
Abstract
Prostate apoptosis response-4 (Par-4), an anticancer protein that interacts with cell surface receptor GRP78, can selectively suppress proliferation and induce apoptosis of cancer cells. The core domain of Par-4 (aa 137–195), designated as SAC, is sufficient to inhibit tumor growth and metastasis without harming normal tissues and organs. Nevertheless, the anticancer effects of SAC have not been determined in ovarian cancer cells. Here, we developed a novel method for producing native SAC in Escherichia coli using a small ubiquitin-related modifier (SUMO) fusion system. This fusion system not only greatly improved the solubility of target protein but also enhanced the expression level of SUMO-SAC. After purified by Ni-NTA affinity chromatography, SUMO tag was cleaved from SUMO-SAC fusion protein using SUMO protease to obtain recombinant SAC. Furthermore, we simplified the purification process by combining the SUMO-SAC purification and SUMO tag cleavage into one step. Finally, the purity of recombinant SAC reached as high as 95% and the yield was 25 mg/L. Our results demonstrated that recombinant SAC strongly inhibited proliferation and induced apoptosis in ovarian cancer cells SKOV-3. Immunofluorescence analysis and competitive binding reaction showed that recombinant SAC could specifically induce apoptosis of SKOV-3 cells through combination with cell surface receptor, GRP78. Therefore, we have developed an effective strategy for expressing bioactive SAC in prokaryotic cells, which supports the application of SAC in ovarian cancer therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Prostate apoptosis response-4 (Par-4), a leucine zipper domain protein, is a tumor suppressor originally identified from rat prostate cancer cells and was later found to be universally expressed among various tissue types [1]. Previous researches have confirmed that the enforced Par-4 expression can suppress tumor growth and induce apoptosis specifically in various tumor cell types of diverse origins, without any cytotoxicity in normal human cells [2]. Serial deletion analyses of the Par-4 sequence have identified the fundamental unit of Par-4 that retained its anticancer activity, designated as SAC, as the core domain comprising 59 amino acids [3]. The SAC domain allows Par-4 to enter the nucleus, activate caspases, inhibit the activity of NF-κB, and selectively kill cancer cells [4], which make it a promising candidate drug for cancer therapy.
Recent studies have identified that the secreted forms of Par-4 and SAC from normal or malignant cancer cells selectively prevent the proliferation of tumor cells [5, 6]. These secreted proteins induce apoptosis of cancer cells by triggering the endoplasmic reticulum (ER) stress-mediated signal transduction pathway through interaction with the cell surface receptor, GRP78 [7,8,9]. Nevertheless, the anticancer effect of SAC has not been determined in ovarian cancer cells. Therefore, recombinant expression of SAC was conducted to facilitate its large-scale production as well as for further studies into its unique properties.
The Escherichia coli expression system is robust, convenient, and inexpensive for protein expression [10, 11]. This prokaryotic system can produce large amounts of protein at a low cost in a short period. However, protein misfolding occurs often, thereby yielding nonfunctional proteins, which are expressed in the form of inclusion bodies [12,13,14,15,16]. These inclusion bodies provide an easy way to isolate and purify the recombinant protein, but in vitro refolding of inactive protein remains a challenge because it is time consuming and inefficient [17].
Therefore, several trials have been performed to improve the solubility of recombinant proteins in prokaryotic expression system. The first method aimed to decrease the protein production rate by reducing the culture temperature and the inducer concentration, but this generally results in reduced protein productivity [18]. Another widely used strategy is to attach a fusion partner, such as glutathione S-transferase (GST), maltose-binding protein (MBP), thioredoxin (Trx), or small ubiquitin-related modifier (SUMO) to the recombinant protein [19,20,21,22,23]. This method has been shown to enhance the expression levels and protein solubility in prokaryotic expression system. Furthermore, affinity tags are often used to simplify the purification of target protein.
Recently, SUMO has emerged as an effective biotechnological tool that enhances the soluble expression of recombinant proteins. It has the great advantage of decreasing proteolytic degradation and simplifying purification owing to its inherent chaperone properties [21, 24]. When attached to the N-terminal of a protein, the fusion protein can be specifically cleaved by SUMO protease on the basis of the tertiary structure of the SUMO tag. This allows the protein of interest to be isolated along with its native N-terminus [25, 26]. Inserting a His-tag into N-terminus of SUMO can simplify purification of the target protein. Therefore, these properties make SUMO an effective fusion strategy for improving heterologous expression in prokaryotic expression system [24, 27].
