxide glutathione peroxidase PHGPx) is a unique intracellular antioxidant enzyme that directly reduces peroxidized phospholipids that have been produced in cell membrane (Imai and Nakagawa 2003). GPx4 knockout mice displayed early embryonic lethal in mice at 7.5 dpc (Imai et al. 2003a) and cell death in several tissues of conditional knockout mice (Imai 2010; Seiler et al. 2008). Before reports about ferroptosis, ablation of GPx4 induces lipid peroxidation-dependent, caspase-independent cell death in embryo and MEF cells (Imai et al. 2009; Seiler et al. 2008). GPx4 recently reported to be a regulator of ferroptosis by RSL3 and erastin, since RSL3 and erastin decreased the activity of GPx4 by direct binding to GPx4 and indirectly loss of glutathione respectively (Yang et al. 2014; Dixon et al. 2012).
On the other hand, mitochondrial GPx4 previously reported to be a suppressor of apoptosis by mitochondrial death pathway, since mitochondrial GPx4 inhibits the release of cytochrome c from mitochondria by reduction of cardiolipin hydroperoxide in apoptosis (Nomura et al. 1999, 2000).
In this review, we summarize recent studies on the lipid peroxidation-dependent cell death such as apoptosis and ferroptosis regulated by organelle-specific GPx4 from lessons of analysis of GPx4 overexpressed cells, knockout cells, and mice.
2 Structure and Expression of Three Types of Organelle-Specific GPx4
The GPxs family consists of various members, including GPx1–8 (Imai 2010; Imai and Nakagawa 2003). GPx1, 2, 3, 4, and 6 are selenoproteins that have selenocysteine at active site in human, except cysteine in GPx6 in mice. GPx5, 7 and 8 have very low glutathione peroxidase activity and thioredoxin-like activity because they have cysteine at active site. GPx3 exists in plasma, and GPx2 is expressed in gastrointestinal tract.
GPx1 and GPx4 are generally expressed in normal tissues. GPx1 can reduce hydrogen peroxide and fatty acid hydroperoxide in cytosol, but not phospholipid hydroperoxide in membrane using glutathione. GPx4 can effectively reduce phospholipid hydroperoxide, fatty acid hydroperoxide, cholesterol hydroperoxide, and thymine hydroperoxide, but ineffectively hydrogen peroxide using glutathione.
Three types of GPx4 are transcribed by different transcriptional start codons and different exons (exon1a and exon1b) from one gene (Imai and Nakagawa 2003; Imai et al. 2006). Only N-terminal sequence including signal sequence for transport to different organelle such as mitochondria, nucleoli, cytosol, and nuclei is different among three types of GPx4, whereas the remaining amino acid sequence on the C-terminal side including the enzymatic active site is exactly the same. Thus, GPx4 contain three types of GPx4, mGPx4 that transported into mitochondria (Arai et al. 1996), nGPx4 that localized in nucleoli (Nakamura et al. 2003) and cGPx4 that distributed in cytosolic and nuclei (Imai et al. 1995). cGPx4 might also strongly associate with membrane in cytosolic sites of organelle (Arai et al. 1996) (Fig. 1).

Structure and distribution of thee types of GPx4. Three types of GPx4, nucleolar, mitochondrial and non-mitochondrial isoforms, are transcribed from one gene by alternative transcription. GPx4 proteins contain nucleolar (34 kDa), non-mitochondrial (20 kDa) and mitochondrial GPx4 (23 kDa) with targeting signal (exon 1b and 1a) at the N-terminal of protein. The parts of C-terminal of three types of GPx4 including selenocysteine (Sec) at the enzymatic active site are the same. GPx4 is one of the selenoproteins including selenocysteine that is encoded by stop codon AUG. Selenocysteine insertion sequence (SECIS) is required for the incorporation of selenocysteine into AUG codon of the GPx4 protein. Right panels showed the distribution of each GPx4-GFP fusion protein in rat basophile leukemia (RBL2H3) cells. Green GFP fluorescence, Blue DAPI staining of nucleus
The expression of cGPx4 mRNA was relatively high in somatic cells while that of nGPx4 mRNA was extremely low. In somatic tissues and cultured cells, the amounts of cGPx4 mRNA were approximately 2.5–12, 600–9000 times higher than those of mGPx4 mRNA and nGPx4 mRNA as determined by TaqMan assay. The expression of mGPx4 mRNA and nGPx4 mRNA was significantly higher only in testis than in other tissues. Expression of mGPx4 mRNA and nGPx4 mRNA is induced significantly in testis during spermatogenesis (Imai et al. 2006).
3 Function of Three Types of Organelle-Specific GPx4 in Cells
Analysis of overexpression of three types of GPx4 in rat basophile leukemia (RBL2H3) cells revealed that three types of GPx4 play organelle-specific independent roles in the modulation of inflammation, signal transduction, and cell death (Imai and Nakagawa 2003; Imai 2010) (Fig. 2).

Functions of three types of GPx4 in rat basophile leukemia cells. Functional analyses of stable transformants overexpressing of three types of GPx4 such as cGPx4 (L9 cells), mGPx4 (M15 cells) and nGPx4 (N63 cells) in rat basophile leukemia cells (RBL2H3 cells) were summarized. GPx4 could directly reduce phospholipid hydroperoxide (PLOOH) to hydroxyl lipid (PLOH) using glutathione (GSH). cGPx4 in the nucleus inhibited the activation of 5-lipoxygenase (5-LOX) by reduction of lipid hydroperoxide (ROOH) to hydroxyl lipid (ROH) as the activator of 5-LOX that is oxidized from inactive Fe (II) to active Fe (III), resulting in the suppression of production of leukotriene B4 (LTB4). cGP4 in the endoplasmic reticulum (ER) inhibit the activation of cyclooxygenase (COX), prostaglandin H2 synthase (PGHS) by reduction of lipid hydroperoxide as the activator of COX that is oxidized from inactive heme Fe (III) to active heme Fe (IV), resulting in the suppression of prostaglandin E2 (PGE2). cGPx4 also suppress the activation of the IgE signaling via p38, resulting in the inhibition of production of platelet-activating factor (PAF). mGPx4 could suppress the release of cytochrome c (cyt.c), the activator of caspase 3 from mitochondria in apoptosis induced by mitochondrial death pathway such as 2-deoxyglucose (2DG), UV and staurosporine (Staur). mGPx4 inhibited the detachment of cytochrome c (cyt.c) from cardiolipin (CL), mitochondrial specific phospholipid and the conformational change of adenine nucleotide transporter (ANT) regulating the opening of permeability transition pore such as voltage-dependent anionic channel (VDAC)-Bax complex by reducing of cardiolipin hydroperoxide (CLOOH). nGPx4 could suppress the oxidative damage of nucleoli induced by doxorubicin (Dox) and actinomycin D (ActD). Three type of organelle-specific GPx4 played an independent and important regulator of local lipid hydroperoxide as a signal molecule. AA arachidonic acid, HPETE hydroperoxyeicosapentanoicacid, PL phospholipid
Overexpression of cGPx4 in the cytosol and nucleus could suppress the production of leukotriene and prostaglandin at the nucleus and endoplasmic reticulum in response to several stimuli, indicating that cGPx4 could suppress the activation of lipoxygenase and cyclooxygenase at the nucleus and endoplasmic reticulum by reducing fatty acid hydroperoxide as activators of lipoxygenase and cyclooxygenase (Imai et al. 1998; Sakamoto et al. 2000).
Overexpression of cGPx4 could suppress the production of platelet-activating factor (PAF) by IgE-antigen stimulation. cGPx4 could suppress the phosphorylation of p38 by lipid hydroperoxide signaling pathway (Sakamoto et al. 2002). However, cGPx4 could not inhibit suppress apoptosis induced by the mitochondrial death pathway (Imai et al. 1996; Arai et al. 1999; Nomura et al. 1999).
On the other hand, overexpression of mGPx4 in the mitochondria could suppress the apoptosis induced by staurosporine, 2-deoxyglucose and UV, whereas mGPx4 could not inhibit the production of leukotriene and prostaglandin (Arai et al. 1999; Nomura et al. 1999). mGPx4 could suppress the release of cytochrome c from mitochondria by inhibition of generation of mitochondria specific phospholipid, cardiolipin hydroperoxide during apoptosis induced by mitochondria death pathway (Nomura et al. 2000).
Overexpression of nGPx4 in the nucleoli could inhibit nucleoli-damaged cell death induced by doxorubicin and actinomycin D, but not staurosporine, 2-deoxyglucose, and UV (Nakamura et al. 2003).
These results demonstrated that three types of organelle-specific GPx4 played an independent and important regulator of local lipid hydroperoxide as a signal molecule, indicating that lipid hydroperoxide generated in the local area of each organelle might function as a signal molecule in inflammation and cell death (Imai 2010).
4 Mitochondrial GPx4 Inhibit the Release of cyt.c from Mitochondria by Cardiolipin Hydroperoxide in Apoptosis by Mitochondrial Death Pathway
Our mGPx4 transformant studies revealed that mGPx4 inhibits apoptosis induced by 2-deoxyglucose, staurosporine, actinomycin D, and UV, but not A23187 and anti-Fas antibody stimulation and mGPx4 could suppress the release of cytochrome c from mitochondria, resulting in inhibition of caspase-3 activation and PS externalization (Arai et al. 1999; Nomura et al. 1999) (Fig. 2).
Cardiolipin, that is located primarily in the mitochondrial inner membrane, has a unique structure containing three glycerol moieties, two phosphate residues, and four fatty acyl chains in the same molecule. Cardiolipin (CL) is critical for maintenance of cristae structure as well as stabilizing mitochondrial electron transport complexes, carrier proteins, and phosphokinases (Imai and Nakagawa 2003; Maguire et al. 2017).
