
Abstract
In the present work, the gene xynB2, encoding a β-xylosidase II of the Glycoside Hydrolase 39 (GH39) family, of Caulobacter crescentus was cloned and successfully overexpressed in Escherichia coli DH10B. The recombinant protein (CcXynB2) was purified using nickel-Sepharose affinity chromatography, with a recovery yield of 75.5 %. CcXynB2 appeared as a single band of 60 kDa on a sodium dodecyl sulfate polyacrylamide gel and was recognized by a specific polyclonal antiserum. The predicted CcXynB2 protein showed a high homology with GH39 β-xylosidases of the genus Xanthomonas. CcXynB2 exhibited an optimal activity at 55 °C and a pH of 6. CcXynB2 displayed stability at pH values of 4.5–7.5 for 24 h and thermotolerance up to 50 °C. The K M and V Max values were 9.3 ± 0.45 mM and 402 ± 19 μmol min−1 for ρ-nitrophenyl-β-d-xylopyranoside, respectively. The purified recombinant enzyme efficiently produced reducing sugars from birchwood xylan and sugarcane bagasse fibers pre-treated with a purified xylanase. As few bacterial GH39 family β-xylosidases have been characterized, this work provides a good contribution to this group of enzymes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The production of fuels and chemical feedstocks from biomass involves the degradation of the cellulosic and hemicellulosic components. Xylan is the major component of hemicellulose, and its breakdown requires the action of different enzymes that are able to hydrolyze several linkages. Endo-1,4-β-xylanase (E.C. 3.2.1.8) and 1,4-β-xylosidase (E.C. 3.2.1.37) play key roles within this context: Endo-1,4-β-xylanase has endo-xylanolytic activity and produces xylooligomers, whereas β-xylosidase catalyzes the removal of β-xylosyl residues from the nonreducing termini of xylobiose and xylooligosaccharides [1].
β-xylosidases are found in different glycoside hydrolase (GH) families, 1, 3, 30, 39, 43, 51, 52, 54, 116, and 120, according to the GH classification system CAZy (Carbohydrate-Active Enzymes Database—http://www.cazy.org), a knowledge-based resource that specializes in the enzymes that build and breakdown complex carbohydrates and glycoconjugates [2]. β-xylosidases belonging to the GH39 family display a typical (α/β)8-barrel fold [3] and perform hydrolysis in a two-stage reaction that consists of glycosylation and water-mediated de-glycosylation steps that lead to a double inversion, with the net retention of the anomeric carbon configuration in the xylose product [4, 5]. Thus, GH39 β-xylosidases can catalyze two types of reactions: hydrolysis and trans-glycosylation [6].
Caulobacter crescentus, a Gram-negative bacterium able to live in oligotrophic environments, expresses numerous genes that encode enzymes involved in biomass metabolism, including five xynB genes that encode β-xylosidases; however, only one gene encodes a protein that belongs to the GH39 family [7]. The occurrence of multiple endo-β-xylanase and β-xylosidase isoforms has been reported in many microorganisms and could be explained by the heterogeneous structure of xylan because not all xyloside linkages are accessible to a single enzyme during the process of hydrolysis. Moreover, the accessibility of linkages changes during the course of catalysis. Thus, a group of enzymes, with each having a specific function, could be a strategy that a microorganism uses to ensure efficient hydrolysis [8].
Microbial β-xylosidases occur in bacteria and fungi, with those of the latter group being the most studied. Despite its important physiological role, relatively little is known about the molecular mechanisms of plant polysaccharide utilization by C. crescentus: to date, there have been two reports concerning the xynB genes of C. crescentus [9, 10]. In the present work, the xynB2 gene (CCNA_02442) was isolated, cloned, and successfully overexpressed in Escherichia coli. The purified recombinant protein was characterized, used to generate a polyclonal antiserum, and efficiently applied in reactions with a fungal xylanase to hydrolyze xylan from birchwood and sugarcane bagasse.
Materials and Methods
Bacterial Strains, Growth Conditions, and Plasmids
C. crescentus strain NA1000, a holdfast mutant derivative of wild-type strain CB15 (Evinger and Agabian), was maintained at 4 °C and grown at 30 °C in PYE complex medium [11]. E. coli strain DH10B was grown in Luria–Bertani (LB) broth at 37 °C [12] and used for the propagation of plasmids and the overexpression of the recombinant protein. The bacterial strains and plasmids used in this study are listed in Table 1.
