
Opinion statement
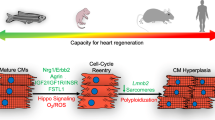
Myocardial infarction is the most common cause of cardiac injury in humans and results in acute loss of large numbers of myocardial cells. Unfortunately, the mammalian heart is unable to replenish the cells that are lost following a myocardial infarction and an eventual progression to heart failure can often occur as a result. Regenerative medicine based approaches are actively being developed; however, a complete blueprint on how mammalian hearts can regenerate is still missing. Knowledge gained from studying animal models, such as zebrafish, newt, and neonatal mice, that can naturally regenerate their hearts after injury have provided an understanding of the molecular mechanisms involved in heart repair and regeneration. This research offers novel strategies to overcome the limited regenerative response observed in human patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemic heart disease is the leading cause of death worldwide and a major burden on the health care resources of industrialized nations [1, 2]. Myocardial infarction (MI) typically leads to the death of about 25 % of the left ventricular cardiomyocytes (approximately one billion cells) and the irreversible formation of fibrotic scar tissue [3]. The human heart is an organ with very low regenerative capacity and homeostatically cardiomyocytes renew at an estimated rate of 1 % annually at the age of 25 to 0.45 % at the age of 75 [4]. This suggests that although the human heart has the potential for limited self-renewal, it cannot sufficiently replace the large numbers of cardiomyocytes lost after MI. For some time, heart transplant has been the only widely successful cure for heart failure, but the lack of available organs and the demanding nature of the surgery and anti-rejection treatments have driven the pursuit of alternative treatments. Several attempts of cardiac repair are under investigation in ongoing clinical trials (reviewed in [5–8]); however, long term beneficial effects have yet to be seen. Studies on animal models that can regenerate hearts naturally have revealed cellular and molecular mechanisms that are missing in the nonregenerative mammals and provide a blueprint for better designed therapeutic approaches to repair the human hearts. In this review we will focus on the description and comparison of heart regeneration in zebrafish, newt, and mouse, and we will discuss about how the knowledge brought about by these model organisms may be used to induce heart regeneration in the adult mammalian heart.
The zebrafish as a model of heart regeneration
The zebrafish (danio rerio) is a teleost (bony fish) of the Cyprinidae family and is able to regenerate its heart after ventricle amputation [9, 10, 11•, 12]. The injury induces the formation of an initial fibrin clot that lasts 7–9 days postamputation (dpa), followed by progressive replacement of the fibrin clot by cardiomyocytes. At 60 dpa the size and shape of the ventricle as well as the contractile properties of beating hearts appear grossly normal [11•, 13•].
Myocardial regeneration
Using cre-lox lineage tracing technology, two studies showed that the newly regenerated cardiomyocytes are derived mainly from the dedifferentiation and proliferation of pre-existing cardiomyocytes at the injury site ([13•, 14•] and reviewed in [9, 10]). This regeneration mechanism involves the disassembly of the sarcomeric structure and the upregulation of the mitotic checkpoint kinase mps1 and the regulator of cell-cycle progression plk1, as well as reactivation of the expression of the embryonic cardiogenic gene gata4 [13•, 14•]. One recent study, using a combination of genetic fate-mapping strategies and a ventricle-specific genetic ablation system, showed that after genetic ablation of the ventricle, atrial cardiomyocytes undergo a transdifferentiation into ventricular cardiomyocytes to participate to the ventricular regeneration [15•]. Endocardial Notch signaling may be essential for this transdifferentiation and contributes to ventricular regeneration non-cell autonomously [15•]. However, contribution of these transdifferentiated atrial cardiomyocytes to injured ventricles is greatly diminished in four-month old adult fish [15•], raising the question of whether transdifferentiation could have a role in adult fish heart regeneration.
At the molecular level, a recent study using a transgenic reporter line (cmlc2:FUCCI zebrafish) and an in vivo chemical screen of modulators of the cardiomyocyte cell cycle identified a number of compounds regulating insulin like growth factor 2, transforming growth factor-β (TGF-β) and sonic hedgehog signaling, that can modulate cardiomyocyte proliferation during heart development and regeneration [16]. Consistent with this report, several TGF- β ligands and receptors were found to be upregulated in response to cryoinjury (see below) of the zebrafish heart. Pharmacologically blocking these receptors resulted in a reduction in cardiomyocyte proliferation and a bulging infarct at the wound site [17]. We independently identified igf2 as a candidate gene enriched during zebrafish heart regeneration via a microarray gene expression study [18]. We further demonstrated that igf2 is expressed in both endocardium and epicardium during heart regeneration after ventricular resection. Blocking Igf signaling using a chemical inhibitor (NVP-AEW-541) or via a dominant-negative Igf1r [19] inhibited DNA synthesis in cardiomyocytes and heart regeneration [20]. Igf signaling specifically regulates proliferation and contribution of the gata4-positive subpopulation of cardiomyocytes [20].
