
Abstract
In the present review, we briefly summarize the biotechnological applications of microbial β-xylosidases in the processing of agro-industrial residues into fuels and chemicals and report the importance of using immobilization techniques to study the enzyme. The advantages of utilizing genes that encode β-xylosidases are readily apparent in the bioconversion of abundant, inexpensive, and renewable resources into economically important products, such as xylitol and bioethanol. We highlight recent research characterizing fungal and bacterial β-xylosidases, including the use of classical biochemical methods such as purification, heterologous recombinant protein expression, and metagenomic approaches to discovery β-xylosidases, with focus on enzyme molecular and kinetic properties. In addition, we discuss the relevance of using experimental design optimization methodologies to increase the efficacy of these enzymes for use with residual biomass. Finally, we emphasize more extensively the advances in the regulatory mechanisms governing β-xylosidase gene expression and xylose metabolism in gram-negative and gram-positive bacteria and fungi. Unlike previous reviews, this revision covers recent research concerning the various features of bacterial and fungal β-xylosidases with a greater emphasis on their biochemical characteristics and how the genes that encode these enzymes can be better exploited to obtain products of biotechnological interest via the application of different technical approaches.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the plant kingdom, the cell wall plays an important role in supporting cell shape by presenting a rigid structure in which cellulose microfibrils are major architectural elements. These structures are formed from a complex matrix of hemicellulose, pectins, glycoproteins, and aromatic substances with waxy features. Lignocellulosic materials including wood, grass, and forest and agricultural residues are chemically composed of cellulose, hemicellulose, and lignin, with varying ratios of these components according to the plant species from which they originate [1]. The human population produces millions of tons of agro-industrial residues annually, and these are largely disposed of in the environment unchecked, generating an excessive accumulation of organic material. These agricultural and industrial wastes are among the largest sources of biomass in the world. Because of the potential for pollution associated with this disposal problem, these residues generate considerable damage to the environment and the economic activities of the agro-industrial sector [2]. Moreover, failure to recover this waste represents the loss of high-value nutrients. With the growing spread of sustainability concepts, the recovery and reuse of by-products and the bioconversion of residues are increasingly required for agribusiness chains [3]. In several countries, food manufacturers produce a large number of by-products, such as cake, bran, bark, and seeds. Because of their low economic cost and wide availability, the use of these by-products as feedstocks for bioprocesses is feasible. Thus, research involving these bioprocesses is growing exponentially and focuses on the search for high value-added products such as highly digestible proteins, enzymes, biofertilizers, and biosurfactants, among other microbial metabolites [4].
In addition, the short replacement cycle and abundance of this biomass make it an interesting alternative energy source that may be able to cope with present and future energy demands [5]. Because of their high specificity, efficiency, safety, and low cost, bacterial implementation strategies employed for utilizing the predominant biopolymers in agro-industrial residues are competitive with traditional physical and chemical methods [6]. However, a detailed analysis of the molecular mechanisms used by bacteria in the hydrolytic attack of plant cell wall polysaccharides showed that the xylan degradation process is complex and often inefficient under natural conditions. Therefore, it is necessary to adopt additional biotechnological and genetic techniques to improve the degradation process of plant compounds in these microorganisms. Microbial genetic engineering may facilitate the production of more efficient enzymes that assist in the conversion of plant biomass [7, 8].
Enzymatic Hydrolysis of Xylan
After cellulose, hemicellulose is considered the second most abundant polysaccharide in plants. Hemicelluloses, which include xylans, are complex, branched heteropolymers that may consist of pentoses (xylose and arabinose), hexoses (mannose, glucose, and galactose), and uronic acids (β-d-glucuronic acid, α-d-4-O-methylglucuronic acid, and α-d-galacturonic acid) [9, 10]. Xylans are hydrolyzed into their monosaccharide components by the synergistic action of different enzymes. This enzyme system is known as the xylanolytic complex and includes mainly β-1,4-d-xylanase and β-d-xylosidases, which act on the main chain of the polysaccharide [11, 12]. Together, xylanases and β-xylosidases hydrolyze xylan-oligosaccharides, thus releasing xylose molecules. Microorganisms can use this monosaccharide as a carbon source for growth [13]. While the hydrolytic ability of endo-xylanases increases with chain length whereas that of exo-xylanases decreases, these enzymes also hydrolyze xylooligosaccharides into xylose [14]. The exo-β-d-xylosidases are enzymes that hydrolyze minor xylooligosaccharides and xylobioses, which are released by the action of endo-xylanases on the xylan, producing xylose. The specificities of these enzymes on small xylose oligosaccharides are high [11].
Other xylanolytic enzymes in the complex are responsible for eliminating specific substituents and include α-l-arabinofuranosidase (EC 3.2.1.55), acetyl xylan esterase (EC 3.1.1.72), α-d-galactosidase (EC 3.2.1.22), α-d-glucuronidase (EC 3.2.1.139), ferulic acid esterase (EC 3.1.1.73), and p-coumaric acid esterase (EC 3.1.1) [14, 15]. These enzymes are known as auxiliary or depolymerizing enzymes and act in combination with xylanase and β-xylosidases in the total xylan hydrolysis process. These auxiliary enzymes are able to act on the bonds between backbone residues and substituent groups or on the connections between substituents [16]. However, xylanases, xylosidases, and β-α-l-arabinofuranosidases are the key enzymes in the hydrolysis of arabinoxylan, which is the main non-starch polysaccharide found in many cereal grains (wheat, sorghum, barley, rice, and rye) and part of high-fiber diets [17–19]. In the case of sugarcane, the major polysaccharides found are those that have a backbone of glucuronic acid with xylose and arabinose branches, i.e., glucuronoarabinoxylan [20]. The prudent use of a good mix of xylan-degrading enzymes may result in cleaner reactions with lower power consumption, first by reducing the use of chemicals and second by facilitating higher yields. These parameters are vital to the economic viability of such an industrial process [9].
Xylanases and β-xylosidases involved in the enzymatic hydrolysis of lignocellulosic biomass are defined by CAZY (carbohydrate-active enzymes database) [21] as glycosyl hydrolases (GHs). GHs are one of the major classes of enzymes that function in the degradation of carbohydrates by hydrolyzing the glycosidic bond between two or more carbohydrates or between a carbohydrate molecule and a non-carbohydrate [22, 23]. Enzyme classification into this group relies on the substrate used, amino acid sequence similarity, three-dimensional structure, and the mechanism of action [24]. To date, there are 133 recognized families described in the CAZY site (http://www.cazy.org/fam/acc_GH.html) [21], which is updated regularly.
The classification of GHs according to substrate specificity is one of the simplest systems used. However, this system is not suitable for enzymes that act on a variety of substrates, which is particularly relevant for complex GHs that act on natural polysaccharides and exhibit broad specificity [25]. Thus, xylanase and β-xylosidase classification by this system is limited due to the heterogeneity and complexity of xylan [14, 15]. Moreover, this system does not take into account the structural characteristics of the enzymes. Consequently, enzymes that act on similar substrates but do not share structural characteristics, mechanisms of action, or evolutionary relationships may be included in the same class [25].
The GH classification system currently employed by Henrissat was suggested in 1991 [15, 26] and classifies families according to the similarity of their amino acid sequences and folding architecture. GHs with conserved domains are classified within the same family. Thus, when the amino acid sequences of two or more GHs are aligned in a fold, these are included in the same category [22]. Because the molecular structure and the action of an enzyme mechanism are related to its primary structure, the system reflects structural and mechanistic features [15].
Based on this classification, it is possible to group different enzymes with specificity for certain substrates, and enzymes that hydrolyze the same substrate can be found in different families. However, the three-dimensional structures of the proteins are more conserved than their amino acid sequences [25]; for this reason, the three-dimensional structures of GHs have been used to organize families into clans, which comprise enzyme families with similar architectures. There are at least 14 clans of GH families designated GH-A to GH-N; of these, GH-A is composed of 19 related families [26, 27].
Biotechnological Applications of β-Xylosidases
The development of technologies that enable the cost-competitive production of enzymes is of great importance not only for the production of biofuels but also for other biotechnological applications in, e.g., the chemical and pharmaceutical sectors, clinical testing, the food and beverage industry, animal fiber feed, the paper industry, and the production of detergents and leather [9, 28]. The major biotechnological applications of β-xylosidases are highlighted in different points as follow:
-
1.
β-Xylosidases have the versatility to be associated with cellulases (EC 3.2.1.4) and pectinases (EC 3.2.1.15) to promote the maceration of tissues during the industrial processing of vegetable fibers, the liquefaction of coffee mucilage, and the extraction of flavorings and dyes in vegetable oils and starch [14, 15].
-
2.
Xylose derived from plant hemicellulose comprises 5 to 20 % of the most widely used sugars for the production of ethanol [29]. Microbial genetic engineering has facilitated a great improvement in the production of enzymes that can assist in the conversion of residual biomass to bioethanol and chemicals. Thus, to reduce the total costs of production, understanding and improving the activities of microorganisms capable of degrading cellulose and hemicellulose into sugar and ethanol is an essential part of biofuel-related research [30].
-
3.
β-Xylosidases are used in association with other polysaccharides in wineries. To reduce the concentration of β-glucans, which are responsible for increased must viscosity and losses during the filtration step of wine clarification [31].
-
4.
In the production of beers, long chains of arabinoxylans are released from cereals, increasing the viscosity of beer and making it cloudy. β-Xylosidases associated to other hemicellulases assist in the solubilization of arabinoxylans by producing smaller oligosaccharides, thus reducing beer viscosity and eliminating beer turbidity [32].
-
5.
The enzymes of the xylanolytic complex are also used in the pulp and paper industry, mainly for the processing of cellulose pulp prior to whitening [12, 33]. Pretreatment with a xylanolytic enzyme complex decreases the consumption of chemicals, particularly chlorine and chlorine dioxide.
-
6.
Xylan-degrading enzymes are also commonly used in animal feed due to the increasing cost of traditional raw materials [34]. The use of enzyme cocktails to reduce the cost of animal feed is one of the most versatile options for increasing profitability in this sector, facilitating the digestion of all the nutrients present [14].
-
7.
In the food industry, a combination of pectinases, β-xylosidase, and hemicellulases, collectively called maceration enzymes, is used in the extraction and clarification of fruit juices, which yields advantages in the performance, functionality, and quality of the final products [12].
-
8.
β-Xylosidases are also used in baking, where they are applied to flour, which contains 2 to 3 % arabinoxylans. These arabinoxylans absorb approximately 30 % of the water added to bread dough, which prevents the development of gluten, reduces the volume of bread, and affects its texture. The application of a mixture of xylan-degrading enzymes to flour leads to the release of water trapped in arabinoxylans, improving dough handling, increasing volume and improving crumb structure in the final product, and significantly increasing the half-life of the bread dough in consumer markets. In addition, the use of these enzymes can effectively replace the additives used in baking as emulsifiers, antioxidants, malted barley, or wheat [32, 35].
-
9.
In the presence of high concentrations of xylose, β-xylosidases with transglycosylation activity lead to the formation of xylooligosaccharides (XOS), which contain two to seven xylose molecules joined by a β-(1–4) bond. XOS can be used as a prebiotic and have a positive impact on human physiological functions, such as reducing cholesterol levels, increasing the bioavailability of calcium, reducing the risk of colon cancer, exerting a cytotoxic effect on human leukemic cells, exerting beneficial effects in type 2 diabetes mellitus, and acting as antimicrobials and antioxidants [36–38]. XOS with two to four xylose residues are preferred for use in foods. In addition to feed additives, XOS may be employed in the pharmaceutical industry [39].
-
10.