Oxidative stress induced peroxidation of CL because CL is rich in polyunsaturated fatty acids, especially linoleic acid. We found that during apoptosis by mitochondrial pathway, oxidation of CL in the mitochondria occurs early events before the release of cytochrome c from mitochondria. Cytochrome c strongly associates with CL, whereas oxidation of CL is a required step for the dissociation of cytochrome c from CL in the inner membrane, since oxidized CL exhibits a reduced binding affinity for cytochrome c over CL. mGPx4 could suppress the peroxidation of cardiolipin in mitochondria and inhibit the dissociation of cytochrome c from mitochondrial inner membrane in apoptosis (Nomura et al. 2000). And mGPx4 could inhibit the change of conformation by loss of activity of adenine nucleotide translocator (ANT) that could regulate the opening of permeability transition pore by cardiolipin hydroperoxide (Imai et al. 2003b). We proposed “cardiolipin hydroperoxide cascade” for the release of cytochrome c from mitochondria in apoptosis in 2003 (Imai and Nakagawa 2003).
Kagan’s group found that pro-apoptotic stimuli induced H2O2 dependent peroxidation of CL by cytochrome c (Kagan et al. 2005). Recent works indicated that oxidation of CL results in migration of CL from inner membrane to the outer mitochondrial membrane (Maguire et al. 2017; Li et al. 2015). Oxidized CL recruits and interacts with Bax to initiate formation of mitochondrial transition pore (Korytowski et al. 2011).
Recently our data showed that mGPx4 could suppress the release of cytochrome c from mitochondria by different mechanisms of Bax- and tBid-induced apoptosis (Imai, unpublished). These data demonstrate that mGPx4 is a regulator of apoptosis by inhibition of CL peroxidation-dependent release of cytochrome c from mitochondria.
5 Suppression of Peroxidation of Phospholipid by Non-mitochondrial GPx4 and Vitamin E Is Essential for the Survival of Cells and Mice
Ablation of all GPx4 gene in mice induced early embryonic lethal at 7.5 dpc (Imai et al. 2003a). GPx4-null embryo could not develop into Inner Mass Cell (ICM), whereas vitamin E could rescue formation of ICM in GPx4-null embryo. Transfection of cDNA for cGPx4 also could recover the formation of ICM in GPx4-null embryo. These results indicated that suppression of generation of lipid hydroperoxide by GPx4 and vitamin E is required for embryo development.
We also succeeded that transgenic complementary rescue method using an all GPx4-loxP transgene rescued embryonic lethality in endogenous all GPx4 KO mice (Imai 2010; Imai et al. 2009). mGPx4 starts codon mutation transgene and nGPx4 start codon mutation transgene could rescue the embryonic lethality of all GPx4 KO mice, whereas cGPx start codon mutation transgene could not rescue the embryonic lethality. Double GPx4 mutation transgene containing double mutation of start codons for mGPx4 and nGPx4 could rescue the lethal phenotype, indicating that cGPx4 is essential for embryo development and normal growth in mice (Imai 2011).
In fact, mGPx4 KO mice and nGPx4 KO mice display normal development except sperm maturation (Schneider et al. 2009; Conrad et al. 2005). These results demonstrated that anti-apoptotic function of mGPx4 is not required for embryonic normal development and programed cell death such as apoptosis in mice and cGPx4 is important role for embryo development.
mGPx4 KO mice showed male infertility by structural damage of mitochondria of spermatozoa, but showed normal production of the number of spermatozoa in testis (Schneider et al. 2009; Imai 2011). On the other hand, spermatocyte—specific all GPx4 KO mice showed severe defect of spermatogenic cells in testis, the significant low level of the number of spermatozoa, a hairpin-like flagella and abnormal structure of mitochondria of spermatozoa, resulting in male infertility (Imai et al. 2009; Fujii and Imai 2014). Administration of vitamin E with spermatocyte-specific all GPx4 KO mice could rescue the defect of spermatogenesis in seminiferous tables in mice, leading to the recover of production of the number of spermatozoa.
Docosahexaenoic acid (DHA) is a long-chain omega-3 polyunsaturated fatty acid that is a critical component of lid structure. DHA plays important roles throughout the body and is essential for maintaining the structure and function of the brain and eye. In the rod cells of retinal photoreceptors for example, DHA within membrane facilitates the conformational change triggered by a light signal. However, DHA is easy for oxidation, since it is polyunsaturated fatty acid. Although photoreceptor cells normally developed and differentiated into rod and cone cells by P12 in photoreceptor cell specific GPx4 KO mice, they rapidly underwent drastic degeneration and completely disappeared by P21. Photoreceptor cell death induced by loss of GPx4 was TUNEL positive, lipid oxidation dependent, and caspase-independent cell death. GPx4 is a critical antioxidant enzyme for the maturation and survival of photoreceptor cells (Ueta et al. 2012). Using in vivo wound repair model by application of n-heptanol on the corona or laser-induced choroidal neovascularization mice model, we clarified that GPx4 plays important role for oxidative homeostasis and cell survival in the corneal epithelial cells and the retinal pigment epithelium (RPE)/choroid tissue (Sakai et al. 2016a; Roggia et al. 2014).
Tamoxifen inducible all GP×4 KO mice induced acute renal failure with ferroptotic cell death (Angeli et al. 2014).
Heart specific all GPx4 KO mice showed embryonic death by cardio sudden death. However heart specific all GPx4 KO mice could normally grow by administration with vitamin E diet. Heart specific all GPx4 KO mice rescued by vitamin E diet show cardio sudden death by exchange to the normal diet (Imai unpublished).
Liver specific all GPx4 KO mice also showed neonatal lethal, however liver specific all GPx4 KO mice normally grow by administration of vitamin E. And change of vitamin E diets to vitamin E deficient diet induces sudden death of Liver specific all GPx4 KO mice (Imai 2011; Carlson et al. 2016).
Endothelium-specific deletion of GPx4 had no obvious impact on normal vascular homeostasis in mice maintained on a normal diet. However when mice were fed a vitamin E depleted diet for 6 weeks before endothelial deletion of GPx4 was induced by tamoxifen, 80% of endothelium GPx4 knockout mice died with detachment of endothelial cells from the basement membrane (Wortmann et al. 2013).
These results demonstrated that suppression of phospholipid peroxidation by cGPx4 and vitamin E is essential for survival in certain cells and mice and imbalance of suppression of phospholipid peroxidation might cause the several diseases (Fig. 3).

Imbalance of membrane oxidation homeostasis cause several diseases in cGPx4 knockout mice and cGPx4 depleted cells. GPx4 knockout mice is early embryonic lethal at 7.5 dpc. GPx4 KO embryo could not form Inner cell Mass (ICM), but addition of vitamin E could rescue it. cGPx4 is required for the survival for the normal embryogenesis, since mGPx4 and nGPx4 KO mice normally grow. Testis, Liver specific GPx4 KO mice display the cell death of spermatogenic cells and hepatocyte, but administration of vitamin E diet could rescue the cell death in tissues of mice. 15-Lipoxygenase (15-LOX) directly could oxidize phospholipid. However, GPx4 depletion in 15-LOX KO mice induce acute renal injury and GPx4 depletion in 15-LOX null-MEF cells induce the lipid peroxidation-dependent cell death, indicating that 15-LOX is not essential for lipid peroxidation-dependent cell death in mice. 15-LOX might be one of the candidate for lipid peroxidation-dependent cell death in 15-LOX expressing cells. Thus, imbalance of membrane oxidation homeostasis (membrane oxidizing system vs. anti membrane oxidizing system) cause the oxidative stress related diseases in animal and human. PLOOH phospholipid hydroperoxide
Cell death by genetic depletion of all GPx4 in each tissue of mice is dependent of loss of cGPx4, since mGPx4 and nGPx4 double KO mice normally survive.
In human, a defect of expression of GPx4 in spermatozoa was found in human infertile male with oligoasthenozoospermia that have the low number and low motility of spermatozoa (Imai et al. 2001). This phenotype in human GPx4 deficient oligoasthenozoospermia were consistent with that of spermatocyte-specific all GPx4 KO mice, indicating that low production of spermatozoa in human infertile patient is due to the defect of spermatogenesis by the deficiency of cGPx4 in testis (Imai et al. 2009).
Sedaghahatian-type spondylometaphyseal dysplasia (SSMD) is a neonatal lethal from spondylometaphyseal dysplasia characterized by severe metaphysical chondrodysplasia with limb shortening, platyspondyly, cardiac conduction, defects, and central nerves system abnormalities. By whole exam sequencing of a child affected with SSMD and her unaffected parents, two rare variants of GPx4 that the mutation results in a frameshift and premature truncation of GPx4 were identified (Smith et al. 2014).
These results indicate that truncating mutation in GPx4 in two families affected with SSMD supports the pathogenic role of mutated GPx4 in this vary rare disease.
GPx4 is one of the selenoproteins in human. Selenocysteine insertion sequence-binding protein 2 (SBP2) is essential for the biosynthesis of selenoproteins including GPx4. Subjects with mutations in the SBP2 gene have decreased levels of many selenoproteins, resulting in a several phenotype with high lipid peroxidation in blood since they have low levels of antioxidant activity such as GPx1 and GPx4. Treatment of the vitamin E for 2 years to the subjects with SBP2 mutation reduced lipid peroxidation product levels to the control subjects, indicating that vitamin E treatment effectively inhibits the generation of lipid peroxidation products in human (Saito et al. 2015).
To clarify the mechanism of cell death by depletion of GPx4, we established GPx4-LoP TG/KO MEF cells (TK cells) from GPx4-loxP TG/KO mice and tamoxifen inducible GPx4 depleted TK cells (ETK cells) by infection of retrovirus of estrogen receptor binding Cre (CreERT2) (Imai et al. 2009; Imai 2010). Conrad’ group already established the tamoxifen inducible GPx4 depleted MEF cells (Pfa1 cells) from GPx4 flox/flox mice (Seiler et al. 2008).
Retrovirus-mediated depletion of GPx4-LoxP TG transgene in EK cells resulted in the loss of all GPx4 protein 2 days and cell death 4 days after infection of Cre-expressing retrovirus. In tamoxifen inducible all GPx4 depleted TK cells and Pfa1 cells, addition of tamoxifen induce loss of all GPx4 24 h and cell death 3 days after tamoxifen treatment. As characteristics of GPx4-deficient cell death in MEF cells and mice, the time required for lethality is very long. Addition of Trolox, vitamin E derivative, or vitamin E could rescue GPx4 depleted cell death by retrovirus transfection in TK cells and addition of tamoxifen in ETK cells and Pfa1cells. Vitamin E rescued GPx4 null-MEF cells could normally grow.