Another key pathway that regulates post-injury cardiomyocyte proliferation is Jak1/Stat3 signaling [21]. Translation profiling of the cardiomyocytes after heart injury revealed dynamic modulation of a number of components of the Jak1/Stat3 pathway. Gain and loss of function approaches demonstrate that this pathway positively regulates cardiomyocyte proliferation and is required for heart regeneration [21]. Cardiomyocyte proliferation is also regulated by p38α MAPK [22]. Using a transgenic line where the cardiomyocytes express a constitutively active (ca)MKK6 (the upstream activator of p38α MAPK), it was shown that p38α MAPK signaling inhibits the cardiomyocyte proliferation during cardiogenesis in embryos and after ventricular amputation in adults [22]. During regeneration zebrafish ventricular cardiomyocytes are able to switch off p38α MAPK signaling allowing proliferation to proceed after injury [22].
Cardiomyocyte proliferation is responsive to external cues such as hypoxia. In zebrafish, drug-induced anemia can cause hypoxia, which leads to elevated levels of HIF1α throughout the heart, which in turn promotes heart regeneration, cardiomyocyte dedifferentiation and proliferation [23]. Conversely, hyperoxia or inhibition of HIFα prevents heart regeneration after ventricular amputation in embryos and adult zebrafish. In rats, hypoxic conditions and cardiac ischemia-induced hypoxia also results in increased of HIFα levels as well [24], but this is not followed by heart regeneration, indicating that the heart regeneration pathways are downstream of the hypoxic response, as suggested by Jopling et al [23]. Microarray studies showed that hypoxia induces the expression of components of the Jak-STAT pathway, which is involved in post-injury cardiomyocyte proliferation [21]. MicroRNAs (miRs) also regulate zebrafish heart regeneration. Levels of the cardiomyocyte specific miR-133 decrease during heart regeneration and ectopic overexpression of miR-133 inhibits cardiomyocyte proliferation and heart regeneration in adult zebrafish by blocking cell cycle genes, including mps1 [25].
Epicardial activation and coronary vessel neovascularization
The non cardiomyocytes cells of the heart also regenerate after amputation and play a crucial role in supporting myocardial regeneration. During regeneration, the epicardial cells undergo a FGF signaling-induced epithelial-mesenchymal transition and proliferate [26]. PDGF signaling is activated in epicardial-derived cells (EPDCs) after injury, which allows the EPDCs to proliferate and contribute to new coronary blood vessel formation [27]. Consistent with these findings, lineage tracing of epicardial cells shows that their fates are limited to non myocardial cell types closely associated with the regenerating vasculature [28]. The activated epicardium also directs myocardial regeneration via the expression of diffusible factors and the modulation of the extracellular environment. Proliferative cardiomyocytes are then guided to the wound site by the release of the diffusible cytokine ligand Cxcl12a from the regenerating epicardium [29]. Using an inhibitor that blocks Cxcr4b (the receptor of Cxcl12a), changes in the migration of the proliferating cardiomyocytes into the wound site were observed [29].
Endocardial activation
Similar to the epicardium, the endocardium is in direct contact with the myocardium and actively signals to cardiomyocytes during zebrafish heart regeneration. Following resection the endocardium undergoes rapid and dramatic transcriptional and morphological changes [30]. Immediately following resection endocardial cells detach from the myocardium and appear more rounded than their usual elongated morphology [30]. The endocardium also expresses retinoic acid (RA) synthesizing enzyme, raldh2 from an early stage. Initially raldh2 is expressed throughout the heart, then later localized to the wound site [30]. This RA-signaling is required for cardiomyocyte proliferation and the endocardial source is later supplemented by expression from the epicardium [30]. Another key factor required in promoting cardiomyocyte proliferation is igf2. Like RA, igf2b is also strongly induced in the endocardium and to a lesser extent in the epicardium following resection and is required for cardiomyocyte proliferation during regeneration [16, 20] (see above).