XOS as prebiotics favor the growth of probiotics including Lactobacillus and Bifidobacterium bifidum sp., which themselves promote a series of benefits to human health, such as reducing constipation and favoring the digestion and absorption of nutrients. In addition, probiotics also assist in preventing gastrointestinal infections by inhibiting the growth of pathogenic microorganisms [40].
-
11.
Xylose can also be used by microorganisms for the production of xylitol, a polyalcohol with a high sweetening power often used in the food industry [41].
-
12.
The immobilization of enzymes specifically is a powerful strategy for improving the stability of these proteins and ensuring high enzymatic activity and greater selectivity for their respective substrates [42]. The biochemical properties exhibited by Aspergillus niger β-xylosidase USP-67, which also has trans-xylosylation activity, were measured when the enzyme was immobilized on PEI-Sepharose, resulting in a 94 % increase in catalytic efficiency and improved stability at high temperatures (the immobilized enzyme showed a half-life of 50 min at 65 °C) versus the same enzyme solubilized in other media [43]. The immobilized A. niger β-xylosidase also exhibited a maximum activity (optimal pH = 4.5) in the presence of elevated concentrations of protons (H+) 10-fold greater than that of the enzyme in soluble form (optimal pH = 5.5). In addition, metal ions were more inhibitory of soluble β-xylosidase than of the enzyme in the immobilized form, so that Zn2+ was able to increase the catalytic ability of the immobilized β-xylosidase to 29 %. The specific activity of the immobilized A. niger β-xylosidase (98.15 U mg−1) was also superior to that of the soluble enzyme (77.96 U mg−1). The immobilized enzyme also had the advantage of not being suppressed by high concentrations of such classic inhibitors of fermentation processes as xylose (100 mM) and glucose (200 mm), further underlining its potential utility for biotechnological processes. Through a slightly different methodology, Bhattacharya and Pletschke [44] studied the β-xylosidase isolated from Bacillus gelatini ABBP-1. The bacterial enzyme was immobilized on magnetic particles, and this condition led to a higher specific activity (4.56 U mg−1) than that of the crude extract (3.35 U mg−1), once again suggesting that immobilization may have a protective effect on the protein’s structure, leading to the optimization of its catalytic efficiency.
β-Xylosidase Properties
Enzymes grouped among the GHs, such as β-xylosidases, act by cleaving the glycosidic bond by two distinct mechanisms: retention of the anomeric carbon configuration or inversion of the anomeric carbon configuration [15]. Most enzymes characterized to date adopt the classic retention mechanism [45]. In retaining GHs [46], a double-displacement mechanism directs the configuration of the anomeric carbon after its catalysis. This mechanism includes glycosylation and transglycosylation. Carboxylic acids are those negatively charged amino acid residues, glutamic or aspartic acids, that constitute the catalytic groups that may be on either side of the glycoside bond. Glycosylation constitutes the first stage of the reaction in which one carboxylic acid group acts to promote acid catalysis of the glycosidic oxygen. The other carboxylic group acts as a base, forming a covalent intermediate complex with the glycosyltransferase enzyme. It is assumed that the transition has a mainly dissociative character, i.e., the breaking of the glycosidic linkage occurs before the attack by the nucleophilic group.
The second step is the transglycosylation reaction, wherein the glycosyl-enzyme complex is hydrolyzed by water, the other carboxylic group acting as a basic catalyst residue to remove protons from the nucleophilic water molecule, thereby forming a new glycosidic bond. However, the yield of transglycosylation reactions is low because the reaction product can be immediately used as a substrate for subsequent enzymatic hydrolysis [46]. A simple shift leads to inverting GHs, which invert the anomeric carbon configuration. Depending on the substrate used, the hydrolysis of the β-glycosidic bond can lead to an alpha or beta configuration. As catalysis involves two carboxylated amino acids of the enzyme that act as acid and base, a water molecule activated by the basic residue anomeric carbon attacks and hydrolyzes the glycosidic bond, thus inverting the configuration [46, 47].
β-Xylosidases are distributed throughout GH families 1, 3, 30, 39, 43, 51, 52, 54, 116, and 120. With the exception of the GH43 family, which operates via inverting the configuration of the anomeric carbon, the other families operate via retention (Table 1) and are classified based on the similarity of conserved amino acid sequences [21, 48]. β-Xylosidases belonging to family GH39 effect hydrolysis in a two-step reaction that consists of glycosylation and deglycosylation steps mediated by water, leading to a double inversion that retains the anomeric carbon configuration from xylose [49, 50]. Thus, GH39 β-xylosidases can also catalyze both hydrolysis and cross-linked glycosylation [51]. The GH39 family belongs to the wider clan GH-A; members of which cleave glycosidic bonds through the retention of the anomeric carbon configuration and have eight repetitions of a β/α three-dimensional structure [22]. In addition to the β-xylosidases, the GH39 family contains the α-l-iduronidases, which are involved in the lysosomal degradation of glycosaminoglycans in higher organisms. Deficiencies in this enzyme can lead to the accumulation of glycosaminoglycans, resulting in mucopolysaccharidosis type I, also known as Hurler and Scheie syndrome [52].
The β-xylosidases are also active against the artificial substrate p-nitrophenyl. Many are specific for xylopyranosides such as p-nitrophenyl-β-d-xylopyranoside (pNPX) or o-nitrophenyl-β-d-xylopyranoside (oNPX). Others are capable of cleaving p-nitrophenyl-α-l-arabinopyranoside (pNPA), p-nitrophenyl-β-l-arabinopyranoside (pNPA-β), p-nitrophenyl-β-d-galactopyranoside (pNPG), or p-nitrophenyl-α-d-glucopyranoside (pNPglu) [53]. Many of these purified enzymes are unable to degrade xylan and have little or no activity against this polymer [12]. However, there are some reports of β-xylosidases that are able to attack xylan slowly to produce xylose [54].
Except in some bacteria [55], prokaryotic β-xylosidases are intracellular, whereas those from fungi are bound to cells or extracellular. Many are monomeric [56, 57], dimeric [58], or tetrameric [59] in their catalytic function. Xylan-degrading enzymes are obtained on an industrial scale largely from bacteria, yeast, and fungi of different genera [12, 14]. The conversion of xylan into monosaccharides may also be performed by acid or enzymatic hydrolysis. However, the former releases toxic waste that can compromise subsequent microbial fermentation, often forming undesirable by-products such as furfural, hydroxymethylfurfural, and other products that inhibit fermentation. The challenge is to select, by bioprospecting, or optimize, by genetic engineering, microorganisms increasingly resistant to different inhibitors.
β-Xylosidases Purified by Conventional Methods
The industrial strain Aspergillus awamori 2B.361U2/1 produces an enzyme pool containing xylanase, β-xylosidase, ferulic acid esterase, and β-glucosidase capable of acting on biomass [60]. The accumulation of such enzymes and the physiological responses of the strain were studied under culture conditions with varying nitrogen sources such as yeast extract, NH4+, NO3−, or urea. The β-xylosidase of A. awamori 2B.361U2/1 exhibited an enzymatic activity of 685 UL−1 and a pH optimum between 6 and 7.8 while maintaining its stability at pH 5.5–6.5 for 3 h. β-Xylosidase purified from Geobacillus thermodenitrificans TSAA1 [61] displayed thermotolerance, alkaline tolerance, and halotolerance, and the host secreted high levels of endo-xylanase and β-xylosidase. This work showed first-hand the extracellular production of an inducible β-xylosidase in a thermostable bacterium grown in a submerged fermentation. From this strain, endo-xylanase and β-xylosidase were identified based on the final product formed, purified and characterized using MALDI-TOF analysis. In this work, the bacterium grew optimally at 65 °C and did not grow at temperatures under 40 °C, suggesting that the strain is an extreme thermophile. Geobacillus species produce a variety of industrially significant extracellular enzymes, including xylanases. The G. thermodenitrificans TSAA1 β-xylosidase was optimally active at 60 °C and pH 7, maintaining its stability from pH 5 to 10 and its thermal stability for 120 min at 60 °C. The estimated protein molecular mass was 63 kDa with a Vmax of 4.16 μmol min−1 mg−1 and a Km of 2.8 mM (Table 2).
In A. niger GS1, the production of β-xylosidase was induced under simultaneous saccharification fermentation (SiSF) conditions using corn pericarp. β-Xylosidase was purified using ammonium sulfate precipitation followed by ion exchange and hydrophobic interaction chromatography [62]. The purified enzyme had a molecular weight of 111 kDa, an isoelectric point of 5.35, and a specific activity of 386.7 mg L−1. The optimal pH was 4.5, while the enzyme was able to maintain its pH stability for only 15 min at pH 5. The optimal temperature was 65 °C, exhibiting full activity after 1 h at 60 °C. The β-xylosidase purified from A. niger GS1 represents the biochemical, structural, and thermodynamic properties typical of other β-xylosidases found in previous reports and is possibly applicable to various biotechnological processes.
The isolate yeast-like fungal strain Aureobasidium pullulans SN090a proved to also produce a β-xylosidase (0.179 IU mL−1) with a pH optimum of 4.0 and a temperature optimum of 27 °C. The thermal stability of the enzyme was maintained for 48 h at 27 °C (Table 2) [64]. The strain Aspergillus tamarii Kita [63] was able to use oat (Avena sativa L.) for the production of β-xylosidase under solid-state fermentation (SSF) or submerged fermentation (SmF) conditions. The enzyme was purified and characterized, and the molecular weight of the enzyme was estimated by SDS-PAGE to be 91 kDa. The maximum production of β-xylosidase by A. tamarii under SSF conditions using oat grains was 33.7 U mL−1 with optimal stability at pH 5.5 for 60 min and at 55 °C for 90 min (Table 2).
By comparing the β-xylosidase features compiled from different reports as shown in Table 2, it can be observed that although the substrate used was varied for three of the four enzymes, the optimal pH at which the enzyme is most efficient against pNPX varied between four and seven. With the exception of the A. pullulans β-xylosidase [64], which had a temperature optimum of 27 °C, the other enzymes had optimal temperatures between 55 and 65 °C.
The β-xylosidases of the GH43 group are highly active and may have numerous practical applications. Highly active, commercially available GH43 β-xylosidases were obtained from four different bacteria, Alkaliphilus metalliredigens, Bacillus pumilus, Bacillus subtilis subsp. subtilis str. 168, and Lactobacillus brevis (ATCC367), and were characterized under different biochemical conditions by Jordan and co-authors [66]. The four β-xylosidases studied maintained more than 80 % of their activities in the presence of pNPA, pNPXα, and xylobiose substrates (Table 3). The enzymes showed functional stability over a wide pH range (4.5 to 10.5) for 1 h. However, the GH43 β-xylosidases displayed a restricted thermal stability range (from 25 to 40 °C), temperatures that favor the growth of mesophilic microorganisms. These properties suggest that GH43 β-xylosidases from different bacteria may be used in enzyme cocktails used to optimize the residual biomass from fermentation processes for mesophilic bacteria and yeast to produce second-generation ethanol.