Retrovirus-infection of mGPx4, cGPx4, nGPx, cGPx4 (cys) and other antioxidants such as GPx1, SOD1, and SOD2 demonstrated that cGPx4 most effectively could rescue the all GPx4 depleted cell death, but not cGPx4 (cys) that mutated a enzymatic active site selenocysteine to cysteine, mGPx4, nGPx4, GPx1, SOD1, and SOD2 (Imai et al. 2009; Imai 2010).
These results demonstrated that the suppression of phospholipid peroxidation by cGPx4 and vitamin E is required for the growth and survival in MEF cells as the same as in mice. GPx4 depletion by genetic system induces lipid peroxidation-dependent, and caspase 3 independent novel cell death, since broad caspase-inhibitor, Z-Bad-FMK could not suppress GPx4 depleted cell death.
Lipid peroxidation by loss of GPx4 was generated in the cytosol sites of organelle, but not in mitochondria, as the signal for novel non-apoptotic cell death (Imai 2010, 2011).
6 Ferroptosis by Oncogenic Mutated Ras-Selective Lethal Compounds
The RAS family of small GTPase (HRAS, NRAS, and KRAS) is commonly mutated in cancer and several groups have searched for small molecules that are selectively lethal to cells expressing oncogenic mutated RAS proteins. Stockewell’s group isolated two classes of novel oncogenic RAS-Selective Lethal (RSL) small molecules named eradicator of RAS and ST (erastin) (Yagoda et al. 2007) and RAS-selective Lethal 3 ((1S, 3R)-RSL3) (Yang and Stockwell 2008). Both compounds killed engineered human tumor cells expressing oncogenic HRASV12 at lower concentrations than isogenic cells expressing wild-type HRAS. Erastin and RSL3 treatment could induce cell death very quickly for 8–12 h in RAS-mutated cancer cells. Erastin and RSL3 treatments do not trigger morphological changes or biochemical processes consistent with apoptosis such as chromatin condensation, DNA ladder formation, and caspase 3 activation. Erastin and RSL3 induced cell death is not inhibited by caspase-inhibitor (Z-VAD-FMK), by a necroptosis inhibitor (necrostatin-1) and by an inhibitor of autophagy (chloroquine, 3-methyladenine). Neither mitochondrial ROS production nor Ca2+ influx is necessary for cell death. Erastin treatment resulted in a unique “dysmorphic” mitochondrial phenotype observable by transmission electron microscopy. Erastin and RSL3 induced cell death is effectively suppressed by the iron chelators, DFO (deferoxamine), and ciclopirox (CPX) as well as by the lipophilic antioxidants, Trolox (a soluble vitamin E analog), butylated hydroxyanisole (BHA), butylated hydroxytoluene (BHT), liproxstatin-1, and ferrostatin-1 (Fer-1). These results indicated that erastin- or RSL3-induced cell death is the iron-dependent oxidative novel non-apoptotic cell death, named “ferroptosis” (Dixon et al. 2012). Ferrostatin-1 (Fer-1) antioxidants was isolated as a ferroptosis specific inhibitor by Stockwell’group. Ferrostatin-1 could suppress the ferroptosis by erastin and RSL3, but not apoptosis and necroptosis (Dixon et al. 2012).
The elucidation of target molecule for Erastin and RSL3 provides that ferroptosis inducer can be divided into two classes of small molecule substrates. Class 1 ferroptosis inducers include erastin, sulfasalazine (SAS), artemisinin and its derivatives, which can inhibit system X −c that transport cystine into cells (Dixon et al. 2012). Class 2 ferroptosis inducers include Ras-selective lethal 3 compound (RSL3), ML162 (DPI17), DPI20, DP112, DP113, etc., which can directly inhibit glutathione peroxidase 4 (GPx4) activity and ultimately lead to an accumulation of lipid peroxides without the decrease of GSH (Yang et al. 2014) (Fig. 4).

Regulation of ferroptosis by oncogenic mutated Ras-Selective Lethal compounds. Ferroptosis is an iron-dependent form of cell death involving the generation of phospholipid peroxidation (PL-Ox) in Ras-mutated cancer cells induced by oncogenic mutated Ras-selective lethal compounds (Type I and Type II RSL) as shown in pink zone. In cancer cells, cystine (Cys-Cys) uptake for maintaining of intracellular glutathione (GSH) via system X −c (pink zone), Iron uptake for production of iron containing enzymes, mitochondrial enzymes, p450 and lipoxygenase (LOX) via transferrin (TF) receptor (TFR) (yellow zone), and Glutamine (Gln) uptake for glutaminolysis in metabolic changes for energy production via SLC1A5 (green zone) were enhanced as compared to normal cells. Glutathione peroxidase 4 (GPx4) could reduce phospholipid hydroperoxide (PLOOH) to hydroxyl phospholipid (PLOH), oxidizing GSH to glutathione disulfide (GSSG) in the process. GPx4 is a key regulator of ferroptosis, since loss of GPx4 activity accumulates ferrous iron (Fe2+)-dependent oxidized phospholipid (PL-Ox), leading to the loss of membrane permeability (LMP) and cell death. Type I ferroptosis inducer, erastin, sulfasalazine and sorafenib, inhibit the cystine transportor, system X −c , resulting in the decrease of GSH, loss of GPx4 activity to ferroptosis. Type II ferroptosis inducer, RSL3 and FIN56 directly bound to GPx4 or induce the degradation of GPx4, resulting in iron-dependent phospholipid peroxidation to ferroptosis without the decrease of GSH. Ferroptosis was inhibited by iron-chelator DFO (deferoxiamine) and CPX (ciclopirox), and anti-lipid peroxidation compounds such as vitamin E, coenzyme Q10 (coQ10), Trolox and ferroptosis specific inhibitor, ferrostatin-1(Fer-1) and liproxstatin-1 (pink zone). Intracellular Fe2+ availability was regulated by degradation of extracellular transferrin (TF) bound Fe3+ incorporated by transferrin receptor (TFR) and intracellular ferritin bound Fe3+ in endosome / lysosome containing ferritinophagy (yellow zone). Sensitivity of phospholipid peroxidation in membrane is regulated by the quality of lipid membrane (Lipoquality) that means the change of the content of polyunsaturated fatty acid (PUFA) such as arachidonic acid (AA) in phospholipid membrane (violet zone). Acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) could modulate the content of AA in phospholipid by remodeling pathway (Lands’ pathway). The decrease of PL-PUFA is correlated to the sensitivity of lipid peroxidation and ferroptosis. Ferroptosis is required for transferrin and glutamine (Gln) in the culture media. Glutamine produces NADPH and ATP during glutaminolysis pathway, but this requirement of glutaminolysis remained to be resolved (green zone). p53 suppress the transcription of system X −c transportor and induction of spermidine/spermine N-acetyltransferase 1 (SAT1), up-regulator of 15-lipoxygenase (15-LOX). Other small molecule inducers of ferroptosis are indicated in red, while suppressors of ferroptosis are in blue. GCL Glutamate cysteine ligase, GSS glutathione synthetase, Cys, cysteine, Glu glutamate, Gln glutamine, α-KG alpha-ketoglutarate, GPNA l-g-glutamyl-p-nirtroanilide, AOA amino-oxyacetate, GLS2 glutaminase, GOT glutamine oxaloacetate aminotransferase, TCA tricarboxylic acd, Mal Malic acid, Pyr pyruvic acid, Lac lactic acid, DFO deferoxamine, CPX ciclopirox. Baf.A1 Bafiromycin A, PepA-Me pepstatin A-methyl ester, STEAP3 six-transmembrane epithelial antigen of the protease 3, DMT1 divalent metal transporter 1, NCOA4 cargo receptor for ferritinophagy, ROSI rosiglitazone, PIO pioglitazone, BSO buthione-(S,R)-sulfoximine, MVA Mevalonate, FPP Farnesyl diphosphate, SQS squalene synthase, SQ squalene
6.1 Inhibition of System X −c Leads to Ferroptosis (Type I)
System X −c is a membrane Na+-dependent cysteine-glutamate exchange transporter, which is a disulfide-linked heterodimer composed of a light-chain subunit (xCT, SLC7A11) and a heavy-chain subunit (CD98hc, SLC3A2) (Sato et al. 1999). While system X −c transports intracellular glutamate to the extracellular space, it transports extracellular cystine into cells, which is the transformed into cysteine for glutathione (GSH) synthesis. Erastin acts as a direct inhibitor of system X −c function (Dixon et al. 2012) (Fig. 3). Erastin treatment leads to significant depletion of intracellular glutathione by inhibition of cellular uptake of cystine, since cystine is a key molecule for glutathione (GSH) synthesis. This effect could be inhibited by β-mercaptoethanol, because β-ME can enhance cysteine uptake through other pathways. Glutamate–cysteine ligase (γ-glutamylcyteine synthetase, GCL) is the rate-limiting first enzyme in the two step synthesis of glutathione (GSH). Blockage of GCL by buthionine-(S,R)-sulfoximine (BSO) can induce depletion of GSH, leading to ferroptosis that is prevented by vitamin E, Ferostatin-1, and DFO. These results indicated that inhibition of GSH synthesis is required to trigger ferroptosis in some cells (Seiler et al. 2008; Angeli et al. 2014). GPx4 can reduce direct toxic lipid peroxide (PLOOH) to nontoxic lipid alcohols (PL-OH) using GSH as a cofactor. Depletion of intracellular glutathione by erastin induced accumulation of lipid peroxidation detected by 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA) and BODIPY-C11. Therefore, inhibition of system X −c by erastin suppresses GPx4 activity to cause accumulation of lethal lipid peroxides and to initiate the execution of ferroptosis. Erastin, sulfasalazine (Gout et al. 2001; Skouta et al. 2014) and sorafenib (Louandre et al. 2013; Lachaier et al. 2014) are ferroptosis inducers through this mechanism.