In this study, we attached the SUMO tag to the N-terminus of SAC to enhance the expression level and protein solubility in E. coli. We simplified the purification procedure and improved the SAC yield via one-step purification. We then tested the recombinant SAC protein for its ability to suppress growth and induce apoptosis in ovarian carcinoma SKOV-3 cell line for the first time. The selectivity and cytotoxicity of SAC demonstrate its potential as a novel candidate for the treatment of ovarian cancer.
Materials and Methods
Materials
Host strains BL21(DE3) were obtained from Tiangen Biotech (Beijing, China). Taq DNA polymerase and restriction endonucleases were purchased from Takara Biotech Co. Ltd. (Shiga, Japan). The pET28a vector was stored in our laboratory. The PCR Purification Kit, Gel Extraction Kit, and Plasmid Mini Kit were from Axygen (Hangzhou, China). The His6-tagged SUMO protease was purchased from Lifesensors (Springfield, NJ, USA). Ni-NTA resin was obtained from Merck (Germany). The human ovarian carcinoma (SKOV-3) cells and the human embryonic kidney 293 (HEK-293) cells were purchased from American Type Culture Collection (Manassas, VA, USA). The purified GRP78 protein was from Sigma (St. Louis, MO, USA). Monoclonal antibodies for Par-4 and GRP78 were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Construction of the pET28a/SUMO-SAC Expression Plasmid
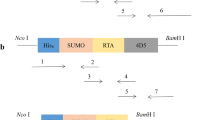
Primers P1 and P2 were used to amplify the SUMO gene, and primers P3 and P4 were used to amplify the SAC gene (GenBank No. AB108448) (Table 1). To obtain the native SAC protein, the gene encoding SUMO and SAC were fused by means of overlapping PCR via primers P1 and P4 to obtain the target gene SUMO-SAC. The gene encoding SUMO-SAC was inserted into the pET28a vector (Fig. 1a). The correct insertion of sequence was confirmed through gene sequencing.

Schematic illustration of the pET28a/SUMO-SAC vector construction and preparation of recombinant SUMO-SAC. a Schematic illustration of expression vector. To obtain the native SAC protein, the gene encoding SUMO and SAC were fused by means of overlapping PCR to obtain the target gene SUMO-SAC. The gene encoding SUMO-SAC was inserted into the pET28a vector. b The solubility and expression level of SUMO-SAC was analyzed on 15% SDS-PAGE. M, molecular weight marker; lanes 1–3, total cellular lysate, supernatant and precipitation from induced E. coli containing pET28a/SUMO-SAC, respectively; lanes 4–6, purified SUMO-SAC eluted by 100, 200, and 500 mM imidazole, respectively
Protein Expression and Purification
The competent cells were transformed with the vector pET28a/SUMO-SAC described above. Single-colony transformant was cultivated in Luria-Bertani medium containing 50 μg/mL kanamycin and incubated at 37 °C and vigorously shaken (200 rpm) to OD600 reached 0.6. Next, 0.1 mM IPTG was added for protein expression at 30 °C for 8 h. The cells were centrifugated, resuspended in buffer A (40 mM PB, pH 7.4, 100 mM NaCl), and disrupted by high-pressure homogenization. The supernatant, which comprised the soluble protein, and the precipitation were checked by 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and the expression level of target protein was monitored with the aid of densitometer scanning.
The samples were loaded onto the Ni-nitrilotriacetic acid resin that is pre-equilibrated with buffer A. The column was washed by 6 column volumes (CVs) of equilibration buffer. The target protein with a His6 tag at the N-terminus was eluted with elution buffer containing different concentrations of imidazole, and the purification process was monitored by the AKTA Explorer. The concentration of SUMO-SAC was quantified by the Bradford protein assay.
Release of SAC by SUMO Protease Cleavage
The purified SUMO-SAC was dialyzed against buffer A to remove the imidazole, and then, the fusion protein was mixed with SUMO protease to release the N-terminal fusion tag. One milligram of SUMO-SAC was incubated with 0.5 μg SUMO protease for 1 h at 37 °C. After cleavage, the mixture was reloaded on the Ni-NTA column to remove the SUMO tag, SUMO-SAC, and SUMO protease, and the target protein SAC was pooled in the flow-through fraction. Western blot was performed to identify the target protein SAC.
Western Blot Analysis
Equal amounts of the samples (10 μg per lane) were loaded onto 15% polyacrylamides gels before transferring onto a nitrocellulose membrane. The membrane was incubated with the Par-4 monoclonal antibody and then incubated with a corresponding secondary antibody. Blots were scanned by enhanced chemiluminescence system.