Isolation and Cloning of the xynB2 Gene
The xynB2 gene (CCNA_02442) was amplified by PCR using 100 ng of total DNA isolated from C. crescentus NA1000 and 50 pmol of each primer, xynB2-F (5′tatgaattcatggcgaacgccggcccc3′) and xynB2-R (5′tattctagactaggccagcggctcgag3′). EcoRI and XbaI sites (underlined) were added to the forward and reverse primers, respectively. The PCR reaction was performed in a Mastercycler Gradient thermocycler (Eppendorf) in the presence of 1.5 mM MgCl2, 2.5 nM dNTPs, and 1.5 U of Taq DNA Polymerase (Promega). A total of 30 cycles of amplification consisted of extension at 94 °C for 45 s, annealing at 58 °C for 45 s and extension at 72 °C for 2 min. The 1.5-kb PCR fragment obtained was cloned in the pJet1.2Blunt (Fermentas) vector and subcloned in the EcoRI/XbaI sites of the expression vector pPROEX-Hta (Invitrogen) that produces an N-terminal histidine-fused translation product. The recombinant clone (pPROEX-xynB2) was sequenced to confirm the identity of the xynB2 gene of C. crescentus; the sequencing was performed by the Sanger method services unit at the Chemical Institute of São Paulo University, São Paulo.
Overexpression and Purification of the Recombinant Protein
E. coli (DH10B) cells carrying pPROEX-xynB2 were incubated in LB medium containing ampicillin (1 mg mL−1) at 37 °C, with shaking at 150 rpm. The cells were grown to mid-log phase (O.D. λ = 0.4–0.6) and induced for 4 h with 1 mM isopropyl-β-thiogalactopyranoside (IPTG) for the overexpression of C. crescentus β-xylosidase II (CcXynB2). Aliquots (5 μL) of the induced cells were collected and used for the extraction of the total proteins, which were then analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) [13]. A 100-mL aliquot of induced recombinant cells was centrifuged for 10 min at 4 °C and 12,000 rpm, and the pellet was treated with 4 mL of FastBreakTM Cell Lysis Reagent 10X (Promega®) containing 0.2 μg mL−1 lysozyme (Sigma), 20 μg mL−1 DNase (Invitrogen), and 1 mM protease inhibitors (GE Healthcare). The mixture was incubated for 30 min at 30 °C and 50 rpm. The lysed cells were centrifuged, and the supernatant was applied to pre-packed nickel-Sepharose columns (GE Healthcare). Three steps of washing with binding buffer (20 mM phosphate buffer (pH 7.4) containing 500 mM NaCl and 20 mM imidazole) were performed before the purification of CcXynB2 with elution buffer (20 mM phosphate buffer (pH 7.4) containing 500 mM NaCl and 500 mM imidazole). The purified recombinant protein CcXynB2 was dialyzed for 48 h against ultrapure water at 4 °C. CcXynB2 was maintained at 4 °C and used for the biochemical characterization and the production of polyclonal antiserum.
CcXynB2 Enzyme Assay and Protein Determination
The β-xylosidase activity of the CcXynB2 enzyme was determined by assaying the amount of ρ-nitrophenol released from the substrate ρ-nitrophenyl-β-d-xylopyranoside (ρNPX; Sigma). A mixture of 250-μL 35 mM ρNPX and 50 mM sodium citrate buffer at pH 5.4 was pre-incubated at 50 °C, and 25 μL of diluted purified enzyme was added. After 10 min, the reaction was stopped by the addition of 1 M Na2CO3, and the absorbance at 410 nm was measured. One unit (U) of β-xylosidase activity was defined as the amount of enzyme that liberated 1 μmol of ρ-nitrophenol per minute. β-xylosidase activity procedure was also carried out at pH 6.0 and a temperature of 55 °C to obtain assay at optimal conditions. The protein concentrations were measured using the Bradford reagent from Bio-Rad, with BSA as the standard.