Heart regeneration in newt and regulation of the extracellular matrix (ECM)
As observed with teolost fish species, adult urodele amphibians such as the newt, Notophthalmus viridescens, display remarkable regenerative capacity of multiple organs and appendages [31]. This regenerative response extends to the heart following ventricular apex resection or a mechanically induced crush injury such that the lost or damaged myocardium is replaced within 3 months [32, 33]. One of the limitations of the newt model is the relatively underdeveloped modes with which to investigate the molecular basis of this process, in part due to difficulties associated with sequencing the complex newt genome [34]. However, more recent application of transcriptional and proteomic approaches have been used to gain valuable insight into the molecular regulation of newt regeneration [33–35]. As opposed to the response of inflammation and metabolic gene expression that is observed in mammals, one of the most robust gene expression changes in the newt are of genes associated with the ECM [35]. The ECM is largely comprised of fibrous proteins and polysaccharides and is required to provide the structural framework for regenerating cardiomyocytes. Components of the ECM (such as collagen and keratin) and molecules that remodel the ECM (such as matrix metalloproteinases) are expressed immediately after injury of the ventricular apex [35, 36]. As well as providing extracellular support to the regenerating myocardium, dynamic changes in the composition of the regenerating ECM could have an important role in signaling to the regenerating myocardium, coordinating the regenerative response. Consistent with this one component of ECM that is expressed in response to injury, Tenascin C, promotes cell cycle reentry of newt cardiomyocytes in vitro [35]. In addition, miR-128 has been found to be important in the regulation of cardiomyocyte proliferative response in the newt [33]. Blockade of this miRNA results in arrest of cardiomyocytes proliferation in response to injury and also results in the deposition and persistence of fibrin and collagen at the wound site suggesting that blocking cardiomyocyte proliferation can affect the remodeling of ECM or that the two share regulatory components [33].
The potential for such a role in heart regeneration of ECM signaling is not a specific newt regenerative response. In the zebrafish, following resection of the apex and formation of a blood clot, an extracellular matrix that is conducive to regeneration has been found to be important for heart regeneration to proceed. A major component of this matrix is the fibronectin that is laid down at the wound site predominantly by the adjacent epicardium following injury [37]. In zebrafish lacking functional fibronectin, regenerating cardiomyocytes fail to integrate into the injury site such that the apex of the heart forms a collagen scar rather than a regenerated myocardium [37].
Heart regeneration in rodents
Several studies in rodents suggest that mammals share a low rate of cardiomyocyte proliferation [38]. For example, the monitoring of tritiated thymidine incorporation to assess DNA synthesis of normal cardiomyocytes from adult mice showed that only one cardiomyocyte in 180,000 incorporates tritiated thymidine (0.0005 %). In cardiomyocytes from hearts injured by focal cauterization of the left ventricular free wall, three cardiomyocytes in 36,000 showed tritiated thymidine incorporation (0.0083 %) [38]. Using genetic fate mapping where the cardiomyocytes express GFP after Cre induced recombination, the percentage of GFP positive cardiomyocytes does not change and remains at about 80 % of all the cardiomyocytes. This suggests that cardiomyocytes are maintained by division of existing cardiomyocytes during normal homeostasis [39]. However, after infarction, this percentage of GFP decreases to about 65 %, suggesting that the main contributor of the limited number of newly regenerated cardiomyocytes is progenitor cells. However, the same group recently used genetic fate mapping with stable isotope labeling and multi-isotope imaging mass spectrometry and demonstrated that pre-existing cardiomyocytes are still the dominant source of cardiomyocyte replacement after MI [40]. This cell cycle activity will lead to polyploidy, multinucleation, and new diploid, mononucleated cardiomyocytes [40]. If the generation of new diploid, mononucleated cardiomyocytes can be enhanced after cardiac injury, perhaps heart repair and regeneration can be achieved.
Unlike adult mice, it was recently shown that the heart of 1-day old neonatal mice regenerates completely after partial surgical resection [41•] or ligation of left anterior descending artery (LAD ligation) [42]. Injured heart appears histologically normal at 21 dpa compared with control mice, and the systolic function is normal 2 months postamputation. Genetic fate mapping showed that the new cardiomyocytes come mainly from pre-existing cardiomyocytes rather than from an unspecified stem cell population [41•, 42]. However, ventricular amputation or LAD ligation of 7-day old mice fails to induce heart regeneration, which coincides with a time point of proliferative arrest in rodents [43], indicating that the regenerative ability of the neonatal mouse heart is lost when cardiomyocytes stop to proliferate. Interestingly, a recent study showed that this postnatal proliferative window of cardiomyocytes coincides with the expression of the homeodomain transcription factor MEIS1, already known to regulate normal embryonic heart development [44]. Meis1 is expressed in 1-day old postnatal mouse (P1) cardiomyocytes and its expression increases from P7 to adulthood. The deletion of Meis1 in knock-out (KO) mice extends the proliferation window of cardiomyocytes, as visualized by the increase in the number of phospho-histone-3 labeled cardiomyocytes at P14. Moreover, in adult KO mice, the number of cardiomyocytes is significantly higher than in control mice, although smaller in size. The conditional deletion of Meis1 specifically in the cardiomyocytes of adult hearts, using cre-lox technology, induces cardiomyocyte cell cycle re-entry after tamoxifen administration. On the other hand, the overexpression of Meis1 inhibits heart regeneration in neonatal mice by reducing the cardiomyocyte proliferative ability. Taken together, these data show that the regenerative ability of neonatal mouse after heart injury depends of the regulation of the cardiomyocyte proliferation by MEIS1.