Recombinant β-Xylosidase Expression
Fungal Recombinant Proteins
In general, recently described recombinant enzymes of fungal origin expressed in bacterial systems have optimal activity between 50 and 60 °C [67]. A few exceptions exhibit optimal activity at or below 40 °C, as in the case of Aspergillus oryzae [68] and Penicillium purpurogenum [48] β-xylosidases (Table 4). Many isolated fungal β-xylosidases are thermally stable for a few hours but not always at the optimal temperature found for activity. As such, the β-xylosidase isolated from Paecilomyces thermophila [72] and expressed in and purified from Pichia pastoris possesses considerable stability because it maintains activity after 72 h of incubation at the optimal temperature of 60 °C. Its molecular weight was estimated to be 52.3 kDa, and the enzyme was most active at pH 7. The enzyme maintained stability from pH 3.0 to 8.0 and at 30 to 80 °C. The enzymatic activity varied according to the substrate used, to name a few, 12.26 and 6.2 U mg−1 in the presence of pNPX and pNPA, respectively. Regarding pH, few β-xylosidases have optimal activity between pH 5 and 7 and are generally tolerant of alkaline environments. Two isolated β-xylosidases of Humicola insolens [75] are of note because they have a wide pH tolerance range, being stable at pH 5 to 12 (Xyl43A) and 6 to 10 (Xyl43B). However, other enzymes have an affinity for acidic environments, such as the β-xylosidases isolated from Neurospora crassa [73], Aspergillus sp. [74], Phanerochaete chrysosporium [70, 71], and Phanerochaete thermophile, which show stability in pH ranges of 4.0–6.0, 3.0–6.0, 3.0–6.0, and 3.0–8.0, respectively (Table 4) [67–75].
Bacterial Recombinant Proteins
Recombinant β-xylosidases of bacterial origin have similar pH ranges for activity, exhibiting optimal activity near 6 and remaining relatively stable between 5 and 7 (Table 5) [81–85], as exemplified by the β-xylosidase isolated from the thermophilic bacterium Thermotoga thermarum [81]. A recombinant protein of 85 kDa from this bacterium was expressed in Escherichia coli BL21 (DE3), and the purified enzyme showed optimal activity at 95 °C and pH 6, remaining stable from pH 5.0 to 7.5 with a half-life of 2 h at 85 °C. In the presence of pNPX, the Km and Vmax of this enzyme were 0.27 mM and 223.3 U mg−1, respectively. In addition, a Km of 0.21 mM and a Vmax of 75 U mg−1 were obtained using pNPA as a chromogenic substrate. Another interesting feature of this recombinant β-xylosidase is its high tolerance for xylose, maintaining 50 % of its original activity at concentrations as high as 1000 mM and exhibiting a high hydrolytic activity against xylooligosaccharides.
A high level of enzymatic activity over a wide pH range is also an important property for enzymes of biotechnological interest. This characteristic was exhibited by the recombinant β-xylosidase of G. thermodenitrificans [85]. This enzyme showed best activity at pH 7 and 60 °C and stability from pH 5 to 8. In addition, the protein retained 70 % residual activity at pH 8.0 and 42 % activity at pH 9.0. This stability can be considered an important advantage compared to other β-xylosidases, which generally have little to no activity at alkaline pHs.
A recombinant β-xylosidase from the bacterium Alicyclobacillus sp. [84] is also noteworthy with an enzyme activity of 564.9 U mL−1. Using the pNPX substrate, this β-xylosidase obtained the highest specific activity reported to date for the GH52 family at 261.1 U mg−1 with a catalytic efficiency of 601.5 mMs−1. Optimal activity was observed between 60 and 65 °C, and the enzyme remained stable between pH 5 and 9, demonstrating its wide pH tolerance. The thermostability of this enzyme is also notable because it retained almost 100 % of its initial activity after 1 h of incubation at 65 °C.
Another attractive enzyme was characterized from the aquatic bacterium Caulobacter crescentus [83]. The xynB2 gene encoding β-xylosidase II was expressed in E. coli, and the purified recombinant protein exhibited a specific activity of 215 U mg−1 (pNPX) under optimal conditions (pH 6.0 at 55 °C). This enzyme also exhibited stability from pH 4.5 to 7.5 and retained more than 50 % of its original activity (immediately after purification) after 6 months of incubation at pH 6.0 and 4 °C, which proves that the enzyme is stable under demanding storage conditions. This enzyme was still able to efficiently produce reducing sugars from birchwood xylan and pulp fibers pretreated with an Aspergillus sugar xylanase, thereby demonstrating in vitro synergistic action with an enzyme from the xylanolytic complex of another microorganism. Another important finding in this article was the observation that the recombinant C. crescentus enzyme was able to efficiently produce reducing sugars from the xylan of sugarcane bagasse fibers, highlighting its likely biotechnological applications. Because few bacterial β-xylosidases from the GH39 family have been characterized, this work provides a major contribution to this group of enzymes.
The molecular structure of the recombinant C. crescentus β-xylosidase II was also studied using crystallographic and small-angle X-ray scattering (SAXS) analyses, establishing that this enzyme is monomeric. In addition, the crystal structure revealed that the enzyme’s attachment area participates in shaping the catalytic interface by moving the β-hairpin catalytic domain [57].
The xynB1 gene, which encodes a bifunctional β-xylosidase-α-l-arabinosidase in C. crescentus, was robustly overexpressed in E. coli. The recombinant protein was characterized and exhibited a β-xylosidase I-specific activity of 1.25 U mg−1 in the presence of oNPX and an α-l-arabinofuranosidase-specific activity of 0.47 U mg−1 using pNPA. The β-xylosidase I activity was higher between pH 3 and 10 with a maximum activity at pH 6 and an optimal temperature of 45 °C, slightly lower than that obtained for the β-xylosidase II from the same bacterium. At the temperature optimum, the enzyme exhibited high stability for 240 min. The enzyme activity was inhibited in the presence of Zn2+ and Cu2+, and the enzyme exhibited a Km and Vmax of 2.89 mM and 0.13 μM min1, respectively, with oNPX (Table 5). The β-xylosidase I model structure indicated that this activity was highly conserved with other GH43 enzymes. The increased number of amino acid residue contacts responsible for maintaining the dimeric structure of the protein indicated that C. crescentus β-xylosidase I is likely more stable as a dimer than as a tetramer [82]. The bacterial L. brevis β-xylosidase has not been highlighted in this section because it is not recombinant; however, its mesophilic properties are included in Table 2 because the enzyme was characterized after purification by traditional biochemical methods [65].
Metagenomic Analysis of β-Xylosidases
New β-xylosidases have also been isolated using a metagenomic approach. Table 6 [89–91] summarizes recent data obtained from the metagenomic analysis of different sources: the soils of a native forest and cultivated land [89], the rumen of a dairy cow [90], and the rumen of a yak [91]. Campos et al. [89] isolated cellulolytic bacterial strains from forest soils and identified new cellulose-encoding genes. They studied two types of forest soils, the first of which was from a native forest located in the Atlantic Forest chosen for its high biodiversity and unique biome. Soil samples were also obtained from a loblolly pine (Pinus taeda) and Eucalyptus grandis cultivated forest. Both sites are located in the subtropical region of Misiones in Argentina. The P. taeda and E. grandis forest soils were chosen because these species have great potential as a source of raw material for lignocellulosic biomass. The results showed that the soil samples from the native forest were the richest in cellulolytic bacteria, and a large biodiversity of prokaryotic species was found.
Gruninger and co-authors [90] used a metagenomic approach to screen the rumen of a dairy cow. These experiments led to the isolation of a multifunctional enzyme with significant β-glucosidase-β-xylosidase-α-arabinofuranosidase activities, Bgxa1. Recombinant Bgxa1 exhibited the highest catalytic activity towards pNPG, followed by pNPA and pNPXf. Additionally, its β-glucosidase catalytic efficiency was 100 times that of β-xylosidase or α-arabinofuranosidase, suggesting that the enzyme is predominantly a glucosidase that can be exploited to produce monosaccharides from different hemicelluloses. Given that the co-fermentation of glucose and xylose offers greater simplicity and potentially less cost for biofuel production, a metagenomic analysis of a yak rumen (Bos grunniens) was also performed [91] to identify enzymes that degrade hemicellulose derived from xylan. In this effort, a bifunctional enzyme exhibiting β-glucosidase/β-xylosidase activity (RuBGX1) from the GH3 family was characterized. Recombinant protein produced from the ruBGX1 gene cloned into an expression vector showed high hydrolytic activity against the substrates pNPG and pNPX. An analysis of their kinetic properties indicated that RuBGX1 has a lower affinity for pNPG (Km of 0.164 mmol L−1) than for pNPX (Km of 0.03 mmol L−1) at 50 °C and pH 6. The β-xylosidase ability of RuBGX1 to hydrolyze xylooligosaccharide substrates was assayed in combination with an endo-xylanase. RuBGX1 showed β-glucosidase and β-xylosidase activities on cello-oligosaccharides and synergistic functions with endo-xylanase to promote the degradation of xylan hemicellulose (Table 6). When all of the results obtained via metagenomic approaches are compared, it can be observed that the recombinant enzymes expressed in E. coli are noteworthy because they have the common characteristic of being multifunctional. Although two of the enzymes belong to the GH43 family and one to the GH3 family, all three enzymes exhibited similar pH optima and optimal temperatures within 5 °C of each other.
Optimization of β-Xylosidase by Experimental Design
Another advantageous tool for optimizing the production of enzymes or simply assessing the effects or impacts that different factors play in enzyme production and enzymatic activity is the application of experimental design methods based on statistics [92]. However, few studies in the literature have aimed to optimize microbial β-xylosidases via experimental design methodology. The fractional factorial design of Plackett and Burman yields a useful approach for the preliminary assessment and selection of variables that are statistically significant, and therefore, this design is generally applied to preliminary studies in the central composite rotational design (CCRD) methodology with surface-plot analysis.
In this context, Banerjee and co-authors [93] optimized the production of β-xylosidase and other enzymes by increasing the concentration of biomass hydrolysis products at two different digestion times, 24 and 48 h, using recombinant enzyme mixtures of Trichoderma reesei expressed in P. pastoris. The optimal composition for the production of glucose (5 %) and xylose (17 %) by action of a β-xylosidase occurred within 48 h of biomass (corn stover) digestion in the presence of 15 mg g−1 of enzyme. Multiple combinations of synthetic fungal enzymes used in these experiments (cellobiohydrolase 1 (CBH1), cellobiohydrolase 2 (CBH2), endo-β-1,4-glucanase (EG1), β-glucosidase, a GH10-family endo-β-1,4-xylanase and β-xylosidase) were optimized for chemical pretreatment of the substrate. The enzyme mixture described above in a 43, 4, 30, 8, 11, and 4 % (w/v) mix, respectively, was responsible for higher glucose release yields (58.2 ± 0.2 %) from corn stover pre-hydrolyzed in alkaline pH.
A fractional factorial planning 25–1, with three central points, was applied to β-xylosidase recovery using cetyl trimethyl ammonium bromide reversed micelles [94]. The factors analyzed included pH, the concentration of surfactant (CTAB), the concentration of hexanol, electrical conductivity, and the concentration of butanol so that only the first three variables had significant effects, in increasing order, on enzyme recovery.
The production of β-xylosidase was further enhanced in a strain of Colletotrichum graminicola [95] in SSF using inexpensive raw materials made up of agro-industrial by-products and/or residues. The culture conditions for the production of β-xylosidase were optimized using CCRD, in which three factors were optimized: peanut hulls at a concentration of 0.5 to 7 % (w/v), fermentation time (6 to 11 days), and initial moisture (1 to 3 mL g−1). Significant effects on the production of β-xylosidase were exerted by cultivation time and initial moisture content, the interaction between them, and the interaction between peanut hull concentration and initial moisture content. The quadratic terms of the three independent variables were significant such that the linear effects of cultivation time and the initial moisture content were most relevant to the optimization process of β-xylosidase production.
Xylose Metabolism in Microorganisms
In microbial natural habitats, there are mixtures of different carbon sources that can potentially be metabolized. Thus, different mechanisms have evolved to allow microbes to selectively absorb specific nutrients and metabolize those carbon sources that promote faster growth, the best success in competition with each other, and adaptation to the environment. For most heterotrophic bacteria and fungi, glucose is the most preferred carbon source.