One-Step Purification of Recombinant SAC
The soluble fraction of the cell lysate containing target fusion protein was loaded onto the nickel ion affinity chromatograph resin. The impure protein was eluted with 6 CVs of equilibration buffer. Then, the diluted SUMO protease (1 μg/mL) was added to aforementioned Ni-NTA column and incubated for 1 h at 37 °C for the removal of SUMO tag. Subsequently, the untagged SAC released into the supernatant was collected from the flow-through fraction.
Cell Proliferation Assay
SKOV-3 cells and HEK-293 cells were inoculated in 96-well plate (7 × 103 cells per well) in RPMI 1640 medium adding 10% serum and then incubated for 24 h at 37 °C with 5% CO2. The SAC protein was diluted into different concentrations. After incubation for 72 h, 10 μL of MTT (5 mg/mL) was added to each well and incubated at 37 °C for 4 h, after which the supernatant was removed. Then, 100 μL DMSO was added and the plate was shook to thoroughly dissolve the formazan. Finally, absorbance was measured at 570 nm by microplate spectrophotometer. The resulting absorbance value is directly proportional to the number of living cells.
Colony Formation
SKOV-3 and HEK-293 cells were inoculated in 6-well plates. After the 24-h incubation, different concentrations of recombinant SAC were added to each well for 2 days. Then, the supernatant was discarded and updated by fresh culture medium every 3 days. Two weeks later, cells were fixed with 4% paraformaldehyde and stained with Giemsa. Colonies > 50 cells were enumerated.
Flow Cytometry Assay
SKOV-3 and HEK-293 cells were inoculated onto 6-well plates and incubated overnight. The purified recombinant SAC was added to each dish at different concentrations. The cells were harvested after 24 h and washed twice with PBS. One hundred microliters binding buffer and 10 μL FITC-labeled Annexin-V were added for 30 min at room temperature. Then, 5 μL propidium iodide was added and incubated for 10 min; subsequently, 300 μL of PBS was added. Flow cytometer (BD FACSCalibur™) was performed immediately to quantify apoptosis.
DAPI Staining
SKOV-3 and HEK-293 cells were inoculated to 6-well plates (5 × 105 cells per well). The medium was discarded the next day and updated by fresh medium containing 200 nM SAC. Two days later, cells were washed three times and fixed with 4% paraformaldehyde and then subjected to staining using DAPI. Visualization of apoptotic cells was observed using confocal microscope.
Immunofluorescence Assay
Immunofluorescence assay was performed to observe the selectivity of SAC on SKOV-3 and HEK-293 cells. Dialysis was applied to change the buffer system of purified SAC using sodium carbonate buffer (pH 9.0). Next, 15 μg of FITC was incubated with 1 mg of SAC at 4 °C overnight in the dark. Then, 50 mM ammonium chloride was used to stop the reaction. Finally, the mixture was dialyzed against PBS buffer to thoroughly remove the free, unbound FITC [28]. Cells were inoculated at 35-mm plates (3 × 105 cells per dish) and incubated with FITC-labeled SAC (5 μM) for 6 h. The nucleus was pre-stained with Hoechst 33342. Then, confocal microscopy was used to visualize the immunofluorescence.
Cell Surface GRP78 Assay
FACS assay was performed to detect cell surface GRP78. SKOV-3 and HEK-293 cells were incubated with purified SAC for 12 h. The cells were not fixed and then subjected to FACS assay using GRP78 primary antibody and R-phycoerythrin-labeled secondary antibody.
Competition of SAC Cytotoxicity
SKOV-3 cells were treated with 200 nM recombinant SAC alone or co-incubated with different concentrations of GRP78 at 37 °C for 24 h. Flow cytometry analysis was performed to measure the apoptosis of SKOV-3 cells [29].
Statistical Analysis
The results were analyzed using the Student’s t test. Data were presented as mean ± standard error (mean ± SE) based on at least three experiments. Statistically significant was considered at P < 0.05.
Results
Construction and Expression of SUMO-SAC
To achieve recombinant expression of SAC in E. coli, several expression plasmids have been constructed. However, overexpression experiments led to a low expression level of SAC and difficulties in purification. Therefore, we cloned the cDNA of SUMO-SAC into pET28a to enhance the SAC expression level and facilitate its purification (Fig. 1a). The host strains that transformed with pET28a/SUMO-SAC were induced by IPTG. The solubility and expression level of the recombinant protein were determined using 15% SDS-PAGE. As shown in Fig. 1b, SUMO-SAC was mostly expressed in the form of soluble protein and its expression level reached 30% of the total supernatant protein.