On the other hand, erastin is also reported to bind to voltage-dependent anion channels (VDAC2 and VDAC3) on the mitochondria to alter membrane permeability and the ion selectivity of the channels, leading to the mitochondrial dysfunction and oxidant release in ferroptosis (Yagoda et al. 2007). Erastin also leads to activation of an endoplasmic reticulum stress response and up-regulation of CHAC1 (cation transporter homolog 1) during ferroptosis (Dioxn et al. 2014). However, this relationship between these events in mitochondria and ER and ferroptosis remained to be elucidated.
The p53 protein has been well characterized for its response to various cellular stresses, including of growth arrest, senescence and apoptosis. p53 inhibits cystine uptake and sensitizes cells to ferroptosis by repressing expression of SLC7A11, a key component of the system X −c transporter with oxidants. p53 binds to the SLC7A11 locus at a specific p53 response element within the 5′ untranslated region (Jiang et al. 2015). p53 (3KR, R117, R161, and R162), an acetylation-defective mutant that fails to induce cell-cycle arrest, senescence and apoptosis, fully retains the ability to regulate SLC7A11 expression and induce ferroptosis upon reactive oxygen species (ROS)-induced stress. Acetylation of K98 of p53 is required for repression of transcription of SLC7A11 and induction of ferroptosis (Wang et al. 2016). p53 (4KR, R98, R117, R161, R162) could not induce tumor suppression. These results involve a new mode of tumor suppression based on p53 regulation of cystine metabolism, ROS responses and ferroptosis.
Addition of high concentration of Glu in neuronal cell lines induces the inactivation of system X −c and inhibition of uptake of cystine, leading to the GSH depletion and oxidative cell death (oxidative glutamate toxicity) named “Oxytosis” (Tan et al. 2001). Oxidative glutamate toxicity can be blocked by Ferrostatin-1, indicating that this cell death is ferroptosis (Dixon et al. 2012; Liu et al. 2015; Kang et al. 2014). GPx4 can suppress the glutamate-induced oxytosis in the retina (Sakai et al. 2015a). However, several reports indicated that mechanism of downstream of ROS accumulation in oxidative glutamate toxicity was different from ferroptosis (Henke et al. 2013; Tobaben et al. 2011). For example, extracellular Ca2+ influx, BH3 interacting domain death agonist (Bid) mediated mitochondrial damage and nuclear translocation of apoptosis inducing factor (AIF) are required for oxidative glutamate toxicity, but not required for ferroptosis. These differences remain to be resolved.
6.2 Inhibition of Chemical Inactivation of GPx4 Leads to Ferroptosis (Type II)
Type II inhibitor of ferroptosis such as (1S,3R)-RSL3 could inhibit ferroptosis without the decrease of GSH, whereas Ferrostatin-1, vitamin E, and DFO suppressed RSL3 induced ferroptosis (Yang and Stockwell 2008) (Fig. 4). GPx4 was identified as a major target protein of (1S,3R)-RSL3 by affinity purification with RSL3 and identification of sequence of amino acids. (1S,3R)-RSL3 contains an electrophilic chloroacetamide and covalently interacts with the active site “selenocysteine” of GPx4 to inhibit its enzymatic activity (Yang et al. 2014, 2016). Treatment of RSL3 induced the peroxidation of phospholipid that is prevented by vitamin E, ferrostatin-1, and DFO. GPx4 overexpression suppresses the RSL3-induced ferroptosis, whereas GPx4 knockdown enhances the sensitivity for RSL3-induced ferroptosis, suggesting that inhibition of GPx4 activity is a major contributor to ferroptosis. Thus, GPx4 is currently believed to be a key regulator in ferroptosis induced by erastin and RSL3.
Another screening of caspase-independent lethal compounds revealed new six specific ferroptosis inducers (Shimada et al. 2016). FIN56, one of six ferroptosis inducer, induced cell death was prevented by vitamin E, DFO, and U0126, MEK inhibitor in Ras-mutated cancer cell lines (BJelR). FIN56 treatment induced cell death for 48 h slowly that RSL3 at 8 h. But FIN56 treatment produces lipid peroxidation 3 h, whereas RSL3 produces it 1 h. FIN56 could not bind to GPx4. But FIN56 promoted degradation of GPx4, whereas a proteasome inhibitor MG132 did not inhibit FIN56 -induced cell death. 5-(tetradecyloxy)-2-furoic Acid (TOFA), an inhibitor of acetyl-CoA carboxylase (ACC), inhibited the degradation of GPx4. TOFA also suppresses FIN56-induced cell death and lipid peroxidation. ACC is an enzyme involved in fatty acid synthesis. But ACC itself was not direct target of FIN56. Chemoproteomics analysis revealed that FIN56 binds and activates squalene synthase (SQS), an enzyme involved in the cholesterol synthesis, in a manner independent of GPx4 degradation. Squalene synthase inhibitors blocked FIN56-induced ferroptosis and increased the mevalonate metabolite such as farnesyl pyrophosphate (FPP) and coenzyme Q10 (CoQ10), an electron carrier in the mitochondrial respiratory chain and endogenous antioxidant. Supplementation of FPP or CoQ10 suppresses the lethality of FIN56 and RSL3. These results indicate that mechanism of cell death by FIN56 involves two distinct pathways, GPx4 degradation pathway that requires the activity of ACC, and activation of SQS, that leads to CoQ10 depletion as antioxidants. GPx4 degradation was also observed in RSL3-induced ferroptosis in MEF cells, indicating that RSL3 might involove another target protein except for GPx4 like FIN56 (Kagan et al. 2016; Shimada et al. 2016) (Fig. 4).
6.3 The Role of Iron Homeostasis in Ferroptosis
Iron is essential for the execution of ferroptosis. Erastin- and RSL3-induced ferroptosis can be inhibited by iron chelator of membrane impermeable (DFO) and membrane permeable (CPX) (Dixon et al. 2012; Yang et al. 2014).
Nutrient starvation in growth medium containing glucose but lacking both amino acids and serum, induces apoptosis in mouse embryonic fibroblasts (MEFs), whereas nutrient starvation medium in the presence of serum change from the apoptotic cell death to ferroptosis. Cystine deficiency of medium in the presence of serum causes ferroptosis in MEF cells, leading to the decrease of intracellular glutathione. This is interesting system that can change apoptosis to ferroptosis with or without serum (Gao et al. 2015). Two serum factors, the iron-carrier protein transferrin and amino acid glutamine, were identified as the inducer of ferroptosis by cystine deficiency and erastin. Ferroptosis by erastin or cystine deficiency is inhibited by genetic silence of transferrin receptor that required for the uptake of transferrin-iron complexes into cells (Gao et al. 2015; Yang and Stockwell 2008). Supplementing the medium with iron-bound transferrin or a bioavailable form of iron (ferric ammonium citrate) accelerates erastin-induced ferroptosis (Gao et al. 2015; Dixon et al. 2012) and GPx4-depleted cell death (Sakai et al. 2016a). These results confirmed that the requirement for iron in ferroptosis (Fig. 4).
l-glutamine (l-Gln) is the most abundant amino acid in the body. Proliferating cells use l-Gln both as a nitrogen source for the biosynthesis of nucleotides, amino acids, and hexamine and as an important carbon source for the tricarboxylic acid (TCA) cycle. Pharmacological inhibition of SLC1A5, l-Gln receptor component by l-g-glutamyl-p-nirtroanilide (GPNA) or RNAi knockdown of these receptor prevented ferroptosis induced by cystine deficiency. Gln is converted into glutamate (Glu) by glutaminases (GLS). An inhibitor of GLS, Compound 968, and RNAi knockdown of GLS2 but not GLS1 blocked ferroptosis induced by erastin or cystine deficiency. Downstream of glutaminolysis, glutamate can be further converted into a-ketoglutarate (a-KG) either transaminase-mediated transamination or by glutamate dehydrogenase (GLUD1)-mediated glutamate deamination. The amino-oxyacetate (AOA), a pan inhibitor of transaminases and RNAi of transaminase GOT1 inhibited ferroptosis induced by erastin and cystine deficiency, but not GLUD1 RNAi could not suppress. The glutamine-fueled intracellular metabolic pathway, glutaminolysis, involved important roles in ferroptosis in cancer cells. Inhibition of glutaminolysis can reduce heart injury triggered by ischemia/reperfusion. These results indicated that glutaminolysis is essential for ferroptosis. NADPH and ATP, which are produced by glutaminolysis might be related to the execution of ferroptosis in cancer cells (Fig. 4).
The Ras-mutated cancer cells accumulate iron by modulating expression levels of ferritin and the transferrin receptor (Yagoda et al. 2007). Iron that is taken up by the transferrin receptor bound to transferrin-Fe(III)2 is routed into the endosome/lysosome pathway. The ferric ion, Fe(III), that is released from transferrin is reduced by an endosomal reductase activity (e.g., six-transmembrane epithelial antigen of the prostate 3) prior to export of ferrous ion, Fe(II), by a transporter such as divalent metal transporter1 (DMT1). Most cellular iron is stored as a component of ferritin in the Fe (III) form (Hentze et al. 2010). Recent studies showed that selective autophagic degradation of ferritin by the cargo receptor NCOA4 that recognized ferritin promotes iron release within lysosomes, and this ferritinophagy is important for cellular iron homeostasis for the increase of free iron (Mancias et al. 2014; Dowdle et al. 2014; Bellelli et al. 2016). The cellular labile iron pool might be used to control intracellular iron homeostasis through its own redox activity or to provide iron constituents for functional proteins such as heme enzymes including P450, cyclooxygenase, NADPH oxidases (NOXs), and non-heme enzyme including lipoxygenase.
Bafiromycin A1 (Baf A1), an inhibitor of vacuolar H+-ATPase, the lysosomal aspartic protease inhibitor pepstatin A-methyl ester (PepA-Me), ammonium chloride (NH4Cl) that acts to neutralize acidic organelle such as lysosome blocked erastin and RSL3-induced ferroptosis and ROS production. PepA-Me caused both an increase in ferritin protein levels and a decrease in iron content, indicating this may block ferroptosis by preventing autophagic degradation of ferritin, whereas Baf A1 and NH4Cl inhibit the iron uptake by transferrin (Torii et al. 2016).