Cardiomyocyte proliferation is also regulated by miRs. Multiple members of miR-15 are upregulated in neonatal stage and contribute to cardiomyocyte mitotic arrest [42]. Overexpression of miR-195 (one of the miR-15 family members) inhibits neonatal heart regeneration, while inhibition of the miR-15 family induces cardiomyocyte proliferation and improves cardiac functions after MI in mice [42]. A high throughput functional screen for human miRs that promote neonatal cardiomyocyte proliferation identified hsa-miR-590-3p or hsa-miR-199-3p as candidates. Injection of viral vectors expressing hsa-miR-590 or hsa-miR-199 in neonatal mouse hearts increased sarcomere disassembly and cell cycle re-entry of cardiomyocytes and the injection at the peri-infarcted area, following MI resulted in reduction of the infarct size and cardiomyocyte proliferation and preserved cardiac functions over time [45]. The miR-17-92 is also necessary and sufficient to promote cardiomyocyte proliferation in vivo in embryonic and neonatal mice, and the overexpression of miR-17-92 enhances cardiomyocyte proliferation in response to heart injury in adults [46].
During embryonic development, organ size control is very important, especially for the heart. In mice, the conditional deletion of the Salv gene induces an increase of cardiomyocyte proliferation and cardiomegaly [47]. Salv encodes an effector of the Hippo signaling, a pathway involved in the control of the size of the imaginal discs in Drosophila by inhibiting cell proliferation and promoting apoptosis. Consistent with this, YAP1 (Yes-associated Protein 1) is a protein negatively regulated by Hippo signaling and its inactivation in KO mice impairs cardiomyocyte proliferation during the embryonic development, whereas its overexpression promotes cardiomyocyte proliferation [48, 49]. YAP1 was shown to increase the activity of the pro-growth IGF and canonical Wnt signaling [48]. Besides this role in cardiac development, YAP1 is also involved in the neonatal mouse heart regeneration after MI [50, 51]. The deletion of the YAP1 gene specifically in the heart impairs cardiac regeneration of neonatal mice after MI, resulting in a deficiency of healthy myocardial tissue throughout the left ventricular free wall at 26 days after MI and an extensive fibrotic infarct. Furthermore, in 7-days old neonatal transgenic mice expressing a constitutively active YAP1 under the control of the cardiomyocyte-specific α-MHC promoter, MI is followed by a complete regeneration of the heart with no fibrosis at P28, compared with the wild-type mice. Thus, overexpression of the YAP1 gene extends the temporal window of regeneration in mice [50].
Neonatal heart regeneration in mice proves that mammals have the potential to regenerate their hearts. The study of the genetic and cellular mechanisms of heart regeneration in neonatal mice should help us to understand why adult mammals lose this ability and allow us to restore this ability in injured hearts from adult mammals. The direct comparison between neonates and adults will reveal the cellular and molecular mechanisms necessary for cardiac regeneration and why these capability are lost in adult mouse compared with the neonatal mouse.
Cryoinjury models to study heart regeneration
Different heart injury models have been used to study regeneration in adult zebrafish and neonatal mice (Table 1).