Purification of Fusion Protein and Release of SAC
On account of the His6-tag being located at the N-terminus of the SUMO tag, Ni-NTA resin was chosen to purify the fusion protein. The impurities were eluted in buffer A, and the target protein was eluted in equilibration buffer containing 100, 200, and 500 mM imidazole, respectively (Fig. 1b). Although the expected molecular weight of SUMO-SAC was 18 kDa, its apparent molecular weight in 15% SDS-PAGE was approximately 23 kDa.
SUMO protease recognizes the protease recognition site as well as the three-dimensional structure of SUMO protein [27, 29], which can be used to verify spatial structure. After many attempts, the optimal conditions for cleaving SUMO-SAC were determined to be 0.5 μg SUMO protease for each milligram of fusion protein incubated for 1 h at 37 °C. As shown in Fig. 2a, the purified SUMO-SAC was incubated with SUMO protease to release the SUMO tag from the fusion protein. This incubation resulted in cleavage of more than 95% of the target protein. When the cleaved mixture was loaded onto the Ni-NTA resin, the untagged SAC remained in the flow-through solution, thereby yielding a single band without any contaminant on the SDS-PAGE gel (Fig. 2a, lane 5). Western blot assay indicated that recombinant SAC reacted specifically with a human Par-4 monoclonal antibody (Fig. 2b).

Purification and identification of recombinant SAC. a SDS-PAGE analysis of SUMO-SAC cleaved by SUMO protease and purified SAC. Lane 1, purified SUMO-SAC; lane 2, purified SUMO-SAC cleaved by SUMO protease; lane 3, released SUMO tag eluted by 500 mM imidazole; lanes 4 and 5, recombinant SAC after separation from SUMO tag. b Western blot analysis of SAC. Lane 1, lysate from uninduced transformants containing pET28a/SUMO-SAC, as negative control; lane 2, purified SAC
One-Step Purification Assay
To simplify the purification procedure, we attempted to purify and cleave the fusion protein in one step. After binding the soluble fraction of the lysate to the Ni-NTA resin, the column was incubated with buffer A containing SUMO protease. After incubation, a new band of 7 kDa, corresponding to SAC, was separated from the Ni-NTA resin and released into the solution (Fig. 3, lane 4). Finally, the purity of recombinant SAC reached as high as 95%. The SUMO tag and SUMO protease containing His6-tag were eluted using imidazole (Fig. 3, lane 5). The typical yield of purified SAC reached 25 mg/L of culture medium.

One-step purification of recombinant SAC. Lanes 1–2, total cellular lysate and supernatant from induced E. coli containing pET28a/SUMO-SAC; lane 3, the flow-through fraction after nickel affinity chromatography; lane 4, the purified recombinant SAC; lane 5, released SUMO tag eluted by 500 mM imidazole
The Activity of Recombinant SAC in Ovarian Cancer
Previous studies demonstrated that the secreted form of SAC from malignant cancer cells selectively induced apoptosis in most cancer cells without any cytotoxicity in normal human cells [30,31,32]. However, the functional relevance of recombinant SAC in ovarian cancer cells has not been determined. In order to explore the antitumor activity of purified SAC in human ovarian cancer cells SKOV-3 and normal cells HEK-293, MTT assay was used to measure the proliferation inhibition. The results indicated that SAC selectively inhibits the proliferation of SKOV-3 cells in a concentration-dependent manner, but without harm to normal cells, HEK-293 (Fig. 4a). The 50% inhibitory concentration of SAC to SKOV-3 cells was 83 nM. Furthermore, we also detected the effect of SAC on colony formation of SKOV-3 and HEK-293 cells. Consistent with the results of MTT assay, fewer colonies were observed in SAC-treated SKOV-3 cells, while no obvious inhibition of colony formation was observed in SAC-treated normal cells HEK-293 (Fig. 4b).

Recombinant SAC inhibits proliferation of cancer cells SKOV-3. a MTT assay. SAC inhibits the growth of ovarian cancer cells SKOV-3 in a dose-dependent manner, whereas without cytotoxicity in normal human cells HEK-293. b Colony formation effects of SAC on SKOV-3 cells and HEK-293 cells. Data are presented as mean ± SE (n = 3). *P < 0.05; **P < 0.01, in comparison to the group treated with PBS
In addition to proliferation inhibition, the antitumor effect of SAC also arises from apoptosis induction [6]. Flow cytometric assays were performed to determine the apoptotic activity of recombinant SAC on cancer cells. SKOV-3 and HEK-293 cells were incubated with recombinant SAC for 24 h (Fig. 5a). The results showed that the degree of apoptosis in SKOV-3 cells upon SAC treatment increased in a concentration-dependent manner whereas no apoptotic activity was observed in normal HEK-293 cells.