Inhibition of ferritinophagy by prevention of autophagy such as ATG13 and ATG3 or knockdown of cargo receptor NCOA4 for ferritinophagy, abrogated the accumulation of ferroptosis-associated cellular labile iron and ROS as well as ferroptosis. These results indicated endosomes/lysosomes and NCOA4 mediated ferritinophagy contribute to ferroptosis by modulating cellular iron homeostasis and ROS production (Gao et al. 2016; Hou et al. 2016) (Fig. 4).
Screening of shRNA suppressors of erastin in U2-OS cells revealed that PHKG2, the catalytic subunit of the PHK (phosphorylase kinase) complex, that activates glycogen phosphorylase (GP) to release glucose-1-phosphate from glycogen. PHK inhibitor, K252a inhibit erastin-induced ferroptosis, but not two glycogen phosphorylase inhibitors could not prevent, indicating that the metabolic pathway of glycogen breakdown is not responsible for the suppression of erastin-induced ferroptosis. Knockdown of PHKG2 suppressed the production of ROS and the availability of ferrous iron, indicating that novel iron regulatory function of PHKG2 is responsible for modulating sensitivity to erastin (Yang et al. 2016). Thus, iron availability to lipoxygenase by PHKG2 and lysosomal ferritin degradation is essential for erastin-induced ferroptosis.
Other pathways regulating the iron availability are also involved in the modulation of ferroptosis. Heat shock protein HSPB1 inhibition or heme oxygenase-1 (HO-1) induction enhanced the sensitivity of erastin-induced ferroptosis by the increase of intracellular free iron (Sun et al. 2015; Kwon et al. 2015).
6.4 Regulation of Phospholipid Peroxidation Modulate the Sensitivity of Ferroptosis
Lipid peroxidation occurs through the mechanism of both an enzymatic lipoxygenase reaction and a non-enzymatic free-radical chain reaction by Fenton reactions. Fenton reaction is defined as the oxidation of organic substrates by a mixture of hydrogen peroxide and ferrous iron. The Fenton reaction is a key reaction in oxidation of phospholipids and no specificity for classes of phospholipids. Singlet molecular oxygen also participates in a non-enzymatic reaction, which yields lipid hydroperoxide (LOOHs) from cholesterol and esterified lipids such as phospholipids and cholesteryl esters (Hauck and Bernlohr 2016). Ferroptosis is an iron-dependent cell death, accumulating lipid peroxidation. GPx4 that could reduce phospholipid hydroperoxide is a regulator of ferroptosis. Vitamin E or Ferrostatin-1 could suppress peroxidation of phospholipid and cell death in ferroptosis. The current proposal model is that after GPx4 activity is lost, lipid hydroperoxide was generated in membrane and attacked with ferrous iron, resulting in formation of lipid radicals and enhancement of lipid peroxidation to the lethal damage of membrane. Erastin and RSL3 treatment resulted in accumulation of several classes of phospholipid hydroperoxide such as phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS) and lysophospholipids and depletion of several polyunsaturated fatty acid (PUFA) such as arachidonic acid (AA, 20:4n-6), indicating that specific oxidized PUFA are cleaved from the glycerophospholipids by phospholipase A2 and subsequently degraded or destroyed in plasma membrane permeability (Angeli et al. 2014; Dixon et al. 2015; Yang et al. 2016) (Fig. 4).
Recent reports demonstrated that depletion of acyl-CoA synthetase long-chain family member 4 (ACSL4) or lysophophatidylcholine acyltransferase 3 (LPCAT3) that encode enzymes required for the reacylation of membrane lysophospholipid such as lyso PC (lysophosphatidylcholine) and lysoPE (Lysophosphatidylethanolamine) with arachidonic acid and other PUFAs (Hashidate-Yoshida et al. 2015), inhibits ferroptosis induced by RSL3 and erastin (Dixon et al. 2015; Doll et al. 2016; Kagan et al. 2016; Yuan et al. 2016). Cell membrane contains several classes of glycerophospholipids, which have numerous structural and functional roles in the cells. Polyunsaturated fatty acids, including arachidonic acid, eicosapentanoic acid, docosahexanoic acid, are located at the sn-2 (but not sn-1)-position of glycerophospholipids in an asymmetrical manner. Using acyl-CoAs as donors, glycerophospholipids are formed by a de novo pathway (Kennedy pathway) and modified by a remodeling pathway (Lands’ pathway) to generate membrane asymmetry and diversity (Sindou and Shimizu 2009). ACSL4 has a marked preference for long-chain PUFAs such as arachidonic acid (AA) (Yan et al. 2015; Cho et al. 2001). The content of PUFA in biomembrane such as phosphatidylcholine (PC) and phosphatidylethanolamine (PE) were complicatedly regulated by lysophophatidylcholine acyltransferase (LPCAT)1-4 and lysophophatidylethanolamine acyltransferase (LPEAT)1-2, remodeling enzymes and phospholipase A2. The remodeling of arachidonic acids in PC, PE, and PI were controlled by acyl-CoA synthetase long-chain family member 4 (ACSL4) and LPCAT2, LPEAT 2, LPCAT3 and LPIAT (Fig. 4).
Conrad’ group showed that ACSL4 KO cells are significantly resistant for RSL3 induced ferroptosis in Pfa1 cells (MEF cells), whereas only a mild protective effect was observed in LPCAT3 KO cells. Loss of ACSL4 gene inhibited RSL3 induced and tamoxifen inducible GPx4 depleted induced ferroptosis and peroxidation of phospholipid. Decrease of arachidonic acid (AA: 20:4) and adrenoyl acid (AdA: 22:4) containing PE (phosphatidylethanolamine) and PI (phosphatidylinositol) were markedly less abundant in ACSL4 KO cells than in WT cells, whereas no decrease was found in phosphatidylcholine and phosphatidylserine (Doll et al. 2016). Kagan’ group showed that loss of ACLS4 suppressed the formation of double and triple-oxidized AA– and AdA containing PE species (15–OOH–AA–PE, 15–OOH–8OH–AA–PE, 15–OOH–9OH–AA–PE and 15–OOH–12OH–PE) in RSL3-induced ferroptosis, whereas the elevation of four oxidized PE was enhanced in RSL3 induced WT cells (Pfa1 cells) and kidneys of GPx4KO mice. Lipoxygenase inhibitor (NCTT-956, PD146176, ML351, and Zileuton) could suppress the RSL3 induced ferroptosis in Pfa1 cells, but not COX inhibitor (Piroxicam) and P450 inhibitor (MSPPOH) could not. Liperfluo fluorescence intensity that was detected for phospholipid hydroperoxide was elevated in endoplasmic reticulum in RSL3-induced ferroptosis. His group demonstrated that oxidation of AA or AdA containing PE by 15-lipoxygenase (15-LOX) in ER is required for the execution for RSL3-induced ferroptosis in Pf1a cells as a ferroptotic death signal (Kagan et al.). Lipidomics analysis by other groups showed that phosphatidylcholine with PUFA was depleted, whereas the levels of ceramide and lysoPC (lysophosphatidylcholine) accumulated during erastin-induced ferroptosis in HT-1080 fibrosarcoma cells. In G401 cells and HT1080 cells, 15-LOX knockdown could suppress the erastin-induced ferroptosis, but not RSL3-induced ferroptosis. Also lipoxygenase inhibitor, Zileuton, Baicalein, and PD146176 could suppress the erastin-induced ferroptosis in G401 cells, but not cyclooxygenase (COX) inhibitor indomethacin (Dixon et al. 2015). ACSL4 knockdown could suppress erastin induced ferroptosis in HepG2 and HL60 cells. ACSL4 overexpression could recover erastin-induced ferroptosis in no ACSL4 expressing erastin-resistant cell lines, LNCaP and K562 cells. Addition of 5-lipoxygenase inhibitor, Zileuton could suppress the erastin-induced ferroptosis in LNCaP cells (Yuan et al. 2016). In these cell lines, 5-lipoxygenase is important for erastin-induced ferroptosis. GPx4 and vitamin E directly could suppress the initial activation of lipoxygenase by lipid hydroperoxide as an activator (Imai et al. 1998). These results indicated that 15-LOX is one of the candidate for initial oxidation of phospholipid in RSL3 and erastin-induced ferroptosis (Fig. 4).
The emerging role of p53 in ferroptosis has been a topic of great interest. But it is unclear how p53 orchestrates its activities in multiple metabolic pathways into tumor suppressive effects (Jiang et al. 2015; Wang et al. 2016). Spermidine/spermine N 1-acetyltransferase 1 (SAT1) gene was identified as a transcription target molecule of p53. SAT1 is a rate-limiting enzyme in polyamine catabolism. Interestingly induction of SAT1 mRNA results in the lipid peroxidation and ferroptosis upon reactive oxygen species induced stress. SAT1 induction was correlated with induction of 15-LOX mRNA. SAT1-induced ferroptosis significantly suppressed in the presence of PD146176, specific inhibitor of 15-LOX. Thus, 15-LOX is the key regulator of ferroptosis induced by SAT1, p53 up-regulator molecule (Ou et al. 2016).
On the other hand, in 15-LOX knockout MEF cells, GPx4 depletion and erastin also could induce ferroptosis. Expression of 15-LOX is especially localized in inflammatory cells and cancer cells. GPx4 depletion in 15-LOX deficient MEF cells also could induce the lipid peroxidation-dependent cell death (Angeli et al. 2014).
These evidences demonstrated that 15-LOX is not essential for the execution for ferroptosis and other oxidation system might be required for lipid oxidation during ferroptosis in 15-LOX deficient cells.
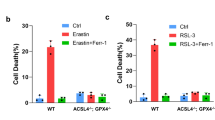
ACSL4 was preferentially expressed in a panel of basal-like breast cancer cell lines and its expression appeared to be strongly correlated with sensitivity to ferroptosis induction by RSL3. GPx4-Acsl4 double knockout cells showed marked resistance to ferroptosis, however RSL3 induced ferroptosis in GPx4-Acsl4 double knockout cells by supplementation of arachidonic acid. Interestingly GPx4-Acsl4 double knockout MEF cells were viable and proliferated normally in cell culture. But we showed that GPx4 knockout MEF cells also was viable and proliferated normally in cell culture with vitamin E. From these results, we proposed that antioxidant balance of lipid peroxidation in phospholipid such as elevation of vitamin E and decrease of PUFA in phospholipid might regulate the fate of cells (Fig. 3). And disruption of imbalance of oxidation of phospholipid might cause lipid peroxidation-dependent cell death such as ferroptosis.