Carbon Catabolite Repression—CCR
In the presence of glucose, the genes required for the utilization of secondary carbon sources are not expressed, and extant enzymes are often inactivated to avoid resource loss. This conserved phenomenon is termed carbon catabolite repression (CCR). This process has been extensively studied in gram-positive and gram-negative bacteria, e.g., B. subtilis and E. coli, respectively. The molecular mechanisms underlying CCR are completely different in these bacteria, although the physiological response to glucose is similar in both organisms [96].
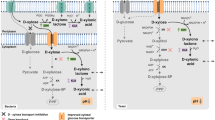
In E. coli, a glucose transporter containing an EIIAGlc domain is the focal processing unit in CCR. This protein belongs to the phosphoenolpyruvate (PEP)/carbohydrate phosphotransferase system (PTS), which is responsible for the uptake and phosphorylation of carbohydrates in some bacteria [97] (Fig. 1). Both phosphotransferases, enzyme I (EI) and histidine protein(HPr), transfer phosphoric groups from PEP to enzymes II (EIIs) that act as sugar transporters. In the absence of glucose, the EIIAGlc domain is phosphorylated and activates adenylate cyclase, which leads to an increase in the intracellular cAMP concentration. The transcriptional inducer factor CRP (cAMP receptor protein) is activated after cAMP binding and positively controls the expression of several secondary catabolic genes, stimulating the upregulation of proteins in the xylan-degrading complex.

Xyloside uptake in E. coli. A glucose transporter containing an EIIAGlc domain is the principal processing unit in CCR. This protein belongs to the phosphoenolpyruvate carbohydrate phosphotransferase system (PTS), which is responsible for the uptake and phosphorylation of xylosides. Enzyme I (EI) and histidine protein (HPr) transfer phosphoryl groups from PEP to enzymes II in the PTS unit that act as sugar transporters. In the absence of glucose and in the presence of xylan, PTS stimulates adenylate cyclase, which leads to an increase in the cAMP concentration, which in turn activates CRP (cAMP receptor protein) and upregulates several secondary catabolic genes, including the xylan-degrading protein complex
In the presence of glucose, EIIAGlc is generally not phosphorylated and is unable to activate adenylate cyclase. The unphosphorylated form of EIIAGlc (PTS) interacts with transporters of secondary carbon sources, causing their inhibition. This mechanism is known as inducer exclusion and allows for the repression of catabolic genes in the presence of glucose [98].
In gram-positive bacteria such as B. subtilis, CCR acts by a different mechanism using the transcription factor CcpA [99], which represses catabolic genes and activates the transcription of genes for overflow breakdown (Fig. 2) [76–79]. The target site for CcpA is the catabolite-responsive element (cre), binding to which is regulated by the presence of the CcpA cofactors HPr (Ser-P) and Crh (Ser-P) [80, 86, 100]. B. subtilis HPr contains regulatory phosphorylation sites (Ser46 and His15) that are phosphorylated during phosphate transfer to the transported sugar. Despite the fact that Crh is homologous to HPr, Crh lacks His15 and is not able join in sugar transport [87]. Two additional components are involved in HPr (Ser-P) dephosphorylation: the phosphorylase activity of HPrK/P [88] and the protein phosphatase PrpC [101]. It is supposed that the two antagonistic activities of HPrK/P are regulated by metabolites. High levels of fructose 1,6-bisphosphate (FBP) and ATP or PPi stimulate the kinase activity, whereas the phosphorylase activity predominates under high concentrations of Pi [102, 103]. Only HPr (Ser-P) is able to interact with CcpA and initiate CCR [96].

CCR in gram-positive bacteria. In B. subtilis, CCR acts via the transcription factor CcpA, which represses catabolic genes and activates the transcription of genes for overflow breakdown. CcpA binds to the catabolite-responsive element (cre) and is regulated by the presence of its cofactors, HPr (Ser-P) and Crh (Ser-P). HPr contains regulatory phosphorylation sites (Ser46) that are phosphorylated during phosphate transfer to the transported sugar. High levels of fructose 1,6-bisphosphate (FBP) and ATP or PPi stimulate the kinase activity, whereas the phosphorylase activity predominates under high concentrations of Pi. Only HPr (Ser-P) is able to interact with CcpA and initiate CCR
Several studies of CCR have been focused on the repression exerted by glucose. Thus, the terms CCR and glucose repression are often used synonymously. However, in both E. coli and B. subtilis, a large number of carbohydrates induce CCR in addition to glucose [104]. In the latter organism, different substrates form a hierarchy in their potential to induce repression via CcpA in the CCR pathway. The individual action of HPr is sufficient to ensure CCR. However, Crh does not have the ability to act as HPr for substrates that cause a drastic repression. The phosphorylation state of HPr in vivo is directly correlated with the strength and the degree of phosphorylation of Ser46 in this enzyme. The central difference between the CCR mechanisms of E. coli and B. subtilis is that in gram-positive bacteria, sugars that are carried by the PTS cause a robust repression. However, the state of phosphorylation of His15 in HPr and its PTS transport activity does not influence the complete mechanism of CCR. It is believed that the hierarchy of carbon substrates in CCR is exclusively determined by the activity of HPrK/P [96].
In B. subtilis, the bicistronic operon xynPB is present and is responsive to the uptake and metabolic yield of xylan degradation [80, 96, 105] (Fig. 3b). An extracellular xylanase converts xylan to β-xylosides taken up by the xynP gene product localized in the cellular membrane. In the cytoplasm, imported xylooligosaccharides are converted to xylose by the action of a β-xylosidase encoded by the xynB gene. Xylose is subsequently converted to xylulose-5-P by enzymes encoded in the xylAB operon downstream of the xynPB operon. All of these genes are negatively regulated by the XylR protein, which binds to operator sites present in the promoter region of each operon and prevents transcription initiation [80, 86–88, 100–106] (Fig. 3a). In the presence of xylose, XylR releases the operator region. Interestingly, B. subtilis cells are not able to grow exclusively on xylose because a xylose-specific transporter is absent in this bacterium [105–108]. However, the membrane protein AraE can slowly take up xylose molecules, which drive the induction of the xynPB and xylAB operons (Fig. 3b). Both xynPB and xylAB are strongly downregulated in the presence of glucose. CCR in B. subtilis is mediated by the cre sites located downstream of the xynPB and xylAB promoter regions. Cre sites are targets of the HPr-Ser46-P/CcpA complex involved in the prevention of transcription initiation (Fig. 3b) [80, 96, 109].

Xylan metabolism in B. subtilis. a In the absence of xylose, the negative regulator XylR binds to operators in the regulatory regions of the xynPB and xylAB operons, leading to the repression of gene expression. Cre sites located upstream of the promoters are downregulated by CcpA, leading to CCR. In contrast, XylR releases operator regions in the presence of xylose, leading to gene upregulation. b The xylan is hydrolyzed by an extracellular xylanase to xylooligosaccharides, which are captured by the XynP carrier. The xylooligosaccharides are degraded to xylose monomers by an intracellular β-xylosidase encoded by the xynB gene. Xylose is subsequently converted to xylulose-5-P by the action of the products of the xylAB operon. The xylulose-5-P is then directed to the pentose phosphate pathway (PPP)
In fact, in B. subtilis, HPr is essential for strong CCR caused by different sugars such as glucose, fructose, and mannitol, whereas Crh is dispensable for the repression exerted by all of these substrates. For example, glucose-6-phosphate acts as an inducer of the XylR repressor, contributing to CCR in B. subtilis. The strength of CcpA-mediated CCR is determined exclusively by the metabolite-controlled activity of HPrK/P. The HPr phosphorylation state in the cells shows that the strength of repression exerted by a specific substrate is well correlated with the levels of HPr (Ser-P). In contrast, in E. coli, the transport activity mediated by PTS (the carbohydrate PTS) has no direct role in the global CCR mechanism. Generally, it is known that the hierarchy of carbon sources in catabolite repression in gram-negative and gram-positive bacteria is regulated by covalent modification, i.e., the phosphorylation state of EIIAGlc (PTS) in E. coli and HPr in B. subtilis (Fig. 2) [96, 104]. In B. subtilis, malate is metabolized like sugar and produces levels of key intermediates that induce CCR in a manner dependent on CcpA and its cofactors [110].
xynB Genes in Caulobacter crescentus
C. crescentus cells are able to express several enzymes involved in the use of lignocellulosic biomass, a skill that allows the use of this bacterium in biotechnological applications [7, 57, 82, 83, 111–113]. Five gene-encoding β-xylosidases, xynB1-5, are present in the genome of this bacterium. The C. crescentus xynB1 and xynB2 genes have been overexpressed in E. coli and their enzymes characterized [57, 82, 83]. In the NA1000 genome, xynB3 (CCNA_00856) was noted to encode a protein with a putative β-xylosidase function, xynB4 (CCNA_02893) a β-xylosidase-α-l-arabinofuranosidase function, and xynB5 (CCNA_03149) a β-glucosidase-β-xylosidase function [112]. Apparently, only xynB4 is organized in an operon in the genome of C. crescentus NA1000. It is possible that xynB4 is expressed as a single transcript (CCNA_02895) together with a gene encoding a putative TonB-dependent receptor protein and the recently characterized xynA1 gene, which encodes a GH10 endo-xylanase, in the NA1000 strain [113]. However, this has yet to be experimentally confirmed. All other xynB genes for β-xylosidases in C. crescentus are coded as an independent mRNA and are likely monocistronic [7, 112].
Studies with the homologous xynB4 and xynB5 genes in C. crescentus CB15 showed that both were induced at the transcriptional level by xylose [114]. Both genes have a conserved motif for xylose induction, a nucleotide sequence (AGAAATGGTACCGGTCTCAT) located in the 5' regulatory region in this bacterium. Transcriptomic analysis has shown that C. crescentus CB15 grown in an M2 medium with glucose and xylose leads to the increased expression of xynB4 and xynB5 (3.78- and 7.11-fold, respectively). However, a DNA chip analysis in the CB15 strain showed that expression of the homologous genes xynB1 and xynB2 (CC0989 and CC2357, respectively) displayed no significant induction in response to xylose [114]. These results are entirely consistent with those reported in C. crescentus NA1000; RT-PCR and transcriptional fusion analyses by Corrêa et al. [7] demonstrated that neither xynB1 nor xynB2 are induced by xylose. In addition, analysis of the non-coding regions of xynB1 and xynB2 using Regulatory Sequence Analysis Tools (RSAT) [115] showed that the conserved motif for xylose induction is not present in these genes [7]. As NA1000 is a mutant derived from the CB15 strain [116], it is expected that many of the functional genomic characteristics of the original strain have been maintained despite adaptation of NA1000 for conjugal function [112].
As mentioned previously, C. crescentus possesses numerous genes that encode hemicellulose-degrading proteins, and xylose metabolism occurs through a process initiated by xylose dehydrogenase (XDH) [117]. In C. crescentus, xylose induces the expression of over 50 genes, including the xyl operon, which expresses the enzymes involved in d-xylose metabolism. Moreover, the XylR repressor is a xylose-sensitive transcriptional factor that belongs to the LacI family and controls the expression of the xyl operon. A regulatory model suggests that in the absence of xylose, XylR binds to the operator sites within xylose-inducible promoters, preventing initiation of the transcription process. In contrast, in the presence of xylose, the sugar binds to XylR and converts the protein into a stable conformation that destabilizes the repressor and reduces its affinity for operator, liberating the promoter to interact with the RNA polymerase core [118].