SAC induces apoptosis of ovarian cancer cells SKOV-3. a Flow cytometry assay to detect apoptosis. The apoptosis degree of SKOV-3 cells upon SAC treatment increased in a concentration-dependent manner whereas no apoptotic activity was observed in normal HEK-293 cells. Data are presented as mean ± SE (n = 3). *P < 0.05; **P < 0.01, in comparison to the group treated with PBS. b DAPI staining. After incubation with 200 nM SAC for 2 days, the nucleus of ovarian cancer cells SKOV-3 showed obvious membrane shrinkage, chromatin condensation, and fragmentation, which was the typical apoptotic morphology
To further define the cancer cell apoptosis morphologically, SKOV-3 cells were stained with DAPI. When SKOV-3 cells were incubated with 200 nM SAC for 2 days, the cells exhibited typical apoptotic morphology in the nuclei. However, no obvious change was observed on HEK-293 cell nucleus (Fig. 5b). These results, combined with those shown in Fig. 2a, suggest that purified recombinant SAC maintained its bioactivity after removal of the fusion partner.
SAC Selectively Binds to Cell Surface GRP78
SKOV-3 and HEK-293 cells were incubated with 5 μM FITC-labeled SAC for 6 h. Using confocal microscopy, we found that SAC was specifically localized on the surface of SKOV-3 cells, but no obvious fluorescence was detected in normal cells HEK-293 (Fig. 6a). Flow cytometry assay performed on unfixed SKOV-3 cells to detect cell surface proteins exhibited a noteworthy increase in GRP78 expression after treatment with SAC (Fig. 6b), indicating that the antitumor effect of SAC correlates with cell surface GRP78.

SAC selectively binds to cell surface GRP78. a Laser scanning confocal microscopy images of SKOV-3 and HEK-293 cells that were incubated with 5 μM FITC-labeled SAC at 37 °C for 6 h. The nucleus was pre-stained with Hoechst 33342. The confocal images were captured at × 1000 magnification. b Recombinant SAC increases GRP78 expression at cancer cell surface. SKOV-3 and HEK-293 cells were incubated with different doses of SAC for 12 h and then collected and subjected to flow cytometry analysis for GRP78. c Competitive binding analysis of the selectivity of SAC. The apoptotic effect of 200 nM SAC alone or co-incubated with different doses of GRP78 on ovarian cancer cells were analyzed by flow cytometry assay. Data are presented as mean ± SE (n = 3). *P < 0.05; **P < 0.01, in comparison to the group without GRP78
It has been reported that the secreted form of Par-4 from transformed cells selectively induces apoptosis in most tumor cells by interacting with the cell surface receptor, GRP78 [7,8,9]. We further explored whether the membrane-localized GRP78 is indispensable for the apoptosis of ovarian cancer SKOV-3 cells mediated by extracellular recombinant SAC. If recombinant GRP78 and SAC are both incubated with SKOV-3 cells, the recombinant GRP78 can compete with cell surface GRP78 to interact with SAC so that the apoptosis of SKOV-3 cells caused by SAC decreased. The results showed that increasing GRP78 concentration led to higher viability of SKOV-3 cells when the cells were treated with 200 nM SAC (Fig. 6c). When the concentration of GRP78 reached 8 μM, the apoptotic effect of SAC on SKOV-3 cells was almost completely inhibited.
Discussion
Previous studies have confirmed that SAC, the fundamental antitumor unit of Par-4 (aa137–195), is a specific anticancer gene in a wide variety of tumor cells [2]. Therefore, SAC is a promising agent for cancer therapy. Recently, a few attempts have been performed for SAC preparation in E. coli. Though SAC was soluble expressed with the TRX fusion tag, no native SAC was isolated from the fusion protein TRX-SAC [5, 6]. Thus, in this study, we described a prokaryotic expression system that can be used to prepare large quantities of native SAC, which exhibits strong anticancer activity.
In our initial trials for SAC expression, low production yield and difficulties in purification were the major problems that had to be overcome. In recent years, a growing number of recombinant proteins have been successfully prepared using the SUMO fusion system [21]. SUMO protein reportedly enhances both expression quantity and solubility of target protein when fused to its N-terminus due to its inherent chaperone properties. The structure of the SUMO tag contains a superficial hydrophilic surface and an interior hydrophobic fraction, which are similar to those found in amphipathic molecules and serves as a conventional molecular chaperone to keep folding intermediates long enough to form the correct steric conformation [24]. In our experiments, we cloned SAC in frame with SUMO and inserted it into pET-28a, then investigated the effect of SUMO tag fusion on the expression of SAC in E. coli. SDS-PAGE analysis indicated that SUMO-SAC was mostly expressed in soluble form.