7 Lipid Peroxidation-Dependent Cell Death by GPx4 Gene Disruption
GPx4 depletion by Cre-LoxP system or knockdown strategy induced lipid peroxidation-dependent cell death in many cells such as T cells (Matsushita et al. 2015), corneal endothelial cells (Uchida et al. 2016), conjunctival epithelial cells (Sakai et al. 2015b), vascular endothelial cells (Sakai et al. 2016b) and keratinocytes (Sengupta et al. 2013). These cell death without anti-cancer drug such as erastin and RSL3 was also rescued by vitamin E or ferrostain-1, anti-lipid peroxidation compounds.
Mechanism of cell death by depletion of GPx4 gene were mainly reported using tamoxifen inducible GPx4 depleted MEF cells (Pfa1 cells) from GPx4 flox/flox mice established by Conrad’ group (Seiler et al. 2008; Angeli et al. 2014; Doll et al. 2016; Kagan et al. 2016). Pfa1 cells have the expression of 15-lipoxygenase (15-LOX). Tamoxifen inducible GPx4 depleted cell death in Pfa1 cells was inhibited by 15-LOX inhibitor, iron-chelator deferoxamine, ferrostatin-1 and vitamin E. RSL3, a direct inhibitor of GPx4, induced ferroptosis in Pfa1 cells also were inhibited by 15-LOX inhibitor, Baicalen, PD146176, and ACSL4 inhibitor, Triacsin C, pioglitazone, iron-chelator deferoxamine, ferrostatin-1 and vitamin E. Active site of 15-LOX contain ferrous irons and GPx4 inhibit the activation of 15-LOX. From these results, Kagan et al. demonstrated that GPx4 depletion in Pfa1 cells initiate 15-Lipoxygenase activation dependent phospholipid peroxidation and induced ferroptosis (Kagan et al. 2016; Doll et al. 2016).
Until now, the mechanism of ferroptosis by both RSL-induced and GPx4 depleted cell death by GPx4 gene disruption in MEF cells are considered to be the same cell death mechanism. However, our established tamoxifen inducible GPx4 depleted MEF cells (ETK cells) did not express 15-LOX, 12-LOX, and 5-LOX (Imai et al. 2009; Imai 2010). In ETK cells, GPx4 depleted cell death by tamoxifen treatment is not inhibited by apoptosis inhibitor Z-BAD-FMK, by knockdown of autophagy regulating protein ATG5 and necroptosis regulator RIP kinase 1, indicating that GPx4 depleted cell death is caspase-independent non-apoptotic cell death. In ETK cells, erastin and RSL3 also induced ferroptosis as reported previously, since these cell deaths could inhibit by iron chelator, ferrostatin-1 and vitamin E. However, GPx4 depleted cell death by tamoxifen treatment in ETK cells could not be inhibited by 15-LOX inhibitor, Baicalen and ACSL4 inhibitor, Triacsin C and pioglitazone, iron-chelator deferoxamine, whereas ferrostatin-1 and vitamin E effectively suppressed the cell death. In ETK cells, erastin and RSL3 induce ferroptosis by 12 h, but tamoxifen treatment induced GPx4 depleted cell death 72–96 h after treatment although GPx4 expression could not be detected by 24 h. Lipid peroxidation was detected 6h after treatment of erastin in ETK cells and suppressed by iron chelator. However, lipid peroxidation by tamoxifen treatment was observed at 26 h early time before cell death at 72 h. Lipid peroxidation 26 h after tamoxifen treatment was suppressed by vitamin E, but not by iron chelator. Addition of scavengers for superoxide, hydrogen peroxide, and overexpression of antioxidant enzyme such as SOD1, SOD2, and GPx1 could not rescue GPx4-depleted cell death in ETK cells (Imai 2010). When vitamin E is added by 26 h after treatment of tamoxifen, GPx4-depleted cell death can be effectively inhibited, but the lethality can not be suppressed after 26 h, indicating iron-independent lipid peroxidation by 26 h is required for GPx4 cell death in ETK cells. Short hairpin RNA (shRNA) mediated knockdown of GPx4 in 15-LOX null-MEF cells is sufficient to induce cell death, and vitamin E also inhibited GPx4 deleted cell death in 15-LOX null-MEF cells. These results indicated that 15-LOX, independent and iron-independent lipid peroxidation is necessary for GPx4-depleted cell death in ETK cells.
Although RSL3 and erastin quickly induced cell death 12 h in ETK cells, GPx4 depletion by tamoxifen slowly induced cell death 72–96 h in ETK cells. One possibility of the differences of time for cell death between GPx4 gene disruption and RSL-compounds is that the existence of another accelerate pathway in ferroptosis by RSL except for GPx4 inactivation, since erastin can bind to VDAC in mitochondria and induce ER stress and RSL can bind to the several proteins except for GPx4. Recent works demonstrated that RSL3 could degrades GPx4 in MEF cells (Pfa1 cells) like FIN56, however whether protein degradation by RSL3 is specific for GPx4 or not remained to be solved (Kagan et al. 2016; Shimada et al. 2016). The other possibility is that the mechanism of GPx4 depleted cell death by GPx4 gene disruption is different from the mechanism of ferroptosis by erastin and RSL3.
In mouse erythroid precursor cells, inactivation of GPx4 induced to the accumulation of toxic lipid intermediates that covalently modify caspase-8 and trigger necroptosis in the absence of death receptor stimulation such as TNFα (Canli et al. 2016). GPx4 inhibition can sensitize cancer cells to apoptosis induced by second mitochondrial-derived activator of caspase (SMAC) mimetics, connecting ferroptosis to apoptotic cell death pathway (Dächert et al. 2016). These results demonstrated that differences of lipid peroxidation producing system such as specific enzyme and random oxidation by Fenton reaction, and producing local site in organelle in cells might execute different cell death signaling, such as apoptosis, ferroptosis, necroptosis, and novel lipid peroxidation cell death.
Thus, GPx4 could regulate several cell death pathways by suppression of phospholipid peroxidation in specific site or local site of organelle (Fig. 2).
8 Ferroptosis in Disease Model
Analysis of role of ferroptosis in pathological cell death has been enabled by the ferroptosis specific small molecule, Ferrostatin-1 (Fer-1) (Dixon et al. 2012), Liproxstatin-1 (Angeli et al. 2014) and vitamin E.
Erastin enhanced chemotherapy drug such as temozolomide, cisplatin, cytarabine/ara-C, and doxorubicin/Adriamycin in certain cancer cells (Yu et al. 2015; Chen et al. 2015; Yamaguchi et al. 2013). In vivo, erastin, piperazine erastin, and RSL3 inhibited tumor growth in a xenograft model (Yang et al. 2014; Sun et al. 2015). In a rat organotypic hippocampal slice culture model, glutamate-induced neurotoxicity was prevented by Fer-1 (Dixon et al. 2012). Fms-like tyrosine kinase3 (FLT-3, also termed CD135) is a cytokine receptor, that is important for the normal development of hematopoietic stem cells and progenitor cells. Inhibitors for Fms-like tyrosine kinase3 (FLT-3) and its downstream signaling molecule phosphoinositide 3-kinase α can suppress lipid peroxidation to inhibit ferroptosis in neuron (Kang et al. 2014).
In a Huntington’s disease model, Fer-1 restored the number of healthy neurons by inhibition of ferroptosis (Skouta et al. 2014). Fer-1 significantly protected the death of developing oligodendrocytes from cystine deprivation (Skouta et al. 2014).
Fer-1 prevented lethality in a model of acute injury of freshly isolated renal tubules, implicating ferroptosis-mediated cell death by acute kidney failure (Skouta et al. 2014). Ferrostatin analog (SRS 16–86) inhibits acute ischemia-reperfusion injury and oxalate nephropathy related acute kidney failure (Linkermann et al. 2014). Inducible knockout of GPx4 in the kidney leads to ferroptosis, which contributes to acute kidney failure in mice (Angeli et al. 2014).
High dose of acetaminophen frequently cause acute liver failure. Fer-1 can inhibit acetaminophen induced ferroptotic cell death (Lorincz et al. 2015). Ischemia/reperfusion-induced liver injury can be prevented in mice by liproxstatin-1 (Angeli et al. 2014). Prevention of glutaminolysis and ferroptosis by compound 968, DFO or Fer-1 inhibits ischemia/reperfusion-induced heart injury ex vivo (Gao et al. 2015).
9 Conclusion and Prospective
Ferroptosis by erastin and RSL3 is an iron-dependent lipid peroxidation induced non-apoptotic cell death in RAS-mutated cancer cells. Erastin inhibits cystine transporter activity and induces the decrease of glutathione and GPx4 activity, resulting in iron or 15-LOX dependent lipid peroxidation induced cell death. 15-LOX is one of the candidates for initial lipid peroxidation in ferroptosis. Fenton reaction by ferrous iron enhances the propagation of phospholipid oxidation and degradation of membrane lipid. Ferroptosis inducer is important for therapy of cancer. Identification of the downstream signaling pathway or executors of 15-LOX independent lipid peroxidation in ferroptosis remained to be solved.
GPx4 could directly reduce phospholipid hydroperoxide in specific organelle and regulate several signal transductions by analysis of GPx4 overexpressing cells.
Overexpression of mGPx4 inhibited apoptosis induced by mitochondrial death pathway. Overexpression of cGPx4 also suppressed the iron-dependent lipid peroxidation in membrane induced by erastin and RSL3, resulting in inhibition of ferroptosis. Where and how lipid peroxidation in organelle such as ER, Golgi, and plasma membrane is generated in ferroptosis by erastin and RSL3 remained to be elucidated.
Indeed, GPx4 is a regulator of ferroptosis by erastin and RSL3. But cell death by GPx4 gene disruption progresses extremely slower than ferroptosis induced by erastin and RSL3. It may be possible that the mechanism of cell death is different between ferroptosis by erastin and RSL3 and GPx4 depleted cell death by GPx4 gene disruption. Differences of site and enzymes of phospholipid oxidation in organelle might show differences of its downstream signal transduction of cell death between ferroptosis by erastin and RSL3 and GPx4 depleted cell death.