Ventricular resection of zebrafish and neonatal mouse hearts removes 10 %–20 % of healthy heart tissues, but the injury displays a different pathogenesis compared with MIs. Cryoinjury makes use of a probe that has been pre-chilled in liquid nitrogen to damage the hearts by freezing and thawing. This induces the loss of cardiac tissue by apoptosis and necrosis and the appearance of a collagen-rich scar, exactly like after MI in mammals, in adult zebrafish [52–54]. Interestingly, different results are reported by these three papers. Fibrin clots but minimal to no collagen scars form in the early stages of cryoinjury as reported by Schnabel et al [54]. Collagen scars form at 7 days postcryoinjury (dpc) then gradually are resolved by 30 dpc as cardiomyocyte proliferation starts from 4 dpc and lasts until 21 dpc (Fig. 1) [52]. Fibrotic scar formation reaches the maximum level by 21 dpc in the report by Gonzalez-Rosa et al [53], and then gets resolved by 130 dpc (Fig. 1). Nevertheless, unlike mammals where the fibrotic scar remains, in zebrafish the scar is either progressively removed and replaced by new heart tissue [52, 53] or does not form [54]. These reports suggested that perhaps different responses might be activated by injuries of different severities. To further investigate the differential regeneration responses, we established a cryoinjury model in neonatal mouse hearts where the severity of injuries can be better controlled (Darehzereshki et al., unpublished data). Indeed, we observed differential levels of scarring at 21, 60, and 120 dpc after nontransmural and transmural injuries done on 1-day old neonatal mice (Darehzereshki et al., unpublished data). Our results suggest that the nature of heart injuries is an important factor to evaluate when considering different strategies of heart repair.

Summary of events during regeneration following cryoinjury in zebrafish. In Chablais et al. [52] cryoinjury of about 20 % of the ventricle induces massive cardiac cell death in the injury area and inflammation at 1-day postcryoinjury (dpc). At 4 dpc myocardium is activated and cardiomyocytes proliferate. At 7 dpc, the dead cells are replaced by a collagen-rich fibrotic scar and a fibrin layer takes place along the inner side of the injury area. At 14 dpc the fibrosis resolves gradually as the cardiomyocytes renewal. At 21 dpc the fibrin layer is strongly reduced. At 30 dpc the fibrosis is almost resolved and at 60 dpc the ventricle is completely regenerated with a normal systolic function. In Gonzalez-Rosa et al. [53] the processes from 1–14 dpc are similar to what are described in Chablais et al. Cryoinjury induces a massive cell death at 1 dpc. At 3 dpc the myocardium is activated and proliferating and a collagen-rich fibrotic scar replaced the dead cells at 7 dpc. At 21 dpc only a few cells remain proliferating in myocardium and the scar is at a more luminal position. However, unlike Chablais’ model, the fibrosis starts resolving from 21 dpc and still persists at 90 dpc. At 130 dpc the heart has fully regenerated, although adopted a rounder shape, leading to limited contractility of the ventral part of the ventricle. A atrium, BA bulbus arteriosus, V ventricle.
Treatment
Developing potential regenerative medicine based treatment for coronary heart diseases and heart failure is still at its infancy. Enhancing neovascularization is important since it allows the injured heart to maintain its remaining cardiac functions. However, heart failure cannot be cured without the replacement of lost cardiomyocytes. Three different strategies have been used to replace lost cardiomyocytes, inducing cardiomyocyte proliferation and enhancing endogenous cardiac regenerative capacity of mammalian hearts; transplanting ectopic cells and direct differentiation of fibroblasts to cardiomyocytes [55–59]. The cell-based and transdifferentiation strategies will be discussed elsewhere.
Inducing cardiomyocyte proliferation and enhancing endogenous capacity of heart regeneration
Cross species comparison has revealed differences in cardiomyocyte responses between zebrafish, neonatal mice, and adult mice after heart injuries. Based on what we have learned from models organisms that can regenerate their hearts naturally after cardiac injuries, a blueprint can be drawn for designing therapeutic strategies via heart regeneration. However, several issues should be kept in mind when considering heart regeneration in model organisms.
Many zebrafish organs have an outstanding capacity to regenerate following injury [60]. Zebrafish retain their ability to regenerate hearts after different kinds of injuries from the embryo to adult. The question is: why do zebrafish retain this regenerative ability until adulthood while mammals appear to lose it after the postnatal stages?
Zebrafish continue to grow throughout their life [61], at first quickly until they reach sexual maturation age (about 2–3 months), and then much more slowly but constantly from adult to death. Thus, the mechanisms involving rapid cell proliferation (such as regeneration) can be reactivated throughout the lifetime of the zebrafish, unlike in mammals wherein growth ends at the adulthood. Interestingly, zebrafish grows faster in low fish density tanks, and under these conditions, cardiomyocyte proliferation is also increased [62]. This fast growth induces expressions of embryonic epicardial markers raldh2 (aldh1a2) and tbx18 in adult epicardial tissue [62]. Another study from the same group showed that the mechanical stress during juvenile heart growth induces expressions of the cardiac stress and injury markers nppa and nppb [63]. This suggests that zebrafish hearts use the same mechanisms for fast heart growth in juveniles and responses to heart injury.