Traditional protease cleavage routes are widely used to cleave the target protein from its fusion partner. These cleavages are typically inaccurate, inefficient, and leave behind a non-natural N-terminal of the target protein. In contrast, SUMO protease can specifically cleave SUMO tag at the Gly-Gly sequence between SUMO and target protein on the basis of the spatial structure of SUMO, and the target protein is then acquired with its native N-terminal without any vector-derived residues after cleavage [25]. In addition, fusing a His6-tag to the N-terminus of SUMO can facilitate purification of the target protein. Therefore, these properties make SUMO an effective fusion strategy for improving heterologous protein expression in E. coli [24, 27]. Our results from the present study determined that the optimal condition for cleaving 1 mg SUMO-SAC required 0.5 μg SUMO protease with incubation for 1 h at 37 °C. This resulted in more than 95% cleavage of the fusion protein.
In an attempt to simplify the purification process, we purified and cleaved SUMO-SAC using a one-step purification strategy. The SUMO protease was incubated with the protein-bound resin, allowing specific and controlled cleavage of the SUMO-SAC directly on the column, thereby releasing purified SAC into the solution. The SUMO tag can thus be removed and SAC purified in a single step. In the end, the purity of SAC reached 95% and the yield of purified recombinant SAC was 25 mg/L in flask fermentation.
Previous studies demonstrated that the secreted form of SAC from both normal and malignant cancer cells specifically exerts a cytotoxic effect in most tumor cells but not normal cells [33, 34]. Nevertheless, the cytotoxicity of SAC had not been determined in ovarian cancer cells. In the present study, an in vitro cytotoxicity analysis indicated that recombinant SAC could inhibit the proliferation and induce cell death in ovarian cancer SKOV-3 cells in a concentration-dependent manner. The 50% inhibitory concentration of SAC to SKOV-3 cells was 83 nM.
It has been reported that cell membrane-located GRP78 is the Par-4 receptor that exhibits cytotoxicity in cancer cells. When Par-4 or SAC binds to cell membrane-located GRP78, it has been shown to produce a robust ER stress loop involving overexpression and transfer of intracellular GRP78 to the cell membrane, in an intracellular Par-4-dependent manner [5, 35,36,37]. Thus, we explored whether the membrane-located GRP78 is dispensable for cytotoxicity mediated by recombinant SAC in ovarian cancer cells SKOV-3. Using confocal microscopy, we found that SAC was selectively localized at the surface of SKOV-3 cells, but nearly no fluorescence was detected in normal cells HEK-293. A flow cytometry assay performed on SKOV-3 cells indicated a significant increase in GRP78 expression on the cell surface. In addition, recombinant GRP78 and SAC were both incubated with ovarian cancer cells to determine the competitive binding effect between recombinant GRP78 and cell surface GRP78 for interaction with SAC in SKOV-3 cells. With increasing GRP78 concentration, the viability of SKOV-3 cells was increased even in the presence of recombinant SAC. The apoptotic effect of SAC on SKOV-3 cells was almost completely suppressed when GRP78 concentration reached 8 μM, indicating that SAC selectively binds to cell surface GRP78 and that the exogenous added GRP78 inhibits the activity of SAC on SKOV-3 cells. Taken together, these results suggest that cell surface GRP78 is essential for the cytotoxicity of recombinant SAC in ovarian cancer cells SKOV-3.
In summary, we were able to express soluble SAC in E. coli using a SUMO tag. The SUMO-SAC could be purified and specifically cleaved by SUMO protease to release native SAC directly on the Ni-NTA affinity column. Recombinant SAC suppressed proliferation and induced apoptosis of ovarian cancer cells SKOV-3 by interacting with cell surface receptor GRP78. Our results demonstrated that the SUMO fusion strategy is a promising candidate to produce and purify bioactive SAC protein. Further studies are required to verify the effectiveness of recombinant SAC in vivo.
References
Hebbar, N., Wang, C., & Rangnekar, V. M. (2012). Mechanisms of apoptosis by the tumor suppressor Par-4. Journal of Cellular Physiology, 227, 3715–3721.
El-Guendy, N., & Rangnekar, V. M. (2003). Apoptosis by Par-4 in cancer and neurodegenerative diseases. Experimental Cell Research, 283, 51–66.
El-Guendy, N., Zhao, Y., Gurumurthy, S., Burikhanov, R., & Rangnekar, V. M. (2003). Identification of a unique core domain of par-4 sufficient for selective apoptosis induction in cancer cells. Molecular and Cellular Biology, 23, 5516–5525.