Polyclonal Antibody Production
The synthesis of the polyclonal antiserum in rabbits was approved by the Animal Ethics Committee of the State University of West Paraná (process 07410). One female rabbit (Oryctolagus cuniculus—New Zealand) was immunized with approximately 500 mg of the purified CcXynB2 in phosphate-buffered saline and 0.5 mL of Freund’s complete adjuvant. After 4 weeks, the rabbit received a second injection containing 1 mg of the antigen in Freund’s incomplete adjuvant; 15 days after that, the rabbit was subjected to cardiac puncture, and the antiserum obtained was used in Western blotting assays.
Western Blot Analysis
Western blotting was performed according to the method of Towbin et al. [14], with the following modifications. Aliquots (2 μL) of the IPTG-induced cells (0, 2, 4 h), 1 μg of CcXynB2, and 1 μg of β-xylosidase I-α-arabinofuranosidase from C. crescentus [10] were resuspended in Laemmli sample buffer. After electrophoresis, the proteins were transferred to a nitrocellulose membrane, and the blot was incubated for 2 h in blocking buffer (10 mM Tris–HCl, pH 7.5) containing 150 mM NaCl, 5 % nonfat dried milk, and 0.05 % sodium azide. The polyclonal antiserum against CcXynB2 was diluted 1:200 in blocking buffer containing 5 % nonfat dried milk, and the blot was incubated for 16 h at 4 °C. The protein blot was then washed with 10 mM Tris–HCl (pH 7.5) and 150 mM NaCl (TBS) containing 0.05 % Tween 20, followed by TBS alone. The blot was incubated with anti-rabbit immunoglobulin G antiserum conjugated with alkaline phosphatase (Sigma), and nitroblue tetrazolium and BCIP (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) were used as the substrates to visualize the reaction.
The Effect of pH and Temperature on Enzyme Activity and Stability
The optimal pH for activity was determined using McIlvaine buffer (pH 3–10) [15] at 50 °C. To determine the pH stability, purified CcXynB2 was incubated in McIlvaine buffer at various pH values (3–10) at 4 °C for 24 h; afterward, the residual activity of the protein was measured at 50 °C and pH 6.0. To determine the optimal temperature, aliquots of the purified recombinant protein were incubated at various temperatures (20–70 °C), and the xylosidase activity was assayed at the optimal pH. To determine the thermostability, the purified protein was incubated in 50 mM McIlvaine buffer (pH 6) at 50, 55, and 60 °C. After 0–240 min of incubation, the samples were cooled in an ice water bath for 15 min, and the activity was measured according to the optimal assay method. The highest residual activity was defined as 100 %.
The Effect of Different Compounds on the Enzyme Activity
The effects of different compounds on the activity of CcXynB2 were tested by measuring the β-xylosidase activity using assay at optimal conditions after incubation of the enzyme at 4 °C for 15 min in the presence of 2 mM of each of the compounds, (NH4)2SO4, HgCl, MgSO4, BaCl2, NH4Cl, iodoacetamide, CuCl, Al2(SO4)3, MnSO4, FeSO4, SnCl2, KCl, NaCl, CaCl2, CuSO4, ZnSO4, EDTA, dithiothreitol (DTT), and β-mercaptoethanol. The residual activities were expressed as the percentage of the activity obtained for the control enzyme (in the absence of the reagents).
The Determination of the Kinetic Parameters
The kinetic parameters of the recombinant enzyme were determined for ρNPX at concentrations ranging from 0.5 to 15 mM in McIlvaine buffer (pH 6) at 55 °C for 10 min. The initial velocities were measured, and a Lineweaver–Burk plot was used to estimate the K M and V Max.
Xylan and Sugarcane Bagasse Hydrolysis by Xylanase and CcXynB2
The enzyme xylanase purified from Aspergillus alliaceus (Abrahão et al., unpublished) and CcXynB2 were evaluated in hydrolysis assays to investigate their ability to metabolize xylan from birchwood (Sigma) and sugarcane bagasse substrates. To perform the tests, the substrates were diluted separately to a final concentration of 1 % (w/v) in 2-mL 50 mM McIlvaine buffer (pH 6.0). In the first step, 1 U of xylanase was added and incubated at 50 °C for 24 h; after this period, the enzyme was inactivated by heat treatment at 100 °C for 5 min. A sample of this mixture was then used to measure the level of reducing sugars produced. After cooling the mixture at 4 °C for 15 min, 2 U of CcXynB2 was added, and the hydrolysis products were measured after 18 h of incubation at 50 °C using the DNS method [16]. The hydrolysis activities of xylanase and CcXynB2 were evaluated based on the amount of reducing sugars produced after the action of each enzyme.