A considerable number of genes and pathways are shared by zebrafish and neonatal mice during embryonic cardiogenesis and regeneration after injury and notably those involved in cardiomyocyte proliferation, the critical step of heart regeneration (Table 2). The main difference between the two models, which makes the comparison relevant between each other, is that the mouse loses its regenerative ability quickly after birth, whereas zebrafish does not. So the effort should be brought to study and understand the mechanisms inactivating this regenerative ability in mice and how it may be reversed. For example, the p38 MAPK pathway regulates negatively the cardiomyocyte proliferation in both normal adult zebrafish and mouse [22, 64]. However, after injury this pathway is switched off only in zebrafish, allowing heart regeneration to proceed [22]. Similarly, the cardiomyocyte proliferation inhibitor miR-133 is downregulated during injury in zebrafish [25]. In mice, miR-133 is involved in the heart size control [65] and its suppression promotes cardiac hypertrophy [65] and cardiomyocyte proliferation [66], indicating that miR-133 negatively regulates cardiomyocyte proliferation like in zebrafish. It remains to be investigated if, unlike in the zebrafish, the expression of miR-133 is maintained in mouse heart following injury and if this prevents these cardiomyocytes from proliferating.
Conclusions
The study of heart regeneration in zebrafish and rodents taught us three main lessons: (1) cardiomyocyte proliferation is the key element; (2) zebrafish and neonatal mouse share a lot of similar molecular and cellular mechanisms; and (3) regeneration involves the re-expression of cardiac developmental genes. The data collected has given us a better understanding of the molecular and cellular mechanisms of heart regeneration and it is now possible to induce and promote heart regeneration in infarcted adult rodents. A promising research field is reprogramming cardiac fibroblasts into cardiomyocyte like cells. Using cardiogenic markers identified in studies of zebrafish and neonatal mouse models, cardiac fibroblasts reprogramming into cardiomyocytes can be induced in vivo in infarcted heart from adult mice to enhance cardiac functions. Unfortunately it is not possible to promote a full and permanent recovery of cardiac function yet and this remains the continuing aim of the regenerative medicine field. Perhaps strategies combining reprogramming and enhancing endogenous regenerative capacity using factors identified from zebrafish, newt and neonatal mice can further improve the cardiac regeneration in mammals.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance
Forouzanfar MH, Moran AE, Flaxman AD, Roth G, Mensah GA, Ezzati M, et al. Assessing the global burden of ischemic heart disease, part 2: analytic methods and estimates of the global epidemiology of ischemic heart disease in 2010. Glob Heart. 2012;7(4):331–42.
Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, et al. Executive summary: heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation. 2013;127(1):143–52.
Murry CE, Reinecke H, Pabon LM. Regeneration gaps: observations on stem cells and cardiac repair. J Am Coll Cardiol. 2006;47(9):1777–85.
Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324(5923):98–102.
Abdelwahid E, Siminiak T, Guarita-Souza LC, Teixeira de Carvalho KA, Gallo P, Shim W, et al. Stem cell therapy in heart diseases: a review of selected new perspectives, practical considerations and clinical applications. Curr Cardiol Rev. 2011;7(3):201–12.
Choi WY, Poss KD. Cardiac regeneration. Curr Top Dev Biol. 2012;100:319–44.
Laflamme MA, Zbinden S, Epstein SE, Murry CE. Cell-based therapy for myocardial ischemia and infarction: pathophysiological mechanisms. Annu Rev Pathol. 2007;2:307–39.
Marban E, Cingolani E. Heart to heart: cardiospheres for myocardial regeneration. Heart Rhythm. 2012;9(10):1727–31.
Kikuchi K, Poss KD. Cardiac regenerative capacity and mechanisms. Annu Rev Cell Dev Biol. 2012;28:719–41.
Lien CL, Harrison MR, Tuan TL, Starnes VA. Heart repair and regeneration: recent insights from zebrafish studies. Wound Repair Regen. 2012;20(5):638–46.
Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298(5601):2188–90.
Raya A, Koth CM, Büscher D, Kawakami Y, Itoh T, Raya RM, et al. Activation of Notch signaling pathway precedes heart regeneration in zebrafish. Proc Natl Acad Sci U S A. 2003;100 Suppl 1:11889–95.
Kikuchi K, Holdway JE, Werdich AA, Anderson RM, Fang Y, Egnaczyk GF, et al. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature. 2010;464(7288):601–5.
Jopling C, Sleep E, Raya M, Martí M, Raya A, Izpisúa Belmonte JC. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 2010;464(7288):606–9. These two papers demonstrate unambiguously that the cardiomyocytes regenerated after injury come from pre-existing cardiomyocytes and not from another cell source.