Zhao, Y., & Rangnekar, V. M. (2008). Apoptosis and tumor resistance conferred by Par-4. Cancer Biology & Therapy, 7, 1867–1874.
Burikhanov, R., Zhao, Y., Goswami, A., Qiu, S., Schwarze, S. R., & Rangnekar, V. M. (2009). The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell, 138, 377–388.
Zhao, Y., Burikhanov, R., Brandon, J., Qiu, S., Shelton, B. J., Spear, B., Bondada, S., Bryson, S., & Rangnekar, V. M. (2011). Systemic Par-4 inhibits non-autochthonous tumor growth. Cancer Biology & Therapy, 12, 152–157.
Shrestha-Bhattarai, T., & Rangnekar, V. M. (2010). Cancer-selective apoptotic effects of extracellular and intracellular Par-4. Oncogene, 29, 3873–3880.
Sayers, T. J. (2011). Targeting the extrinsic apoptosis signaling pathway for cancer therapy. Cancer Immunology, Immunotherapy, 60, 1173–1180.
Chaudhry, P., Singh, M., Parent, S., & Asselin, E. (2012). Prostate apoptosis response 4 (Par-4), a novel substrate of caspase-3 during apoptosis activation. Molecular and Cellular Biology, 32, 826–839.
Braun, P., Hu, Y., Shen, B., Halleck, A., Koundinya, M., Harlow, E., & LaBaer, J. (2002). Proteome-scale purification of human proteins from bacteria. Proceedings of the National Academy of Sciences of the United States of America, 99, 2654–2659.
Zafar, A., Aftab, M. N., ud Din, Z., Aftab, S., Iqbal, I., & ul Haq, I. (2016). Cloning, purification and characterization of a highly thermostable amylase gene of Thermotoga petrophila into Escherichia coli. Applied Biochemistry and Biotechnology, 178, 831–848.
Baneyx, F. (1999). Recombinant protein expression in Escherichia coli. Current Opinion in Biotechnology, 10, 411–421.
Mahamad, P., Boonchird, C., & Panbangred, W. (2016). High level accumulation of soluble diphtheria toxin mutant (CRM197) with co-expression of chaperones in recombinant Escherichia coli. Applied Microbiology and Biotechnology, 100, 6319–6330.
Tian, H., Zhao, Y., Chen, N., Wu, M., Gong, W., Zheng, J., Fernig, D. G., Jungbauer, A., Wang, D., Li, X., & Jiang, C. (2016). High production in E. coli of biologically active recombinant human fibroblast growth factor 20 and its neuroprotective effects. Applied Microbiology and Biotechnology, 100, 3023–3034.
Rueda, F., Cano-Garrido, O., Mamat, U., Wilke, K., Seras-Franzoso, J., García-Fruitós, E., & Villaverde, A. (2014). Production of functional inclusion bodies in endotoxin-free Escherichia coli. Applied Microbiology and Biotechnology, 98, 9229–9238.
Gao, J., & Wang, H. (2015). Prokaryotic expression, refolding and purification of high-purity mouse Midkine in Escherichia coli. Applied Biochemistry and Biotechnology, 176, 454–466.
Kong, B., & Guo, G. L. (2011). Enhanced in vitro refolding of fibroblast growth factor 15 with the assistance of SUMO fusion partner. PloS One, 6, e20307.
Fan, J., Huang, L., Sun, J., Qiu, Y., Zhou, J., & Shen, Y. (2015). Strategy for linker selection to enhance refolding and bioactivity of VAS-TRAIL fusion protein based on inclusion body conformation and activity. Journal of Biotechnology, 209, 16–22.
Kapust, R. B., & Waugh, D. S. (1999). Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Science, 8, 1668–1674.
Li, Y. (2013). Production of human antimicrobial peptide LL-37 in Escherichia coli using a thioredoxin-SUMO dual fusion system. Protein Expression and Purification, 87, 72–78.
Zhang, M., Qiu, Z., Li, Y., Yang, Y., Zhang, Q., Xiang, Q., Su, Z., & Huang, Y. (2013). Construction and characterization of a recombinant human beta defensin 2 fusion protein targeting the epidermal growth factor receptor: in vitro study. Applied Microbiology and Biotechnology, 97, 3913–3923.
Dian, C., Eshaghi, S., Urbig, T., McSweeney, S., Heijbel, A., Salbert, G., & Birse, D. (2002). Strategies for the purification and on-column cleavage of glutathione-S-transferase fusion target proteins. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences, 769, 133–144.