Statistical Analyses
The experiments were performed in triplicate, with the use of replicates to avoid inconsistent data. The statistical program Origin 6.0 graph (Data Analysis and Technical Graph) was used for the statistical analysis of the data by scatter plot analysis and for plotting the graphs.
Results and Discussion
Cloning, Expression, and Purification of Recombinant CcXynB2
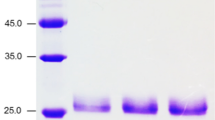
A 1,500-bp DNA fragment encoding the xynB2 gene of C. crescentus was amplified by PCR and cloned into pJet1.2Blunt. The recombinant β-xylosidase II of C. crescentus (CcXynB2) was expressed in E. coli as a protein fusion with a histidine tag obtained by ligating the complete xynB2 gene into the expression vector pPROEX-Hta digested with EcoRI/XbaI (pPROEX-xynB2). This plasmid was then used to transform E. coli strain DH10B. High expression of the enzyme was observed by SDS-PAGE analysis (Fig. 1a, lanes 3–5). CcXynB2 was purified using nickel-Sepharose affinity chromatography (Fig. 1a, lanes 7–8). The purified CcCynB2 protein appeared as a single band on an SDS-PAGE gel, with a molecular mass of approximately 60 kDa, consistent with the deduced molecular mass of the N-terminal His-tag recombinant enzyme (58 kDa).

β-Xylosidase II overexpression in E. coli (DH10B) and Western blotting. a SDS-PAGE showing the steps of purification of recombinant CcXynB2: 1 10-kDa protein ladder; 2 protein extract from strain DH10B harboring pPROEX-xynB2; 3–5 total protein extract induced with IPTG (1 mM) for 2, 3, and 4 h, respectively; 6 cell lysate before nickel-Sepharose affinity purification. Aliquots of 2 μL of the recombinant CcXynB2 after the first (7) and the second (8) elution with imidazole–phosphate buffer. The arrow indicates CcXynB2. b Western blot performed with antiserum against CcXynB2. Aliquots of 2 μL of the total protein extract induced with IPTG (1 mM) for 0 (lane 1), 3 (lane 2), and 4 h (lane 3); 1 μg of purified CcxynB2 (lane 4); and 1 μg of purified C. crescentus β-xylosidase I-α-l-arabinofuranosidase (lane 5) were separated by SDS-PAGE
Sequence analysis predicted that the protein encoded by C. crescentus xynB2 (CCNA_02442) is a β-xylosidase belonging to the GH39 family. The 500-amino acid protein sequence shows a higher degree of identity with bacterial GH39 β-xylosidases from Xanthomonas. The predicted C. crescentus β-xylosidase protein is 68 and 67 % identical to its counterparts from the gammaproteobacteria Xanthomonas axonopodis and Xanthomonas citri, respectively. A specific polyclonal antiserum was obtained through the immunization of a rabbit using recombinant enzyme. The protein extracts induced with IPTG and purified CcXynB2 were recognized by the specific antiserum in a Western blot experiment (Fig. 1b). However, the bifunctional enzyme β-xylosidase I-α-arabinofuranosidase from C. crescentus was not recognized by the CcXynB2 antiserum. In fact, the two enzymes have β-xylosidase activity [9, 10], but they present low similarity among themselves, 30 % homology and 13 % identity. Therefore, CcXynB2 antiserum can be applied in future gene expression experiments using total C. crescentus protein.
Biochemical Characterization of the Recombinant β-Xylosidase II
CcXynB2 was purified with a recovery yield of 75.5 %, and the specific activity of the enzyme was 215 U mg−1 to ρNPX (data not shown). Xylosidases in general have been described as bifunctional or even multifunctional [17, 18]. However, CcXynB2 was unable to cleave ρ-NF-arabinofuranoside or ρ-NF-β-d-glucopyranoside (data not shown), indicating a high specificity of the recombinant enzyme. CcXynB2 was able to hydrolyze typical substrates of β-xylosidases such as xylobiose, xylotriose, and xylopentaose [9]. Recently, crystallographic and SAXS analysis of the GH39 C. crescentus β-xylosidase II was conducted [9] and showed that CcXynB2 is a monomer in solution because the shortened C-terminus prevents the formation of a stable tetramer. Unlike β-xylosidases from Geobacillus stearothermophillus [3] and Thermoanaerobacterium saccharolyticum [19], the only two other GH39 members with known 3-D structures have a tetrameric arrangement.