Among all of the β-xylosidase-encoding genes in C. crescentus NA1000, xynB2 (CCNA_02442), encoding the β-xylosidase II enzyme, is the best studied thus far [7, 57, 83]. Because the xynB2 gene is not regulated by xylose, it was cloned under the control of an artificial promoter to generate a PxylX O-xynB2 strain that overexpressed the enzyme in the presence of xylose [7]. In addition, a null mutant, Δ-xynB2, was created by two homologous recombination events in which a spectinomycin resistance cassette interrupted the chromosomal gene xynB2. Interestingly, the absence of the β-xylosidase II enzyme in C. crescentus cells led to upregulation of the xynB genes, inducing global β-xylosidase activity. A transcriptional analysis of the genes xynB1 (via RT-PCR analysis) and xynB2 (using a lacZ transcriptional fusion) revealed a significant induction of these genes in the xynB2-null mutant when this strain was cultured in the presence of various agro-industrial residues. In addition, a high β-xylosidase activity was also observed in the null mutant strain compared to the wild-type strain. In contrast, overexpression of the xynB2 gene caused downregulation of β-xylosidase activity in this bacterium (Fig. 4).

The xynB2 gene in C. crescentus-encoded β-xylosidase II. a The overexpression of the xynB2 gene downregulates total β-xylosidase activity and transcriptionally represses the xynB1 and xynB2 genes. b In contrast, depletion of the xynB2 gene upregulates global β-xylosidase activity in the bacterium, inducing transcription of the xynB1 and xynB2 genes. The GH39-family β-xylosidase II that acts as a monomer in C. crescentus is able to degrade hemicellulose but may have also evolved a regulatory function (for details, see the text)
The predicted amino acid sequence of the β-xylosidase II protein was analyzed to determine if its structure could aid in understanding its probable involvement in the regulation of the xynB genes of C. crescentus. The results indicate that residues 19–134 of the protein also have sequence similarity to the ETS family of transcription factors, which are associated with some C2H2 zinc finger proteins. In this sequence, motifs were also found that contain a DNA-binding sequence specific to transcription factor activity, as well as for dependent transcriptional regulatory DNA. Although these computer-modeling analyses have noted that the protein in question does in fact have homology to β-xylosidases, a structural domain known to bind to nucleic acids has also been identified. The secondary structure of this domain resembles ribonuclease RNA ligands with significant structural similarity. All the bioinformatic data associated with these experimental results suggests a regulatory role for β-xylosidase II in C. crescentus, despite its efficient β-xylosidase activity (Fig. 4).
It is known that members of the GH39 family, which includes C. crescentus β-xylosidase II, are tetrameric. However, crystal structure analyses of the recombinant protein showed that the C. crescentus β-xylosidase II is monomeric [57]. Taken together, the data from the gene expression studies, the computational modeling, and the structural analyses of the recombinant protein suggest that this enzyme may have evolved a regulatory function that involves a DNA-binding mechanism while still retaining a highly active and stable β-xylosidase function [7, 83]. This presumed regulatory function remains to be further investigated through binding assays. The xynB2-null mutant exhibits high β-xylosidase activity in the presence of spectinomycin and different agro-industrial residues, and this property can be used in biotechnological processes dependent on the use of pentoses, e.g., for the production of fuels and chemicals from biomass [119].
The C. crescentus xynB5 gene was overexpressed in E. coli, and the recombinant protein purified belonging to GH3 family denominated BglX-V-Ara with β-glucosidase-β-xylosidase-α-l-arabinosidase activity, thus proving to be multifunctional for the three different chromogenic substrates tested (ρNPG, ρNPX, and ρNPA). The product of the xynB5 gene is able to act on different substrates and not only differs in structural and functionally features compared to the xynB1 and xynB2 genes but also because it can act on a greater range of compounds that are not broken down by the products of the xynB1 and xynB2 genes [120]. Interestingly, the ability to metabolize a variety of complex compound, in a manner that is independent of the catalytic efficiency of a particular enzyme, is extremely relevant to biotechnological applications [121].
xynB Gene Regulation in Fungi
The β-xylosidase of A. oryzae is encoded by the xylA gene and is induced by xylan and xylose. The promoter region contains the XlnR consensus binding sequence (5′-GGCTA/GA-3′) [92], which is homologous to sequences found in A. niger and is responsible for mediating XlnR induction in vivo. All of the genes controlled by A. niger XlnR contain one or more copies of the consensus core sequence (5′-GGCTAA-3′) in their promoter regions. Each gene varies in the copy number (from one to four) and orientation of the binding site sequence [122].
In A. niger, a loss-of-function mutation in the xlnR gene encoding the transcriptional activator XlnR and a strain with multiple copies of this gene were investigated to define which genes are controlled by XlnR [123]. The transcriptional activator XlnR regulates the transcription of the xlnB, xlnC, and xlnD genes encoding the main xylanolytic enzymes endo-xylanases B and C and β-xylosidase, respectively. In addition, the transcription of the genes encoding the accessory enzymes involved in xylan degradation, including α-glucuronidase A, acetylxylan esterase A, arabinoxylan arabinofuranohydrolase A, and feruloyl esterase A, was found to be regulated by XlnR. XlnR also activates the transcription of two endo-glucanase-encoding genes, eglA and eglB, indicating that transcriptional regulation by XlnR extends beyond genes encoding xylanolytic enzymes [124] (Fig. 3).
As in A. niger, there is an xlnR gene in Aspergillus nidulans encoding a Zn2Cys6 transcriptional activator necessary for the synthesis of the main xylanolytic enzymes, i.e., endo-xylanases X22, X24, and X34 and β-xylosidase XlnD [124, 125]. However, the expression of xlnR is not sufficient for the induction of the xylanolytic complex protein-encoding genes; the presence of xylose is required. The catabolite repressor CreA indirectly represses xlnA (which encodes X22) and xlnB (which encodes X24), as well as exerts direct repression on xlnA. CreA-mediated indirect repression occurs through repression of xlnR, i.e., the xlnR gene promoter is repressed by glucose, and this repression is abolished in creA-null mutant strains. In addition, deregulated expression of xlnR completely alleviates the glucose repression of xlnA and xlnB. Thus, CreA and XlnR form a transcriptional cascade regulating A. nidulans xylanolytic genes [126].
The β-xylosidase-encoding gene bxl1 has been cloned from the thermophilic filamentous fungus Talaromyces emersonii. The gene bxl1 consists of an ORF of 2388 nucleotides without introns that encodes a putative protein of 796 amino acids. The β-xylosidase produced contains a signal peptide of 21 amino acids that yields a mature protein of 775 amino acids with a predicted molecular mass of 86.8 kDa. The deduced amino acid sequence of bxl1 displays considerable homology with the primary structures of other GH3-family β-xylosidases, β-glucosidases, or gene products from A. niger, A. nidulans, A. oryzae, and T. reesei. The bxl1 gene is induced by xylan and by the combination of birchwood xylan with the synthetic, non-metabolizable substrate methyl-β-d-xylopyranoside. d-Xylose was able to induce bxl1 gene expression in low concentrations and repress induction of the gene at high concentrations. The upstream regulatory sequence of the bxl1 gene contains six CreA binding sites, indicating that repression by d-glucose may be mediated at least in part by this catabolic repressor [127].
The effect of several carbon sources on the expression levels of bxl1 was examined, and a temporal induction of β-xylosidase transcription is observed when the fungus is induced on medium supplemented with beechwood xylan. Initial levels are low but increase to a maximum after 36 h of growth. Transcription of the gene then decreases to a steady-state level and returns again to a maximum expression at 72 h of growth. As in bacteria, some paradoxical data exist in fungi about the role of d-xylose as an inducing carbon source [127]. In some cases, d-xylose does not affect β-xylosidase expression, such as the xynB1-xynB2 genes of C. crescentus NA1000 [7]. However, in other studies, including those using T. reesei [128, 129], A. niger [125], A. nidulans [130], or the xynB3-xynB5 genes of C. crescentus CB15 [113], d-xylose has been shown to induce transcription of β-xylosidases. In T. emersonii, birchwood xylan was able to induce significant levels of β-xylosidase expression at specific time intervals when supplemented in growth medium with low-concentration d-xylose (0.1 %, w/v). The expression level of the gene was greatly enhanced by the addition of xylose, and the temporal expression was replaced with high levels of expression at all time points studied. However, expression was inhibited when 3 % d-xylose (w/v) was used as a sole carbon source. These data corroborate the observation that in some microorganisms, high concentrations of d-xylose repress β-xylosidase transcription [131].
Understanding the regulation and expression of the xylanolytic genes has numerous practical applications. An example in this field is the production of xylitol [41]. Transcription-level gene regulation was optimized for promoter strength and plasmid copy number. Because Saccharomyces cerevisiae is not able to utilize xylan or xylose as a carbon source for growth [132], different sugars (including glucose, fructose, mannose, galactose, or maltose, as well as the non-sugars tryptone, yeast extract, and xylan) were selected as a co-substrate for cell growth and the regeneration of the reduced cofactor required in the xylitol conversion. The fermentation process was optimized to improve the yield of xylitol [133].
Conclusions and Future Perspectives
Studies of β-xylosidases have historically been more focused on eukaryotic microorganisms such as fungi and yeasts. Thus, the search for new species (or new strains of already known species) with highly productive xylan-degrading enzymes from unexplored biomes has accelerated considerably. These explorations are an important effort, considering the wide metabolic versatility presented by microorganisms and the richness of global biodiversity. The advent of new genetic engineering techniques and the deposition of complete bacterial genome sequences in the last 10 years has led to growing numbers of reported prokaryotic β-xylosidases, as apparent in a cursory perusal of the CAZY website. The biotechnological applications of β-xylosidases are obvious, e.g., in the production of biofuels from waste biomass as an alternative to increasingly insufficient fossil fuel production. Presently, parallel efforts in different areas of science—biochemistry, molecular biology, computational biology, molecular biophysics, chemical engineering, and statistics—have supported uniformly significant data to elucidate the structure, biochemical properties, mechanisms of action, optimization, metabolic evolution, and applicability of microbial β-xylosidases. However, little is known concerning the mechanisms that control the expression of genes encoding β-xylosidases in microorganisms, particularly in bacteria that have a large number of genes and predicted protein sequences deposited in the database due to various prokaryotic genome sequencing efforts. Approaches based on nucleic acid analysis, such as metagenomics, have contributed to the surprising discovery of new genes encoding multifunctional β-xylosidases. Nevertheless, it is evident that knowledge of the regulatory mechanisms of β-xylosidase gene expression is essential for engineering strains able to operate under the increasingly difficult conditions imposed by the constraints of numerous biotechnological segments of interest.
Abbreviations
- pNPX:
-
p-Nitrophenyl-β-d-xylopyranoside
- oNPX:
-
o-Nitrophenyl-β-d-xylopyranoside
- pNPG:
-
p-Nitrophenyl-β-d-glucopyranoside
- pNPA:
-
p-Nitrophenyl-α-l-arabinopyranoside
- pNPAf :
-
p-Nitrophenyl-α-d-arabinopyranoside
- SSF:
-
Solid-state fermentation
- SmF:
-
Submerged fermentation
- SiSF:
-
Simultaneous saccharification fermentation
- CCR:
-
Carbon catabolic repression
References
Rennie, E. A., & Scheller, H. V. (2014). Xylan biosynthesis. Current Opinion in Biotechnology, 26, 100–107.
Prade, R. A. (1996). Xylanases: from biology to biotechnology. Biotechnology and Genetic Engineering Reviews, 13, 101–131.
Laufenberg, G., Kunz, B., & Nystroem, M. (2003). Transformation of vegetable waste into value added products: (A) the upgrading concept, (B) practical implementations. Bioresource Technology, 87, 167–198.