Zhang R, Han P, Yang H, Ouyang K, Lee D, Lin YF, et al. In vivo cardiac reprogramming contributes to zebrafish heart regeneration. Nature. 2013;498(7455):497–501. This paper shows that atrial cardiomyocytes are able to transdifferentiate into ventricular cardiomyocytes and participate in ventricle regeneration in fish embryos and larvae.
Choi WY, Gemberling M, Wang J, Holdway JE, Shen MC, Karlstrom RO, et al. In vivo monitoring of cardiomyocyte proliferation to identify chemical modifiers of heart regeneration. Development. 2013;140(3):660–6.
Chablais F, Jazwinska A. The regenerative capacity of the zebrafish heart is dependent on TGFbeta signaling. Development. 2012;139(11):1921–30.
Lien CL, Schebesta M, Makino S, Weber GJ, Keating MT. Gene expression analysis of zebrafish heart regeneration. PLoS Biol. 2006;4(8):e260.
Kamei H, Ding Y, Kajimura S, Wells M, Chiang P, Duan C. Role of IGF signaling in catch-up growth and accelerated temporal development in zebrafish embryos in response to oxygen availability. Development. 2011;138(4):777–86.
Huang Y, Harrison MR, Osorio A, Kim J, Baugh A, Duan C, et al. Igf signaling is required for cardiomyocyte proliferation during zebrafish heart development and regeneration. PLoS One. 2013;8(6):e67266.
Fang Y, Gupta V, Karra R, Holdway JE, Kikuchi K, Poss KD. Translational profiling of cardiomyocytes identifies an early Jak1/Stat3 injury response required for zebrafish heart regeneration. Proc Natl Acad Sci U S A. 2013;110(33):13416–21.
Jopling C, Suñe G, Morera C, Izpisua Belmonte JC. p38alpha MAPK regulates myocardial regeneration in zebrafish. Cell Cycle. 2012;11(6):1195–201.
Jopling C, Suñé G, Faucherre A, Fabregat C, Izpisua Belmonte JC. Hypoxia induces myocardial regeneration in zebrafish. Circulation. 2012;126(25):3017–27.
Jürgensen JS, Rosenberger C, Wiesener MS, Warnecke C, Hörstrup JH, Gräfe M, et al. Persistent induction of HIF-1alpha and -2alpha in cardiomyocytes and stromal cells of ischemic myocardium. FASEB J. 2004;18(12):1415–7.
Yin VP, Lepilina A, Smith A, Poss KD. Regulation of zebrafish heart regeneration by miR-133. Dev Biol. 2012;365(2):319–27.
Lepilina A, Coon AN, Kikuchi K, Holdway JE, Roberts RW, Burns CG, et al. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell. 2006;127(3):607–19.
Kim J, Wu Q, Zhang Y, Wiens KM, Huang Y, Rubin N, et al. PDGF signaling is required for epicardial function and blood vessel formation in regenerating zebrafish hearts. Proc Natl Acad Sci U S A. 2010;107(40):17206–10.
Kikuchi K, Gupta V, Wang J, Holdway JE, Wills AA, Fang Y, et al. tcf21+ epicardial cells adopt non-myocardial fates during zebrafish heart development and regeneration. Development. 2011;138(14):2895–902.
Itou J, Oishi I, Kawakami H, Glass TJ, Richter J, Johnson A, et al. Migration of cardiomyocytes is essential for heart regeneration in zebrafish. Development. 2012;139(22):4133–42.
Kikuchi K, Holdway JE, Major RJ, Blum N, Dahn RD, Begemann G, et al. Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev Cell. 2011;20(3):397–404.
Brockes JP, Kumar A. Plasticity and reprogramming of differentiated cells in amphibian regeneration. Nat Rev Mol Cell Biol. 2002;3(8):566–74.
Borchardt T, Braun T. Cardiovascular regeneration in non-mammalian model systems: what are the differences between newts and man? Thromb Haemost. 2007;98(2):311–8.
Witman N, Murtuza B, Davis B, Arner A, Morrison JI. Recapitulation of developmental cardiogenesis governs the morphological and functional regeneration of adult newt hearts following injury. Dev Biol. 2011;354(1):67–76.
Looso M, Preussner J, Sousounis K, Bruckskotten M, Michel CS, Lignelli E, et al. A de novo assembly of the newt transcriptome combined with proteomic validation identifies new protein families expressed during tissue regeneration. Genome Biol. 2013;14(2):R16.
Mercer SE, Odelberg SJ, Simon HG. A dynamic spatiotemporal extracellular matrix facilitates epicardial-mediated vertebrate heart regeneration. Dev Biol. 2013;382(2):457–69.