The effects of pH and temperature on the activity of CcXynB2 were evaluated using ρNPX as the substrate. The pH curve displayed a maximal activity of β-xylosidase at pH 6 (Fig. 2a). An identical pH optimum was observed for the GH39 β-xylosidase from Thermoanaerobacterium sp. [20]. A pH maximum of 6.5 was observed for the GH39 xylosidase from Bacillus halodurans C-125 [21]. More than 50 % of the enzyme activity was maintained after the incubation of CcXynB2 for 24 h at 4 °C in the pH range of 4.5 to 7.5 (Fig. 2b). C. crescentus β-xylosidase II possesses stability at slightly acidic to neutral pH. The determination of the pH optimum and stability is very important from a biotechnological standpoint as these parameters indicate the situations in which waste treatment enzymes might be practical. Because enzymatic digestion is generally performed using several enzymes, knowledge of the basic characteristics of each is useful to facilitate their synergy and the efficiency of their reuse for various plant cell wall hemicelluloses.

a Effect of pH on CcXynB2 activity. C. crescentus recombinant β-xylosidase II was incubated with ρNPX in McIlvaine buffer at pH values from 3 to 10 at 50 °C for 10 min. b Stability of CcXynB2 at different pH values in McIlvaine buffer solution at 4 °C for a period of 0 (filled circle) to 24 h (empty circle). The data presented in a and b are representative of three independent experiments, and the standard deviations are shown as vertical bars
CcXynB2 demonstrated more than 50 % of its original activity (immediately after purification) after long periods (6 months) of incubation at pH 6.0 and 4 °C (data not shown). In fact, Santos et al. [9] showed that CcXynB2 has a longer loop from the auxiliary domain, which makes a number of polar and hydrophobic contacts with the parental (α/β)8-barrel domain, modifying the accessibility and molecular topography of the catalytic interface. These interactions also maintain the accessory domain tightly linked to the catalytic core, which might be important for enzyme function and stability.
CcXynB2 exhibited an optimal activity at 55 °C (Fig. 3a) and showed half-lives of 240 min at 50 °C, 160 min at 55 °C, and 40 min at 60 °C (Fig. 3b). These results suggest that CcXynB2 is stable up to 50 °C and displays a modest thermotolerance, which indicates that it might be sufficiently robust for certain industrial applications. GH39 β-xylosidase from B. halodurans C-125 remained stable over the pH range of 3.5 to 8.5, and it was optimally active at 55 °C [6].

a Evaluation of the CcXynB2 activity at different incubation temperatures. The enzyme was incubated at different temperatures (range 20 to 75 ± 2 °C) and assayed at the optimal pH. b Test of the enzymatic stability exhibited by CcXynB2. The experiment was conducted at the three best temperatures determined in the experiment shown in a: 50 (filled circle), 55 (empty circle), and 60 °C (filled square). The activity assay was performed at 50 °C with ρNPX and a 10-min time reaction. The graphs in a and b represent the means of three experiments, and the vertical bars indicate the standard deviations
The Michaelis–Menten constants were determined for ρNPX: the K M and V Max values were 9.3 ± 0.45 mM and 402 ± 19 μmol min−1, respectively (data not shown). The K M value obtained for CcXynB2 is consistent with the report of Smaali et al. [6] for the GH39 β-xylosidase from B. halodurans C-125 (K M of 8.61 mM). However, the K M of 0.09 mM obtained for the GH39 β-xylosidase from Thermoanaerobacterium sp. [20] indicates a variable affinity for the same substrate between enzymes belonging to the GH39 family in bacteria.