Couto, S. R., & Sanromán, M. A. (2006). Application of solid-state fermentation to food industry—a review. Journal of Food Engineering, 76, 291–302.
Terenzi, H. F., Jorge, J. A., & Amorin, D. S. (2005). Xylanases from fungi: properties and industrial applications. Applied Microbiology and Biotechnology, 67, 577–591.
Yoon, K. Y., Woodams, E. E., & Hang, Y. D. (2006). Enzymatic production of pentoses from the hemicellulose fraction of corn residues. LWT—Food Science and Technology, 39, 388–392.
Corrêa, J. M., Mingori, M. R., Gandra, R. F., Loth, E. A., Seixas, F. A. V., & Simão, R. C. G. (2014). Depletion of the xynB2 gene upregulates β-xylosidase expression in C. crescentus. Applied Biochemistry and Biotechnology, 172, 1085–1097.
Verma, D., & Satyanarayana, T. (2012). Molecular approaches for ameliorating microbial xylanases. Bioresource Technology, 117, 360–367.
Juturu, V., & Wu, J. C. (2012). Microbial xylanases: engineering, production and industrial applications. Biotechnology Advances, 30, 1219–1227.
Dodd, D., & Cann, I. K. O. (2009). Enzymatic deconstruction of xylan for biofuel production. Global Change Biology Bioenergy, 18, 2–28.
Sunna, A., & Antranikian, G. (1997). Xylanolytic enzymes from fungi and bacteria. Critical Reviews in Biotechnology, 17, 39–67.
Polizeli, M. L. T. M., Rizzatti, A. C. S., Monti, R., Terenzi, H. F., Jorge, J. A., & Amorim, D. S. (2005). Xylanases from fungi: properties and industrial applications. Applied Microbiology and Biotechnology, 67, 577–591.
Finell, J., Jokela, J., Leisola, M., & Riekkola, M. L. (2002). Total hydrolysis of xylotetrose and xylobiose by soluble and cross-linked crystalline xylanase II from Trichoderma reesei. Biocatalysis and Biotransformation, 20, 281–290.
Juturu, V., & Wu, J. C. (2014). Microbial exo-xylanases: a mini review. Applied Biochemistry and Biotechnology, 174, 81–92.
Collins, T., Gerday, C., & Feller, G. (2005). Xylanases families and extremophiles. FEMS Microbiology Reviews, 29, 3–23.
De Vries, R. P., & Visser, J. (2001). Aspergillus enzymes involved in degradation of plant cell wall polysaccharides. Microbiology and Molecular Biology Reviews, 65, 497–522.
Tuncer, M., & Ball, A. S. (2003). Co-Operative actions and degradation analysis of purified xylan-degrading enzymes from Thermomonospora fusca BD25 on oat spelt xylan. Journal of Applied Microbiology, 94, 1030–1035.
Sorensen, H. R., Pedersen, S., & Meyer, A. S. (2007). Synergistic enzyme mechanisms and effects of sequential enzyme additions on degradation of water insoluble wheat arabinoxylan. Enzyme and Microbial Technology, 40, 908–918.
Saghir, S., Iqbal, M. S., Hussain, M. A., Koschella, A., & Heinze, T. (2008). Structure characterization and carboxymethylation of arabinoxylan isolated from ispaghula (Plantago ovata) seed husk. Carbohydrate Polymers, 74, 309–317.
Beg, Q. K., Kapoor, M., Mahajan, L., & Hoondal, G. S. (2001). Microbial xylanases and their industrial applications: a review. Applied Microbiology and Biotechnology, 56, 326–338.
Lombard, V., Golaconda, R. H., Drula, E., Coutinho, P. M., & Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZY) in 2013. Nucleic Acids Research, 42, D490–D495.
Henrissat, B., Callebau, I., Fabrega, S., Lehn, P., Mornon, J. P., & Davies, G. (1995). Conserved catalytic machinery and the prediction of a common fold for several families of glycosyl hydrolases. Proceedings of the National Academy of Sciences of the United States of America, 92, 7090–7094.
Henrissat, B., & Davies, G. (2000). Glycoside hydrolases and glycosyltransferases. Families, modules, and implications for genomics. Plant Physiology, 124, 1515–1519.
Henrissat, B., & Davies, G. (1997). Structural and sequence-based classification of glycoside hydrolases. Current Opinion in Structural Biology, 7, 637–644.
Davies, G., & Henrissat, B. (1995). Structures and mechanisms of glycosil hydrolases. Structure, Cambridge, 3, 853–859.
Henrissat, B. A. (1991). Classification of glycosyl hydrolases based on amino acid sequence similarities. The Biochemical Journal, 280, 309–316.
Coutinho, P. M., & Henrissat, B. (1999). In carbohydrate-active enzymes: an integrated database approach. In H. J. Gilbert, G. Davies, H. Henrissat, & B. Svensson (Eds.), Recent advances in carbohydrate bioengineering (pp. 3–12). Cambridge: The Royal Society of Chemistry.
Kirk, O., Borchert, T. V., & Fuglsang, C. C. (2002). Industrial enzyme applications. Current Opinion in Biotechnology, 13, 345–351.
Screenath, H. K., & Jeffries, T. W. (2000). Production of ethanol form wood hydrolysate by yeasts. Bioresource Technology, 72, 253–260.
Liu, C. F., Sun, R. C., Qin, M., Hang, A. P., Ren, J. L., Xub, F., & Wu, S. B. (2006). Chemical modification of ultrasound-pretreated sugarcane bagasse with maleic anhydride. Industrial Crops and Products, 26, 212–219.
Van Rensburg, P., Strauss, M. L., Lambrechts, M. G., Cordero Otero, R. R., & Pretorius, I. S. (2007). The heterologous expression of polysaccharidase-encoding genes, with oenological relevance in Saccharomyces cerevisiae. Journal of Applied Microbiology, 103, 2248–2257.
Kulkarni, N., Shendye, A., & Mala, R. (1999). Molecular aspects of xylanases. FEMS Microbiology Reviews, 23, 411–456.
Medeiros, R. G., Hanada, R., & Ferreira-Filho, E. X. (2003). Production of xylan-degrading enzymes from Amazon forest fungal species. International Biodeterioration and Biodegradation, 52, 97–100.
Ahuja, S., Ferreira, G., & Moreira, A. (2004). Utilization of enzymes for environmental applications. Critical Reviews in Biotechnology, 24, 125–154.
Camacho, N. A., & Aguiar, O. G. (2003). Production, purification and characterization of a low molecular mass xylanase from Aspergillus sp. and its application in bakery. Applied Biochemistry and Biotechnology, 104, 159–172.
Mussato, S. L., & Mancilla, I. M. (2007). Non-digestible oligosaccharides: a review. Carbohydrate Polymers, 68, 587–597.
Sheu, W. H. H., Lee, I. T., Chen, W., & Chan, Y. C. (2008). Effects of xylooligosaccharides in type 2 diabetes mellitus. Journal of Nutritional Science and Vitaminology, 54, 396–401.
Chen, L. L., Zhang, M., Zhang, D. H., Chen, X. L., Sun, C. Y., Zhou, B. C., & Zhang, Y. Z. (2009). Purification and enzymatic characterization of two β-endoxylanases from Trichoderma Sp K9301 and their actions in xylooligosaccharide production. Bioresource Technology, 100, 5230–5236.
Kallel, F., Driss, D., Chaabouni, S. E., & Ghorbel, R. (2015). Biological activities of xylooligosaccharides generated from garlic straw xylan by purified xylanase from Bacillus mojavensis UEB-FK. Applied Biochemistry and Biotechnology, 175, 950–964.
Gibson, G. R. (2004). Prebiotics. Best Practice & Research. Clinical Gastroenterology, 18, 287–298.
Camargo, D., Sene, L., Saraiva Variz, S., & Felipe, M. G. A. (2015). Xylitol bioproduction in hemicellulosic hydrolysate obtained from sorghum forage biomass. Applied Biochemistry and Biotechnology, 175, 3628–3642.
Mateo, C., Palomo, J. M., Fernandez-Lorente, G., Guisan, J. M., & Fernandez-Lafuente, R. (2007). Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzyme and Microbial Technology, 40, 1451–1463.
Benassi, V. M., Silva, T. M., Pessela, B. C., Guisan, J. M., Mateo, C., Lima, M. S., Jorge, J. A., & Polizeli, M. L. T. M. (2013). Immobilization and biochemical properties of a β-xylosidase activated by glucose-xylose from Aspergillus niger USP-67 with transxylosylation activity. Journal of Molecular Catalysis B: Enzymatic, 89, 93–101.
Bhattacharya, A., & Pletschke, B. I. (2014). Magnetic cross-linked enzyme aggregates (CLEAs): a novel concept towards carrier free immobilization of lignocellulolytic enzymes. Enzyme and Microbial Technology, 61–62, 17–27.
Koshland, D. E. (1953). Stereochemistry and the mechanism of enzymatic reactions. Biological Reviews of the Cambridge Philosophical Society, 28, 416–436.
McCarter, J. D., & Withers, S. G. (1994). Mechanisms of enzymatic glycoside hydrolysis. Current Opinion in Structural Biology, 4, 885–892.
Motomitsu, K., Yuji, H., Shinya, F., Masafumi, H., Takane, K., & Kenji, Y. (2009). Conversion of inverting glycoside hydrolases into catalysts for synthesizing glycosides employing a glycosynthase strategy. Trends in Glycoscience and Glycotechnology, 21, 23–39.
Ravanal, M. C., Alegría-Arcos, M., Gonzales-Nilo, F. D., & Eyzaguirre, J. (2013). Penicillium purpurogenum produces two GH family 43 enzymes with β-xylosidase activity, one monofunctional and the other bifunctional: biochemical and structural analyses explain the difference. Archives of Biochemistry and Biophysics, 540, 117–124.
Whiters, S. G. (2001). Mechanisms of glycosyl transferases and hydrolases. Carbohydrate Polymers, 44, 325–337.
Czjzek, M., David, A. B., Bravman, T., Shoham, G., Henrissat, B., & Shoham, Y. (2005). Enzyme substrate complex structures of a GH39 β-xylosidase from Geobacillus stearothermophilus. Journal of Molecular Biology, 353, 838–846.
Smaali, I., Rémond, C., & O’donohue, M. J. (2006). Expression in Escherichia coli and characterization of β-xylosidases GH39 and GH-43 from Bacillus halodurans C-125. Applied Microbiology and Biotechnology, 73, 582–590.
Unger, E. G., Durrant, J., Anson, D. S., & Hopwood, J. J. (1994). Recombinant alpha-l-iduronidase: characterization of the purified enzyme and correction of mucopolysaccharidosis type I fibroblast. The Biochemical Journal, 304, 43–49.
Kitamoto, N., Yoshino, S., Ohmiya, N., & Tsukagoshi, N. (1999). Sequence analysis, overexpression, and antisense inhibition of a β-xylosidase gene, xylA, from Aspergillus oryzae KBN616. Applied and Environmental Microbiology, 65, 20–24.
Dekker, R. F., & Richards, G. N. (1976). Hemicellulases: their occurrence, purification, properties, and mode of action. Advances in Carbohydrate Chemistry and Biochemistry, 32, 277–352.
Kim, Y. A., & Yoon, K. H. (2010). Characterization of a Paenibacillus woosongensis β-xylosidase-α-arabinofuranosidase produced by recombinant Escherichia coli. Journal of Microbiology and Biotechnology, 20, 1711–1716.
Tsujibo, H., Takada, C., Tsuji, A., Kosaka, M., Miyamoto, K., & Inamori, Y. (2001). Cloning, sequencing, and expression of the gene encoding an intracellular β-D-xylosidase from Streptomyces thermoviolaceus Opc-520. Bioscience, Biotechnology, and Biochemistry, 65, 1824–1831.