The phenotype of tissue specific GPx4 KO mice and cells is recovered with treatment of vitamin E. These results demonstrated that imbalance between lipid oxidation system and lipid peroxidation suppression system such as GPx4 and vitamin E causes several diseases in mice and human.
Abbreviations
- GPx4:
-
Glutathione peroxidase
- mGPx4:
-
Mitochondrial GPx4
- cGPx4:
-
Non-mitochondrial GPx4
- nGPx4:
-
Nucleolar GPx4
- tBid:
-
Truncated Bid
- CL:
-
Cardiolipin
- TG:
-
Transgenic
- MEF:
-
Mouse embryonic fibroblast
- RSL:
-
Ras-Selective Lethal
- Tam:
-
Tamoxifen
- GSH:
-
Glutathione
- LOX:
-
Lipoxygenase
- DFO:
-
Deferoxamine
- Fer-1:
-
Ferrostatin-1
- LOX:
-
Lipoxygenase
- PC:
-
Phosphatidylcholine
- PE:
-
Phosphatidylethanolamine
- ACSL4:
-
Acyl-CoA synthetase long-chain family member 4
- AA:
-
Arachidonic acid
References
Angeli JPF, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, Basavarajappa D, Rådmark O, Kobayashi S, Seibt T, Beck H, Neff F, Esposito I, Wanke R, Förster H, Yefremova O, Heinrichmeyer M, Bornkamm GW, Geissler EK, Thomas SB, Stockwell BR, O’Donnell VB, Kagan VE, Schick JA, Conrad M (2014) Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16:1180–1191
Arai M, Imai H, Sumi D, Imanaka T, Takano T, Chiba N, Nakagawa Y (1996) Import into mitochondria of phospholipid hydroperoxide glutathione peroxidase requires a leader sequence. Biochem Biophys Res Commun 227:433–439
Arai M, Imai H, Koumura T, Yoshida M, Emoto K, Umeda M, Chiba N, Nakagawa Y (1999) Mitochondrial phospholipid hydroperoxide glutathione peroxidase plays a major role in preventing oxidative injury to cells. J Biol Chem 274:4924–4933
Bellelli R, Federico G, Matte’ A, Colecchia D, Iolascon A, Chiariello M, Santoro M, De Franceschi L, Carlomagno F (2016) NCOA4 deficiency impairs systemic iron homeostasis. Cell Rep 14:411–421
Canli Ö, Alankuş YB, Grootjans S, Vegi N, Hültner L, Hoppe PS, Schroeder T, Vandenabeele P, Bornkamm GW, Greten FR (2016) Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood 127:139–148
Carlson BA, Tobe R, Yefremova E, Tsuji PA, Hoffmann VJ, Schweizer U, Gladyshev VN, Hatfield DL, Conrad M (2016) Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol 9:22–31
Chen L, Li X, Liu L, Yu B, Xue Y, Liu Y (2015) Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-gamma-lysae function. Oncol Rep 33:1465–1474
Cho YY, Kang MJ, Sone H, Suzuki T, Abe M, Igarashi M, Tokunaga T, Ogawa S, Takei YA, Miyazawa T, Sasano H, Fujino T, Yamamoto TT (2001) Abnormal uterus with polycysts, accumulation of uterine prostaglandins, and reduced fertility in mice heterozygous for acyl-CoA synthetase 4 deficiency. Biochem Biophys Res Commun 284:993–997
Conrad M, Moreno SG, Sinowatz F, Ursini F, Kölle S, Roveri A, Brielmeier M, Wurst W, Maiorino M, Bornkamm GW (2005) The nuclear form of phospholipid hydroperoxide glutathione peroxidase is a protein thiol peroxidase contributing to sperm chromatin stability. Mol Cell Biol 25:7637–7644
Dächert J, Schoeneberger H, Rohde K, Fulda S (2016) RSL3 and erastin differentially regulate redox signaling to promote Smac mimetic-induced cell death. Oncotarget. doi:10.18632/oncotarget.11687. (Epub ahead of print)
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B III, Stockwell BR (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–1072
Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, Stockwell BR (2014) Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 23:e02523
Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, Stockwell BR (2015) Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol 10:1604–1609
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, Prokisch H, Trümbach D, Mao G, Qu F, Bayir H, Füllekrug J, Scheel CH, Wurst W, Schick JA, Kagan VE, Angeli JP, Conrad M (2016) ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. doi:10.1038/nchembio.2239. (Epub ahead of print)
Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, Menon S, Wang Z, Honda A, Pardee G, Cantwell J, Luu C, Cornella-Taracido I, Harrington E, Fekkes P, Lei H, Fang Q, Digan ME, Burdick D, Powers AF, Helliwell SB, D’Aquin S, Bastien J, Wang H, Wiederschain D, Kuerth J, Bergman P, Schwalb D, Thomas J, Ugwonali S, Harbinski F, Tallarico J, Wilson CJ, Myer VE, Porter JA, Bussiere DE, Finan PM, Labow MA, Mao X, Hamann LG, Manning BD, Valdez RA, Nicholson T, Schirle M, Knapp MS, Keaney EP, Murphy LO (2014) Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol 16:1069–1079
Fujii J, Imai H (2014) Redox reactions in mammalian spermatogenesis and the potential targets of reactive oxygen species under oxidative stress. Spermatogenesis 4:e979108
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X (2015) Glutaminolysis and transferrin regulate ferroptosis. Mol Cell 59:298–308
Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X (2016) Ferroptosis is an autophagic cell death process. Cell Res 26:1021–1032
Gout PW, Buckley AR, Simms CR, Bruchovsky N (2001) Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x −c cystine transporter: a new action for an old drug. Leukemia 215:1633–1640
Hashidate-Yoshida T, Harayama T, Hishikawa D, Morimoto R, Hamano F, Tokuoka SM, Eto M, Tamura-Nakano M, Yanobu-Takanashi R, Mukumoto Y, Kiyonari H, Okamura T, Kita Y, Shindou H, Shimizu T (2015) Fatty acid remodeling by LPCAT3 enriches arachidonate in phospholipid membranes and regulates triglyceride transport. Elife. 4. doi:10.7554/eLife.06328
Hauck AK, Bernlohr DA (2016) Oxidative stress and lipotoxicity. J Lipid Res 57:1976–1986
Henke N, Albrecht P, Bouchachia I, Ryazantseva M, Knoll K, Lewerenz J, Kaznacheyeva E, Maher P, Methner A (2013) The plasma membrane channel ORAI1 mediates detrimental calcium influx caused by endogenous oxidative stress. Cell Death Dis 4:e470
Hentze MW, Muckenthaler MU, Galy B, Camaschella C (2010) Two to tango: regulation of mammalian iron metabolism. Cell 142:24–38
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ III, Kang R, Tang D (2016) Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12:1425–1428
Imai H (2010) New strategy of functional analysis of PHGPx knockout mice model using transgenic rescue method and Cre-LoxP system. J Clin Biochem Nutr 46:1–13
Imai H (2011) Disruption of homeostasis of suppression of phospholipid peroxidation in biomembrane case novel cell death in disease. Oleoscience 11:15–23
Imai H, Nakagawa Y (2003) Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med 34:145–169
Imai H, Sumi D, Hanamoto A, Arai M, Sugiyama A, Nakagawa Y (1995) Molecular cloning and functional expression of a cDNA for rat phospholipid hydroperoxide glutathione peroxidase: 3′-untranslated region of the gene is necessary for functional expression. J Biochem 118:1061–1067
Imai H, Sumi D, Sakamoto H, Hanamoto A, Arai M, Chiba N, Nakagawa Y (1996) Overexpression of phospholipid hydroperoxide glutathione peroxidase suppressed cell death due to oxidative damage in rat basophile leukemia cells (RBL-2H3). Biochem Biophys Res Commun 222:432–438
Imai H, Narashima K, Arai M, Sakamoto H, Chiba N, Nakagawa Y (1998) Suppression of leukotriene formation in RBL-2H3 cells that overexpressed phospholipid hydroperoxide glutathione peroxidase. J Biol Chem 273:1990–1997
Imai H, Suzuki K, Ishizaka K, Ichinose S, Oshima H, Okayasu I, Emoto K, Umeda M, Nakagawa Y (2001) Failure of the expression of phospholipid hydroperoxide glutathione peroxidase in the spermatozoa of human infertile males. Biol Reprod 64:674–683
Imai H, Hirao F, Sakamoto T, Sekine K, Mizukura Y, Saito M, Kitamoto T, Hayasaka M, Hanaoka K, Nakagawa Y (2003a) Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem Biophys Res Commun 305:278–286
Imai H, Koumura T, Nakajima R, Nomura K, Nakagawa Y (2003b) Protection from inactivation of the adenine nucleotide translocator during hypoglycaemia-induced apoptosis by mitochondrial phospholipid hydroperoxide glutathione peroxidase. Biochem J 371:799–809
Imai H, Saito M, Kirai N, Hasegawa J, Konishi K, Hattori H, Nishimura M, Naito S, Nakagawa Y (2006) Identification of the positive regulatory and distinct core regions of promoters, and transcriptional regulation in three types of mouse phospholipid hydroperoxide glutathione peroxidase. J Biochem 140:573–590
Imai H, Hakkaku N, Iwamoto R, Suzuki J, Suzuki T, Tajima Y, Konishi K, Minami S, Ichinose S, Ishizaka K, Shioda S, Arata S, Nishimura M, Naito S, Nakagawa Y (2009) Depletion of selenoprotein GPx4 in spermatocytes causes male infertility in mice. J Biol Chem 284:32522–32532
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W (2015) Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520:57–62
Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG (2005) Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 1:223–232
Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, Kapralov AA, Amoscato AA, Jiang J, Anthonymuthu T, Mohammadyani D, Yang Q, Proneth B, Klein-Seetharaman J, Watkins S, Bahar I, Greenberger J, Mallampalli RK, Stockwell BR, Tyurina YY, Conrad M, Bayır H (2016) Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. doi:10.1038/nchembio.2238. (Epub ahead of print)
Kang Y, Tiziani S, Park G, Kaul M, Paternostro G (2014) Cellular protection using Flt3 and PI3 Kα inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nat Commun 5:3672
Korytowski W, Basova LV, Pilat A, Kernstock RM, Girotti AW (2011) Permeabilization of the mitochondrial outer membrane by Bax/truncated Bid (tBid) proteins as sensitized by cardiolipin hydroperoxide translocation: mechanistic implications for the intrinsic pathway of oxidative apoptosis. J Biol Chem 286:26334–26343