To determine the activity of β-xylosidase II under different conditions of salt and other chemical agents, CcXynB2 was incubated for 15 min at 4 °C in the presence of various reagents (Table 2). The results showed that the enzyme was partially inactivated by the salts (NH4)2SO4 and MnSO4 and the reducing agent β-mercaptoethanol; the enzyme showed a further inhibition of activity when in contact with MgSO4, BaCl2, NH4Cl, iodoacetamide, CuCl, Al2(SO4)3, FeSO4, SnCl2, NaCl, CaCl2, CuSO4, EDTA, and DTT. In contrast, the enzyme showed an increased activity when incubated in the presence of KCl, suggesting that potassium somehow provides a beneficial cofactor for the catalytic site, which improved the performance.
Xylan and Sugarcane Bagasse Hydrolysis by Xylanase and CcXynB2
The ability of CcXynB2 to hydrolyze xylan from birchwood and sugarcane bagasse residue was observed after the separate incubation of the recombinant protein and a xylanase purified from A. alliaceus (Abrahão et al., unpublished; Fig. 4). The relative percentage of xylan from birchwood and sugarcane bagasse hydrolysis products, previously treated with xylanase, increased 2.5- and 6.5-fold, respectively, after incubation with CcXynB2 for 18 h (Fig. 4). In the absence of CcXynB2, no additional reducing sugars were observed in the reactions after 18 h of incubation. This result suggests that the C. crescentus β-xylosidase II could be utilized in the pre-degradation of hemicellulose fiber, releasing sugars of five carbons that can be used in fermentation processes.

Percentage of xylan and sugarcane bagasse hydrolysis by the successive action of xylanase purified from A. alliaceus and CcXynB2. Birchwood xylan (1 %, w/v; white bars) and sugarcane bagasse (1 %, w/v; gray bars) were incubated for 24 h in a water bath at 50 °C with xylanase (1 U mL−1) purified from A. alliaceus (Abrahão et al., unpublished). After this period, aliquots of the sample were boiled for 5 min and re-incubated or not with CcXynB2 (2 U mL−1) for 18 h at 50 °C. Samples of the reactions were used for the determination of reducing sugars at 0, 24, and 42 h of incubation time. The asterisk indicates the denaturation time of xylanase and CcXynB2 added to the reaction. Standard deviations are shown as vertical bars
The benefit of using a combination of different enzymes has been reported [23]. The synergism between the enzymes produced by Trichoderma and Aspergillus on sugarcane bagasse allowed a high degree of hemicellulose chain depolymerization, making the bioconversion of lignocellulosic biomass into sugars more efficient than that obtained by chemical methods. In addition, a balance in the formation of inhibitory compounds that hinder the later performance of the fermentation process of sugars was observed.
Conclusion
In this work, a β-xylosidase II (CcXynB2) gene was cloned and successfully overexpressed in E. coli. CcXynB2 showed a high homology with the GH39 enzymes of Xanthomonas. CcXynB2 demonstrated a high activity after 24 h of incubation at pH values ranging from 4.5 to 7.5 and was stable at temperatures up to 50 °C. An important finding in this study is that CcXynB2 was able to produce reducing sugars efficiently from birchwood xylan and sugarcane bagasse fibers pre-treated with a purified xylanase. Because few bacterial GH39 β-xylosidases have been characterized, this work provides a good contribution to this group of enzymes.
References
Ahmed, S., Riaz, S., & Jamil, A. (2009). Molecular cloning of fungal xylanases: an overview. Applied Microbiology and Biotechnology, 84, 19–35.
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., & Henrissat, B. (2009). The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Research, 37, D233–D238.
Czjzek, M., David, A. B., Bravman, T., Shoham, G., Henrissat, B., & Shoham, Y. (2005). Enzyme-substrate complex structures of a GH39 β-xylosidase from Geobacillus stearothermophillus. Journal of Molecular Biology, 353, 838–846.
White, A., & Rose, D. R. (1997). Mechanism of catalysis by retaining β-glycosyl hydrolases. Current Opinion in Structural Biology, 7, 645–651.
Whiters, S. G. (2001). Mechanisms of glycosyl transferases and hydrolases. Carbohydrate Polymers, 44, 325–337.
Smaali, I., Rémond, C., & O’Donohue, M. J. (2006). Expression in Escherichia coli and characterization of β-xylosidases GH39 and GH-43 from Bacillus halodurans C-125. Applied Microbiology and Biotechnology, 73, 582–590.