Santos, C. R., Polo, C. C., Corrêa, J. M., Simão, R. C. G., Seixas, F. A. V., & Murakami, M. T. (2012). Accessory domain changes accessibility and molecular topography of the catalytic interface in monomeric GH39 β-xylosidases. Acta Crystallographica Section D: Biological Crystallography, 68, 1339–1345.
Contreras, L. M., Gómez, J., Prieto, J., Clemente-Jiménez, J. M., Las Heras-Vázquez, F. J., Rodríguez-Vico, F., Blanco, F. J., & Neira, J. L. (2008). The family 52 β-xylosidase from Geobacillus stearothermophilus is a dimer: structural and biophysical characterization of a glycoside hydrolase. Biochimica et Biophysica Acta, 1784, 1924–1934.
Sakka, K., Yoshikawa, K., Kojima, Y., Karita, S., Ohmiya, K., & Shimada, K. (1993). Nucleotide sequence of the Clostridium stercorarium xylA gene encoding a bifunctional protein with β-D-xylosidase and α-L-arabinofuranosidase activities, and properties of the translated product. Bioscience, Biotechnology, and Biochemistry, 57, 268–272.
Gottschalk, L. M. F., Paredes, R. S., Teixeira, R. S. S., Silva, A. S., & Bon, E. P. S. (2013). Efficient production of lignocellulolytic enzymes β-xylanase, β-xylosidase, ferulic acid esterase and β-glucosidase by the mutant strain Aspergillus awamori 2B.361 U2/1B. Brazilian Journal of Microbiology, 44, 569–576.
Anand, A., Kumar, V., & Satyanarayana, T. (2013). Characteristics of thermostable endoxylanase and β-xylosidase of the extremely thermophilic bacterium Geobacillus thermodenitrificans TSAA1 and its applicability in generating xylooligosaccharides and xylose from agro-residues. Extremophiles, 17, 357–366.
Díaz-Malváez, F. I., García-Almendárez, B. E., Hernández-Arana, A., Amaro-Reyes, A., & Regalado-González, C. (2013). Isolation and properties of β-xylosidase from Aspergillus niger GS1 using corn pericarp upon solid state fermentation. Process Biochemistry, 48, 1018–1024.
El-Gindy, A. A., Saad, R. R., & Fawzi, M. E. (2015). Purification of β-xylosidase from Aspergillus tamarii using ground oats and a possible application on the fermented hydrolysate by Pichia stipitis. Annals of Microbiology, 65, 965–974.
Nasr, S., Soudi, R. M., Salmanian, H. A., & Ghadam, P. (2013). Partial optimization of endo-1, 4-β-xylanase production by Aureobasidium pullulans using agro-industrial residues. Iranian Journal of Basic Medical Sciences, 16, 1245–1253.
Lasrado, L. D., & Gudipati, M. (2013). Purification and characterization of beta-D-xylosidase from Lactobacillus brevis grown on xylooligosaccharide. Carbohydrate Polymers, 92, 1978–1983.
Jordan, B. D., Wagschal, K., Grigorescu, A. A., & Braker, D. J. (2013). Highly active β-xylosidases of glycoside hydrolase family 43 operating on natural and artificial substrates. Applied Microbiology and Biotechnology, 97, 4415–4428.
Teng, C., Jia, H., Yan, Q., Zhou, P., & Jiang, Z. (2011). High-level expression of extracellular secretion of a β-xylosidase gene from Paecilomyces thermophila in Escherichia coli. Bioresource Technology, 102, 1822–1830.
Suzuki, S., Fukuoka, M., Ookuchi, H., Sano, M., Ozeki, K., Nagayoshi, E., Takii, Y., Matsushita, M., Tada, S., Kusumoto, K., & Kashiwagi, Y. (2010). Characterization of Aspergillus oryzae glycoside hydrolase family 43 β-xylosidase expressed in Escherichia coli. Journal of Bioscience and Bioengineering, 109, 115–117.
Chen, Z., Jia, H., Yang, Y., Yan, Q., Jiang, Z., & Teng, C. (2012). Secretory expression of a β-xylosidase gene from Thermomyces lanuginosus in Escherichia coli and characterization of its recombinant enzyme. Letters in Applied Microbiology, 55, 330–337.
Huy, N. D., Thayumanavan, P., Kwon, T.-H., & Park, S.-M. (2013). Characterization of a recombinant functional xylosidase-arabinofuranosidase from Phanerochaete chrysosporium. Journal of Bioscience and Bioengineering, 116, 152–159.
Huy, N. D., Nguyen, C. L., Jeong-Woo Seo, J.-W., Kim, D.-H., & Park, S.-M. (2015). Putative endoglucanase PcGH5 from Phanerochaete chrysosporium is a β-xylosidase that cleaves xylans in synergistic action with endo-xylanase. Journal of Bioscience and Bioengineering, 119, 416–420.
Juturu, V., & Wu, J. C. (2013). Heterologous expression of β-xylosidase gene from Paecilomyces Thermophila in Pichia Pastoris. World Journal of Microbiology and Biotechnology, 29, 249–255.
Kirikyali, N., & Connerton, I. F. (2014). Heterologous expression and kinetic characterization of Neurospora Crassa β-xylosidase in Pichia pastoris. Enzyme and Microbial Technology, 57, 63–68.
Wongwisansri, S., Promdonkoy, P., Matetaviparee, P., Roongsawang, N., Eurwilaichitr, L., & Tanapongpipat, S. (2013). High-level production of thermotolerant β-Xylosidase of Aspergillus sp. bcc125 in Pichia pastoris: characterization and its application in ethanol production. Bioresource Technology, 132, 410–413.
Yang, X., Shi, P., Huang, H., Luo, H., Wang, Y., Zhang, W., & Yao, B. (2014). Two xylose-tolerant gh43 bifunctional β-xylosidase/α-arabinosidases and one GH11 xylanase from Humicola insolens and their synergy in the degradation of xylan. Food Chemistry, 148, 381–387.
Lulko, A. T., Buist, G., Kok, J., & Kuipers, O. P. (2007). Transcriptome analysis of temporal regulation of carbon metabolism by CcpA in Bacillus subtilis reveals additional target genes. Journal of Molecular Microbiology and Biotechnology, 12, 82–95.
Moreno, M. S., Schneider, B. L., Maile, R. R., Weyler, W., & Saier, M. H., Jr. (2001). Catabolite repression mediated by the CcpA protein in Bacillus subtilis, novel modes of regulation revealed by whole-genome analyses. Molecular Microbiology, 39, 1366–1381.
Blencke, H. M., Homuth, G., Ludwig, H., Mäder, U., Hecker, M., & Stülke, J. (2003). Transcriptional profiling of gene expression in response to glucose in Bacillus subtilis: regulation of the central metabolic pathways. Metabolic Engineering, 5, 133–149.
Yoshida, K., Kobayashi, K., Miwa, Y., Kang, C. M., Matsunaga, M., Yamaguchi, H., Tojo, S., Yamamoto, M., Nishi, R., Ogasawara, N., Nakayama, T., & Fujita, Y. (2001). Combined transcriptome and proteome analysis as a powerful approach to study genes under glucose repression in Bacillus subtilis. Nucleic Acids Research, 29, 683–692.
Galinier, A., Deutscher, J., & Martin-Verstraete, I. (1999). Phosphorylation of either Crh or Hpr mediates binding of CcpA to the Bacillus subtilis xyn cre and catabolite repression of the xyn operon. Journal of Molecular Biology, 286, 307–314.
Shi, H., Li, X., Gu, H., Zhang, Y., Huang, Y., Wang, L., & Wang, F. (2013). Biochemical properties of a novel thermostable and highly xylose-tolerant β-xylosidase-α-arabinosidase from Thermotoga thermarum. Biotechnology for Biofuels, 6, 27.
Graciano, L., Corrêa, J. M., Gandra, R. F., Seixas, F. A., Kadowaki, M. K., Sampaio, S. C., Silva, J. L., Osaku, C. A., & Simão, R. C. G. (2012). The cloning, expression, purification, characterization and modeled structure of Caulobacter crescentus β-xylosidase I. World Journal of Microbiology and Biotechnology, 28, 2879–2888.
Corrêa, J. M., Graciano, L., Abrahão, J., Loth, E. A., Gandra, R. F., Kadowaki, M. K., Henn, C., & Simão, R. C. G. (2012). Expression and characterization of a GH39 β-xylosidaseII from Caulobacter crescentus. Applied Biochemistry and Biotechnology, 168, 2218–2229.
Zhang, S., Wang, H., Shi, P., Xu, B., Bai, Y., Luo, H., & Yao, B. (2014). Cloning, expression, and characterization of a thermostable β-xylosidase from thermoacidophilic Alicyclobacillus sp. A4. Process Biochemistry, 49, 1422–1428.
Jain, I., Kumar, V., & Satyanarayana, T. (2014). Applicability of recombinant β-xylosidase from the extremely thermophilic bacterium Geobacillus thermodenitrificans in synthesizing alkylxylosides. Bioresource Technology, 170, 462–469.
Schumacher, M. A., Seidel, G., Hillen, W., & Brennan, R. G. (2007). Structural mechanism for the fine-tuning of CcpA function by the small molecule effectors glucose 6-phosphate and fructose 1,6-bisphosphate. Journal of Molecular Biology, 368, 1042–1050.
Galinier, A., Haiech, J., Kilhoffer, M. C., Jaquinod, M., Stulke, J., Deutscher, J., & Martin-Verstraete, I. (1997). The Bacillus subtilis Crh gene encodes a Hpr-like protein involved in carbon catabolite repression. Proceedings of the National Academy of Sciences of the United States of America, 94, 8439–8444.
Mijakovic, I., Poncet, S., Galinier, A., Monedero, V., Fieulaine, S., Janin, J., Nessler, S., Marquez, J. A., Scheffzek, K., Hasenbein, S., Hengstenberg, W., & Deutscher, J. (2002). Pyrophosphate-producing protein dephosphorylation by hpr kinase/phosphorylase, a relic of early life? Proceedings of the National Academy of Sciences of the United States of America, 99, 13442–13447.
Campos, E., Alvarez, M. J. N., Di Lorenzo, G. S., Gonzalez, S., Rorig, M., Talia, P., Grasso, D. H., Sáez, F., Secade, P. M., Perdices, M. B., & Cataldi, A. A. (2014). Purification and characterization of a GH43 β-xylosidase from Enterobacter sp. identified and cloned from forest soil bacteria. Microbiological Research, 169, 213–220.
Gruninger, R. J., Gong, X., Forster, R. J., & Mcallister, T. A. (2014). Biochemical and kinetic characterization of the multifunctional β-glucosidase/β-xylosidase/α-arabinosidase, Bgxa1. Applied Microbiology and Biotechnology, 98, 3003–3012.
Zhou, J., Bao, L., Chang, L., Liu, Z., You, C., & Lu, H. (2011). Beta-xylosidase activity of a GH3 glucosidase⁄xylosidase from yak rumen metagenome promotes the enzymatic degradation of hemicellulosic xylans. Letters in Applied Microbiology, 54, 79–87.
Vásquez, M. P., Da Silva, J. N. C., De Souza-Júnior, M. B., & Pereira-Júnior, N. (2007). Enzymatic hydrolysis optimization to ethanol production by simultaneous saccharification and fermentation. Applied Biochemistry and Biotechnology, 137, 141–153.
Banerjee, G., Car, S., Scott-Craig, J. S., Borrusch, M. S., & Walton, J. D. (2010). Rapid optimization of enzyme mixtures for deconstruction of diverse pretreatment-biomass feedstock combinations. Biotechnology for Biofuels, 3, 1–15.