Piatkowski T, Mühlfeld C, Borchardt T, Braun T. Reconstitution of the myocardium in regenerating newt hearts is preceded by transient deposition of extracellular matrix components. Stem Cells Dev. 2013;22(13):1921–31.
Wang J, Karra R, Dickson AL, Poss KD. Fibronectin is deposited by injuryactivated epicardial cells and is necessary for zebrafish heart regeneration. Dev Biol. 2013;382(2):427–35.
Soonpaa MH, Field LJ. Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res. 1998;83(1):15–26.
Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, et al. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med. 2007;13(8):970–4.
Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493(7432):433–6.
Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331(6020):1078–80. This paper shows for the first time that neonatal mouse is able to regenerate its heart after injury, showing that mammals have the ability of cardiac regeneration.
Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, et al. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci U S A. 2013;110(1):187–92.
Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol. 1996;28(8):1737–46.
Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, et al. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature. 2013;497(7448):249–53.
Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492(7429):376–81.
Chen J, Huang ZP, Seok HY, Ding J, Kataoka M, Zhang Z, et al. mir-17–92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res. 2013;112(12):1557–66.
Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332(6028):458–61.
von Gise A, Lin Z, Schlegelmilch K, Honor LB, Pan GM, Buck JN, et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci U S A. 2012;109(7):2394–9.
Xin M, Kim Y, Sutherland LB, Qi X, McAnally J, Schwartz RJ, et al. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci Signal. 2011;4(196):ra70.
Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci U S A. 2013;110(34):13839–44.
Del Re DP, Yang Y, Nakano N, Cho J, Zhai P, Yamamoto T, et al. Yesassociated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J Biol Chem. 2013;288(6):3977–88.
Chablais F, Veit J, Rainer G, Jaźwińska A. The zebrafish heart regenerates after cryoinjury-induced myocardial infarction. BMC Dev Biol. 2011;11:21.
González-Rosa JM, Martín V, Peralta M, Torres M, Mercader N. Extensive scar formation and regression during heart regeneration after cryoinjury in zebrafish. Development. 2011;138(9):1663–74.
Schnabel K, Wu CC, Kurth T, Weidinger G. Regeneration of cryoinjury induced necrotic heart lesions in zebrafish is associated with epicardial activation and cardiomyocyte proliferation. PLoS One. 2011;6(4):18503.
Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142(3):375–86.
Nam YJ, Song K, Luo X, Daniel E, Lambeth K, West K, et al. Reprogramming of human fibroblasts toward a cardiac fate. Proc Natl Acad Sci U S A. 2013;110(14):5588–93.
Nam YJ, Song K, Olson EN. Heart repair by cardiac reprogramming. Nat Med. 2013;19(4):413–5.
Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, et al. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012;485(7400):593–8.
Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature. 2012;485(7400):599–604.
Gemberling M, Bailey TJ, Hyde DR, Poss KD. The zebrafish as a model for complex tissue regeneration. Trends Genet. 2013;29(11):611–20.
Tsai SB, Tucci V, Uchiyama J, Fabian NJ, Lin MC, Bayliss PE, et al. Differential effects of genotoxic stress on both concurrent body growth and gradual senescence in the adult zebrafish. Aging Cell. 2007;6(2):209–24.
Wills AA, Holdway JE, Major RJ, Poss KD. Regulated addition of new myocardial and epicardial cells fosters homeostatic cardiac growth and maintenance in adult zebrafish. Development. 2008;135(1):183–92.
Gupta V, Gemberling M, Karra R, Rosenfeld GE, Evans T, Poss KD. An injury-responsive gata4 program shapes the zebrafish cardiac ventricle. Curr Biol. 2013;23(13):1221–7.
Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, et al. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19(10):1175–87.
Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13(5):613–8.
Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, et al. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008;22(23):3242–54.
Acknowledgements
This work was done with the support of the California Institute of Regenerative Medicine (TG2-01168), and NIH (NHLBI).
Compliance with Ethics Guidelines
Conflict of Interest
Dr. Laurent Gamba, Dr. Michael Harrison, and Dr. Ching-Ling Lien each declare no potential conflicts of interest relevant to this article.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Regenerative Medicine and Stem-cell Therapy
Rights and permissions
About this article
Cite this article
Gamba, L., Harrison, M. & Lien, CL. Cardiac Regeneration in Model Organisms. Curr Treat Options Cardio Med 16, 288 (2014). https://doi.org/10.1007/s11936-013-0288-8
Published:
DOI: https://doi.org/10.1007/s11936-013-0288-8