Marks, M. E., Castro-Rojas, C. M., Teiling, C. D. U. L., Kapatral, V., Walunas, T. L., & Crosson, S. (2010). The genetics basis of laboratory adaptation in Caulobacter crescentus. Journal of Bacteriology, 192, 3678–3688.
Juturu, V., & Wu, J. C. (2011). Microbial xylanases: engineering, production and industrial applications. Biotechnology Advances. doi:10.1016/j.biotechadv.2011.11.006.
Santos, C. R., Polo, C. C., Corrêa, J. M., Simão, R. C. G., Seixas, F. A. V., & Murakami, M. T. (2012). Accessory domain changes accessibility and molecular topography of the catalytic interface in monomeric GH39 Beta-xylosidases. Acta Crystallographica Section D, 68, 1339–1345.
Graciano, L., Corrêa, J. M., Gandra, R. F., Seixas, F. A. V., Kadowaki, M. K., Sampaio, S. C., et al. (2012). The cloning, expression, purification, characterization and modeled structure of Caulobacter crescentus β-xylosidase I. World Journal of Microbiology and Biotechnology, 28(9), 2879–2888.
Poindexter, J. S. (1964). Biological properties and classification of the Caulobacter group. Bacteriological Reviews, 28, 231–295.
Sambrook, J., Fritsch, E. F., & Maniatis, T. (1989). Molecular cloning: a laboratory manual (2nd ed.). Cold Spring Harbor: Cold Spring Harbor Laboratory.
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head bacteriophage T4. Nature, 226, 680–685.
Towbin, H., Staehelin, T., & Gordon, J. (1979). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proceedings of the National Academy of Sciences of the United States of America, 76, 4350–4354.
McIlvaine, T. C. (1921). A buffer solution for colorimetric comparison. Journal of Biological Chemistry, 49, 183–186.
Miller, G. L. (1959). Use of dinitrosalicylic acid reagent for determination of reducing sugars. Analytical Chemistry, 31(3), 426–428.
Tuncer, M. (2000). Characterization of β-xylosidase and α-L-arabinofuranosidase activities from Thermomonospora fusca BD25. Turkish Journal of Biology, 24, 753–767.
Mai, J., Juergen, W., & Lorenz, W. W. (2000). Cloning, sequencing, and characterization of the bifunctional xylosidase–arabinosidase from the anaerobic thermophile Thermoanaerobacter ethanolicus. Gene, 247, 137–143.
Yang, J. K., Yoon, H. J., Ahn, H. J., Lee, B. I., Pedelacq, J. D., Liong, E. C., et al. (2004). Crystal structure of β-D-xylosidase from Thermoanaerobacterium saccharolyticum, a family 39 glycoside hydrolase. Journal of Molecular Biology, 335, 155–165.
Wagschal, K., Franqui-Espiet, D., Lee, C. C., Robertson, G. H., & Wong, D. W. (2005). Enzyme-coupled assay for β-xylosidase hydrolysis of natural substrates. Applied and Environmental Microbiology, 71, 5318–5323.
Wagschal, K., Franqui-Espiet, D., Lee, C. C., & Wong, D. W. (2008). Cloning, expression and characterization of a glycoside hydrolase family 39 xylosidase from Bacillus halodurans C-125. Applied Biochemistry and Biotechnology, 146, 69–78.
Evinger, M., & Agabian, N. (1977). Envelope-associated nucleoid from Caulobacter crescentus stalked and swarmer cells. Journal of Bacteriology, 132, 294–301.
Gottschalk, L. M. F., Oliveira, R. A., & Bom, E. P. S. (2010). Cellulases, xylanases, β-glucosidase and ferulic acid esterase produced by Trichoderma and Aspergillus act synergistically in the hydrolysis of sugarcane bagasse. Biochemical Engineering Journal, 51, 72–78.
Acknowledgments
This work was supported by grants from the Fundação Araucária (convênio 893/2012), Fundo Paraná SETI, CNPq, and Fundação Parque Tecnológico Itaipu (PTI C&T/FPTI-BR).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Corrêa, J.M., Graciano, L., Abrahão, J. et al. Expression and Characterization of a GH39 β-Xylosidase II from Caulobacter crescentus . Appl Biochem Biotechnol 168, 2218–2229 (2012). https://doi.org/10.1007/s12010-012-9931-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-012-9931-1