Hasmann, F. A., Pessoa, A., Jr., & Roberto, I. C. (2001). Screening of variables in β-xylosidase recovery using cetyl trimethyl ammonium bromide reversed micelles. Applied Biochemistry and Biotechnology, 91, 719–728.
Zimbardi, A. L. R. L., Sehn, C., Meleiro, L. P., Souza, F. H. M., Masui, D. C., Nozawa, M. S. F., Guimarães, L. H. S., Jorge, J. A., & Furriel, R. P. M. (2013). Optimization of β-glucosidase, β-xylosidase and xylanase production by Colletotrichum graminicola undersolid-state fermentation and application in raw sugarcane trash saccharification. International Journal of Molecular Sciences, 14, 2875–2902.
Singh, K. D., Schmalisch, M. H., Stulke, J., & Gorke, B. (2008). Carbon catabolite repression in Bacillus subtilis, quantitative analysis of repression exerted by different carbon sources. Journal of Bacteriology, 190, 7275–7284.
Deutscher, J., Francke, C., & Postma, P. W. (2006). How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiology and Molecular Biology Reviews, 70, 939–1031.
Gorke, B., & Stulke, J. (2008). Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nature Reviews Microbiology, 6, 613–624.
Fujita, Y., Miwa, Y., Tojo, S., & Hirooka, K. (2007). Carbon catabolite control and metabolic networks mediated by the ccpa protein in Bacillus subtilis. In Y. Fujita (Ed.), Global regulatory networks in Bacillus subtilis (pp. 91–110). Trivandrum: Transworld Research Network.
Schumacher, M. A., Seidel, G., Hillen, W., & Brennan, R. G. (2006). Phosphoprotein Crh-Ser46-P displays altered binding to CcpA to effect carbon catabolite regulation. The Journal of Biological Chemistry, 281, 6793–6800.
Singh, K. D., Halbedel, S., Görke, B., & Stülke, J. (2007). Control of the phosphorylation state of the hpr protein of the phosphotransferase system in Bacillus subtilis, implication of the protein phosphatase PrpC. Journal of Molecular Microbiology and Biotechnology, 13, 165–171.
Reizer, J., Hoischen, C., Titgemeyer, F., Rivolta, C., Rabus, R., Stulke, J., Karamata, D., Saier, M. H., Jr., & Hillen, W. (1998). A novel protein kinase that controls carbon catabolite repression in bacteria. Molecular Microbiology, 27, 1157–1169.
Ramstrom, H., Sanglier, S., Leize-Wagner, E., Philippe, C., Van Dorsselaer, A., & Haiech, J. (2003). Properties and regulation of the bifunctional enzyme Hpr kinase/phosphatase in Bacillus subtilis. The Journal of Biological Chemistry, 278, 1174–1185.
Hogema, B. M., Arents, J. C., Bader, R., Eijkemans, K., Yoshida, H., Takahashi, H., Aiba, H., & Postma, P. W. (1998). Inducer exclusion in Escherichia coli by non-PTS substrates: the role of the PEP to pyruvate ratio in determining the phosphorylation state of enzyme IIAGlc. Molecular Microbiology, 30, 487–498.
Lindner, C., StuLke, J., & Hecker, M. (1994). Regulation of xylanolytic enzymes in Bacillus subtilis. Microbiology, 140, 753–757.
Dahl, M. K., Degenkolb, J., & Hillen, W. (1994). Transcription of the xyl operon is controlled in Bacillus subtilis by tandem overlapping operators spaced by four base-pairs. Journal of Molecular Biology, 243, 413–424.
Schmiedel, D., & Hillen, W. (1996). A Bacillus subtilis 168 mutant with increased xylose uptake can utilize xylose as sole carbon source. FEMS Microbiology Letters, 135, 175–178.
Schmiedel, D., & Hillen, W. (1996). Contributions of xylR, CcpA, and cre to diauxic growth of Bacillus megaterium and to xylose isomerase expression in the presence of glucose and xylose. Molecular and General Genetics, 250, 259–266.
Kraus, A., Hueck, C., GaRtner, D., & Hillen, W. (1994). Catabolite repression of the Bacillus subtilis xyl operon involves a cis element functional in the context of an unrelated sequence, and glucose exerts additional XylR-dependent repression. Journal of Bacteriology, 176, 1738–1745.
Meyer, F. M., Jules, M., Mehne, F. M. P., Le Coq, D., Landmann, J. J., Görke, B., Aymerich, S., & Stülke, J. (2011). Malate-mediated carbon catabolite repression in Bacillus subtilis involves the HPrK/CcpA pathway. Journal of Bacteriology, 193, 6939–6949.
Nierman, W. C., Feldblyum, T. V., Laub, M. T., Paulsen, I. T., Nelson, K. E., Eisen, J., et al. (2001). Complete genome sequence of Caulobacter crescentus. Proceedings of the National Academy of Sciences of the United States of America, 98, 4136–4141.
Marks, M. E., Castro-Rojas, C. M., Teiling, C. D. U. L., Kapatral, V., Walunas, T. L., & Crosson, S. (2010). The genetics basis of laboratory adaptation in Caulobacter Crescentus. Journal of Bacteriology, 192, 3678–3688.
Graciano, L., Corrêa, J. M., Vieira, F. G. N., Bosetto, A., Loth, E. A., Kadowaki, M. K., Gandra, R. F., & Simão, R. C. G. (2015). Cloning and expression of the xynA1 gene encoding a xylanase of the GH10 group in Caulobacter crescentus. Applied Biochemistry and Biotechnology, 175, 3915–3929.
Hottes, A. K., Meewan, M., Yang, D., Arana, N., Romero, P., Mcadams, H. H., et al. (2004). Transcriptional profiling of Caulobacter crescentus during growth on complex and minimal media. Journal of Bacteriology, 186, 1448–1461.
Turatsinze, J. V., Thomas-Chollier, M., Defrance, M., & Van Helden, J. (2008). Using RSAT to scan genome sequences for transcription factor binding sites and cis regulatory modules. Nature Protocols, 3, 1578–1588.
Evinger, M., & Agabian, N. (1977). Envelope-associated nucleoid from Caulobacter crescentus stalked and swarmer cells. Journal of Bacteriology, 132, 294–301.
Stephens, C., Christen, B., Fuchs, T., Sundaram, V., Watanabe, K., & Jenal, U. (2007). Genetic analysis of a novel pathway for d-xylose metabolism in Caulobacter crescentus. Journal of Bacteriology, 189, 2181–2185.
Stephens, C., Christen, B., Watanabe, K., Fuchs, T., & Jenal, U. (2007). Regulation of D-xylose metabolism in Caulobacter crescentus by a LacI-type repressor. Journal of Bacteriology, 189, 8828–8834.
Jordan, D. B., & Wagschal, K. (2010). Properties and applications of microbial β-D-xylosidases featuring the catalytically efficient enzyme from Selenomonas ruminantium. Applied Microbiology and Biotechnology, 86, 1647–1658.
Justo, P. I., Corrêa, J. M., Maller, A., Kadowaki, M. K., da Conceição-Silva, J. L., Gandra, R. F., & Simão, R. C. G. (2015). Analysis of the xynB5 gene encoding a multifunctional GH3-BglX β-glucosidase-β-xylosidases-α-arabinosidase member in Caulobacter crescentus. Antonie Van Leeuwenhoek, 108, 993–1007.
Lagaert, S., Pollet, A., Courtin, C. M., & Volckaert, G. (2014). β-Xylosidases and α-L-arabinofuranosidases: accessory enzymes for arabinoxylan degradation. Biotechnology Advances, 32, 316–332.
Marui, J., Tanaka, A., Mimura, S., Graaff, L. H., Visser, J., Kitamoto, N., Kato, M., Kobayashi, T., & Tsukagoshi, N. (2002). A transcriptional activator, Aoxlnr, controls the expression of genes encoding xylanolytic enzymes in Aspergillus Oryzae. Fungal Genetics and Biology, 35, 157–169.
Van Peij, N. N. M. E., Visser, J., & De Graaf, L. H. (1998). Isolation and analysis of xlnR, encoding a transcriptional activator co-ordinating xylanolytic expression in Aspergillus niger. Molecular Microbiology, 27, 131–142.
Van Peij, N. N. M. E., Gielkens, M. M. C., De Vries, R. P., Visser, J., & De Graaff, L. H. (1998). The transcriptional activator xlnR regulates both xylanolytic and endoglucanase gene expression in Aspergillus niger. Applied and Environmental Microbiology, 64, 3615–3619.
Van Peij, N. N., et al. (1997). β-Xylosidase activity, encoded by xlnD, is essential for complete hydrolysis of xylan by Aspergillus niger but 584 F.J. not for induction of the xylanolytic enzyme spectrum. European Journal of Biochemistry, 245, 164–173.
Tamayo, E. N., Villanueva, A., Hasper, A. A., Graaff, L. H., Ramón, D., & Orejasm. (2008). CreA mediates repression of the regulatory gene xlnR which controls the production of xylanolytic enzymes in Aspergillus nidulans. Fungal Genetics and Biology, 45, 984–993.
Reen, F. J., Murray, P. G., & Tuohy, M. G. (2003). Molecular characterisation and expression analysis of the first hemicellulase gene (Bxl1) encoding β-xylosidase from the thermophilic fungus Talaromyces emersonii. Biochemical and Biophysical Research Communications, 305, 579–585.
Hrmová, M., Biely, P., & Vršanská, M. (1986). Specificity of cellulase and β-xylanase induction in Trichoderma reesei Qm9414. Archives of Microbiology, 144, 307–311.
Kristufek, D., Zeilinger, S., & Kubicek, C. P. (1995). Regulation of β-xylosidase formation by xylose in Trichoderma Reesei. Applied Microbiology and Biotechnology, 42, 713–771.
Perez-Gonzalez, J. A., van Peij, N. N., Bezoen, A., MacCabe, A. P., Ramón, D., & de Graaff, L. H. (1998). Molecular cloning and transcriptional regulation of the Aspergillus nidulans xlnD gene encoding a β-xylosidase. Applied and Environmental Microbiology, 64, 1412–1419.
Margolles-Clark, E., Ilmen, M., & Pentilla, M. (1997). Expression patterns of ten hemicellulase genes of the filamentous fungus Trichoderma reesei on various carbon sources. Journal of Biotechnology, 57, 167–179.
Gong, C. S., Chen, L. F., Flickinger, M. C., Chiang, L. C., & Tsao, G. T. (1981). Production of ethanol from D-xylose by using D-xylose-isomerase and yeasts. Applied and Environmental Microbiology, 41, 430–436.
Li, Z., Qu, H., Li, C., & Zhou, X. (2013). Direct and efficient xylitol production from xylan by Saccharomyces cerevisiae through transcriptional level and fermentation processing optimizations. Bioresource Technology, 149, 413–419.
Acknowledgments
R.C.G. Simão was partially supported by grants from the Araucaria Foundation (process 630/2014). A. Bosetto and L. Graciano were Coordination fellows of Improvement of Higher Education Personnel (CAPES). E.L. Santos was a fellow of the Brazilian National Council for Scientific and Technological Development (CNPq). P.I. Justo is a professional funded by Diagnostics of America SA (DASA). S.S. Venzon is a professional funded by the Federal Technological University of Paraná.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Bosetto, A., Justo, P.I., Zanardi, B. et al. Research Progress Concerning Fungal and Bacterial β-Xylosidases. Appl Biochem Biotechnol 178, 766–795 (2016). https://doi.org/10.1007/s12010-015-1908-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-015-1908-4