Kwon MY, Park E, Lee SJ, Chung SW (2015) Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 6:24393–24403
Lachaier E, Louandre C, Godin C, Saidak Z, Baert M, Diouf M, Chauffert B, Galmiche A (2014) Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res 34:6417–6422
Li XX, Tsoi B, Li YF, Kurihara H, He RR (2015) Cardiolipin and its different properties in mitophagy and apoptosis. J Histochem Cytochem 63:301–311
Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz PS, Weinlich R, Vanden Berghe T, Vandenabeele P, Pasparakis M, Bleich M, Weinberg JM, Reichel CA, Bräsen JH, Kunzendorf U, Anders HJ, Stockwell BR, Green DR, Krautwald S (2014) Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A 111:16836–16841
Liu Y, Wang W, Li Y, Xiao Y, Cheng J, Jia J (2015) The 5-lipoxygenase inhibitor zileuton confers neuroprotection against glutamate oxidative damage by inhibiting ferroptosis. Biol Pharm Bull 38:1234–1239
Lorincz T, Jemnitz K, Kardon T, Mandl J, Szarka A (2015) Ferroptosis in involved in acetaminophen induced cell death. Pathol Oncol Res 21:1115–1121
Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Mazière JC, Chauffert B, Galmiche A (2013) Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer 133:1732–1742
Maguire JJ, Tyurina YY, Mohammadyani D, Kapralov AA, Anthonymuthu TS, Qu F, Amoscato AA, Sparvero LJ, Tyurin VA, Planas-Iglesias J, He RR, Klein-Seetharaman J, Bayır H, Kagan VE (2017) Known unknowns of cardiolipin signaling: the best is yet to come. Biochim Biophys Acta 1862:8–24
Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC (2014) Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509:105–109
Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M (2015) T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med 212:555–568
Nakamura T, Imai H, Tsunashima N, Nakagawa Y (2003) Molecular cloning and functional expression of nucleolar phospholipid hydroperoxide glutathione peroxidase in mammalian cells. Biochem Biophys Res Commun 311:139–148
Nomura K, Imai H, Koumura T, Arai M, Nakagawa Y (1999) Mitochondrial phospholipid hydroperoxide glutathione peroxidase suppresses apoptosis mediated by a mitochondrial death pathway. J Biol Chem 274:29294–29302
Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y (2000) Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J 351:183–193
Ou Y, Wang SJ, Li D, Chu B, Gu W (2016) Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A 113:E6806–E6812
Roggia MF, Imai H, Shiraya T, Noda Y, Ueta T (2014) Protective role of glutathione peroxidase 4 in laser-induced choroidal neovascularization in mice. PLoS ONE 9:e98864
Saito Y, Shichiri M, Hamajima T, Ishida N, Mita Y, Nakao S, Hagihara Y, Yoshida Y, Takahashi K, Niki E, Noguchi N (2015) Enhancement of lipid peroxidation and its amelioration by vitamin E in a subject with mutations in the SBP2 gene. J Lipid Res 56:2172–2182
Sakai O, Uchida T, Roggia MF, Imai H, Ueta T, Amano S (2015a) Role of glutathione peroxidase 4 in glutamate-induced oxytosis in the retina. PLoS ONE 10:e0130467
Sakai O, Uchida T, Imai H, Ueta T, Amano S (2015b) Role of glutathione peroxidase 4 in conjunctival epithelial cells. Invest Ophthalmol Vis Sci 56:538–543
Sakai O, Uchida T, Imai H, Ueta T (2016a) Glutathione peroxidae 4 plays an important role in oxidative homeostasis and wound repair in corneal epithelial cells. FEBS Open Bio 6:1238–1247
Sakai O, Yasuzawa T, Sumikawa Y, Ueta T, Imai H, Sawabe A, Ueshima S (2016b) Role of GPx4 in human vascular endothelial cells, and the compensatory activity of brown rice on GPx4 ablation condition. Pathophysiology S0928–4680(16):30068–30072
Sakamoto H, Imai H, Nakagawa Y (2000) Involvement of phospholipid hydroperoxide glutathione peroxidase in the modulation of prostaglandin D2 synthesis. J Biol Chem 275:40028–40035
Sakamoto H, Tosaki T, Nakagawa Y (2002) Overexpression of phospholipid hydroperoxide glutathione peroxidase modulates acetyl-CoA, 1-O-alkyl-2-lyso-sn-glycero-3-phosphocholine acetyltransferase activity. J Biol Chem 277:50431–50438
Sato H, Tamba M, Ishii T, Bannai S (1999) Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem 274:11455–11458
Schneider M, Förster H, Boersma A, Seiler A, Wehnes H, Sinowatz F, Neumüller C, Deutsch MJ, Walch A, Hrabé de Angelis M, Wurst W, Ursini F, Roveri A, Maleszewski M, Maiorino M, Conrad M (2009) Mitochondrial glutathione peroxidase 4 disruption causes male infertility. FASEB J. 23:3233–3242
Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Rådmark O, Wurst W, Bornkamm GW, Schweizer U, Conrad M (2008) Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 8:237–248
Sengupta A, Lichti UF, Carlson BA, Cataisson C, Ryscavage AO, Mikulec C, Conrad M, Fischer SM, Hatfield DL, Yuspa SH (2013) Targeted disruption of glutathione peroxidase 4 in mouse skin epithelial cells impairs postnatal hair follicle morphogenesis that is partially rescued through inhibition of COX-2. J Invest Dermatol 133:1731–1741
Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, Stockwell BR (2016) Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol 12:497–503
Shindou H, Shimizu T (2009) Acyl-CoA: lysophospholipid acyltransferases. J Biol Chem 284:1–5
Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, Stockwell BR (2014) Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 136:4551–4556
Smith AC, Mears AJ, Bunker R, Ahmed A, MacKenzie M, Schwartzentruber JA, Beaulieu CL, Ferretti F, FORGE Canada Consortium, Majewski J, Bulman DE, Celik FC, Boycott KM, Graham GE (2014) Mutations in the enzyme glutathione peroxidase 4 cause Sedaghatian-type spondylometaphyseal dysplasia. J Med Genet 51:470–474
Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, Wang H, Cao L, Tang D (2015) HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 34:5617–5625
Tan S, Schubert D, Maher P (2001) Oxytosis: a novel form of programmed cell death. Curr Top Med Chem 1:497–506
Tobaben S, Grohm J, Seiler A, Conrad M, Plesnila N, Culmsee C (2011) Bid-mediated mitochondrial damage is a key mechanism in glutamate-induced oxidative stress and AIF-dependent cell death in immortalized HT-22 hippocampal neurons. Cell Death Differ 18:282–292
Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, Suzuki T, Mori M, Yoshimoto Y, Takeuchi T, Yamada K (2016) An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J 473:769–777
Uchida T, Sakai O, Imai H, Ueta T (2016) Role of glutathione peroxidase 4 in corneal endothelial cells. Curr Eye Res 15:1–6
Ueta T, Inoue T, Furukawa T, Tamaki Y, Nakagawa Y, Imai H, Yanagi Y (2012) Glutathione peroxidase 4 is required for maturation of photoreceptor cells. J Biol Chem 287:7675–7682
Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, Gu W (2016) 2016 Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep 17(2):366–373
Wortmann M, Schneider M, Pircher J, Hellfritsch J, Aichler M, Vegi N, Kölle P, Kuhlencordt P, Walch A, Pohl U, Bornkamm GW, Conrad M, Beck H (2013) Combined deficiency in glutathione peroxidase 4 and vitamin E causes multiorgan thrombus formation and early death in mice. Circ Res 113:408–417
Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, Smith R, Lessnick SL, Sahasrabudhe S, Stockwell BR (2007) RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447:864–868
Yamaguchi H, Hsu JL, Chen CT, Wang YN, Hsu MC, Chang SS, Du Y, Ko HW, Herbst R, Hung MC (2013) Caspase-independent cell death is involved in the negative effect of EGF receptor inhibitors on cisplatin in non-small cell lung cancer cells. Clin Cancer Res 19:845–854
Yan S, Yang XF, Liu HL, Fu N, Ouyang Y, Qing K (2015) Long-chain acyl-CoA synthetase in fatty acid metabolism involved in liver and other diseases: an update. World J Gastroenterol 21:3492–3498
Yang WS, Stockwell BR (2008) Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 15:234–245
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR (2014) Regulation of ferroptotic cancer cell death by GPX4. Cell 156:317–331
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR (2016) Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113:E4966–E4975
Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, Zeh HJ, Kang R, Tang D (2015) The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol 2:e1054549
Yuan H, Li X, Zhang X, Kang R, Tang D (2016) Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun 478:1338–1343
Acknowledgements
We thank H. Nakano and S. Nagata for helpful comments on the manuscript. We also thank members of Department of Hygienic Chemistry, School of Pharmaceutical Sciences, Kitasato University for helpful discussion. This work was supported in part by Grants-in-Aid from Scientific Research (C) (26460075) from JSPS KAKENHI and Scientific Research on Innovative Areas (15H01386 and 16H01367) from a MEXT (Ministry of Education, Culture, Sports, Science and Technology), Japan, and research grants from Iijima Tojyuro Memorial Food Science Foundation and Kitasato University Research Grant for Young Researchers.
Competing Interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing AG
About this chapter
Cite this chapter
Imai, H., Matsuoka, M., Kumagai, T., Sakamoto, T., Koumura, T. (2016). Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. In: Nagata, S., Nakano, H. (eds) Apoptotic and Non-apoptotic Cell Death. Current Topics in Microbiology and Immunology, vol 403. Springer, Cham. https://doi.org/10.1007/82_2016_508
Download citation
DOI: https://doi.org/10.1007/82_2016_508
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-23912-5
Online ISBN: 978-3-319-23913-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)