Chaubey, N., & Ghosh, S. S. (2013). Molecular cloning, purification and functional implications of recombinant GST tagged hGMCSF cytokine. Applied Biochemistry and Biotechnology, 169, 1713–1726.
Ichikawa, Y., Kagawa, W., Saito, K., Chikashige, Y., Haraguchi, T., Hiraoka, Y., & Kurumizaka, H. (2013). Purification and characterization of the fission yeast telomere clustering factors, Bqt1 and Bqt2. Protein Expression and Purification, 88, 207–213.
Ye, T., Lin, Z., & Lei, H. (2008). High-level expression and characterization of an anti-VEGF165 single-chain variable fragment (scFv) by small ubiquitin-related modifier fusion in Escherichia coli. Applied Microbiology and Biotechnology, 81, 311–317.
Li, Y. (2013). Recombinant production of crab antimicrobial protein scygonadin expressed as thioredoxin and SUMO fusions in Escherichia coli. Applied Biochemistry and Biotechnology, 169, 1847–1857.
Upadhyay, S. K., Saurabh, S., Rai, P., Singh, R., Chandrashekar, K., Verma, P. C., Singh, P. K., & Tuli, R. (2010). SUMO fusion facilitates expression and purification of garlic leaf lectin but modifies some of its properties. Journal of Biotechnology, 146, 1–8.
Xu, R., Dong, Y., Wang, L., Tao, X., Sun, A., & Wei, D. (2014). TAT-RhoGDI2, a novel tumor metastasis suppressor fusion protein: expression, purification and functional evaluation. Applied Microbiology and Biotechnology, 98, 9633–9641.
Lv, X., Zhang, J., Xu, R., Dong, Y., Sun, A., Shen, Y., & Wei, D. (2016). Gigantoxin-4-4D5 scFv is a novel recombinant immunotoxin with specific toxicity against HER2/neu-positive ovarian carcinoma cells. Applied Microbiology and Biotechnology, 100, 6403–6413.
Lee, T. J., Jang, J. H., Noh, H. J., Park, E. J., Choi, K. S., & Kwon, T. K. (2010). Overexpression of Par-4 sensitizes TRAIL-induced apoptosis via inactivation of NF-κB and Akt signaling pathways in renal cancer cells. Journal of Cellular Biochemistry, 109, 885–895.
Lee, T. J., Lee, J. T., Kim, S. H., Choi, Y. H., Song, K. S., Park, J. W., & Kwon, T. K. (2008). Overexpression of Par-4 enhances thapsigargin-induced apoptosis via downregulation of XIAP and inactivation of Akt in human renal cancer cells. Journal of Cellular Biochemistry, 103, 358–368.
Jagtap, J. C., Dawood, P., Shah, R. D., Chandrika, G., Natesh, K., Shiras, A., Hegde, A. S., Ranade, D., & Shastry, P. (2014). Expression and regulation of prostate apoptosis response-4 (Par-4) in human glioma stem cells in drug-induced apoptosis. PloS One, 9, e88505.
Meynier, S., Kramer, M., Ribaux, P., Tille, J. C., Delie, F., Petignat, P., & Cohen, M. (2015). Role of PAR-4 in ovarian cancer. Oncotarget, 6, 22641–22652.
Kline, C. L., & Irby, R. B. (2011). The pro-apoptotic protein prostate apoptosis response protein-4 (Par-4) can be activated in colon cancer cells by treatment with Src inhibitor and 5-FU. Apoptosis, 16, 1285–1294.
Zhang, X. X., Li, H. D., Zhao, L., Song, H. J., Wang, G., Guo, Q. J., Luan, Z. D., & Su, R. J. (2013). The cell surface GRP78 facilitates the invasion of hepatocellular carcinoma cells. BioMed Research International, 2013, 917296.
Li, Z., Zhang, L., Zhao, Y., Li, H., Xiao, H., Fu, R., Zhao, C., Wu, H., & Li, Z. (2013). Cell-surface GRP78 facilitates colorectal cancer cell migration and invasion. The International Journal of Biochemistry & Cell Biology, 45, 987–994.
Irby, R. B., & Kline, C. L. (2013). Par-4 as a potential target for cancer therapy. Expert Opinion on Therapeutic Targets, 17, 77–87.
Funding
This work was funded by the National Natural science Foundation of China (No. 21646005/B060806) and China Postdoctoral Science Foundation funded project (No. 2016M601529).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Zhang, J., Sun, A., Dong, Y. et al. Recombinant Production and Characterization of SAC, the Core Domain of Par-4, by SUMO Fusion System. Appl Biochem Biotechnol 184, 1155–1167 (2018). https://doi.org/10.1007/s12010-017-2599-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-017-2599-9