
Abstract
Purpose of review
Dermatomyositis is an idiopathic inflammatory myopathy with a variety of systemic and cutaneous manifestations. The myositis-specific autoantibodies (MSAs) are associated with phenotypic features and provide a tool for sub-classification of dermatomyositis patients. This review focuses on recent work characterizing the clinical features that accompany the MSAs in dermatomyositis.
Recent findings
There is increasing recognition of the distinct clinical and pathological phenotypes associated with each MSA. Most of these features display considerable overlap between MSA groups. Despite this, there are notable differences between the typical combinations of cutaneous and systemic manifestations, response to therapy, prognosis, and disease sequelae that define each dermatomyositis MSA group.
Summary
The MSAs may ultimately improve diagnosis and sub-classification of dermatomyositis patients. However, more work is needed to understand the pathologic basis for much of the heterogeneity found within these subgroups.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
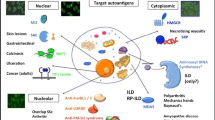
Dermatomyositis (DM) is an idiopathic inflammatory myopathy with a range of systemic and cutaneous manifestations that leads to significant variability in clinical presentation. The original DM classification criteria were limited to muscle involvement and a limited number of cutaneous manifestations [1, 2], but the significant heterogeneity among DM patients is now recognized and amyopathic DM is a defined subgroup in the 2017 EULAR/ACR DM criteria [3]. Not only are there a variety of cutaneous findings [4], but pulmonary disease, joint disease, muscle disease, and malignancy are also all variably present in DM patients. The inconsistent clinical presentation creates both a diagnostic and patient management challenge for clinicians. By definition, myositis-specific autoantibodies (MSAs) are found only in patients with idiopathic inflammatory myopathies (dermatomyositis, polymyositis, inclusion body myositis, and necrotizing myopathies). Interestingly, most MSAs found in DM are not found in other inflammatory myopathies and, furthermore, are each associated with characteristic clinical features along the DM spectrum (Fig. 1a). Recent work in larger DM cohorts has helped to characterize these autoantibodies and their associated phenotypes. Recognition of the phenotypes associated with MSA subgroups can guide clinical care and ensure that patients at increased risk of a more severe cutaneous or systemic disease course have appropriate treatment and follow-up.


Myositis-specific autoantibody clinical phenotypes. a Clinical phenotypes associated with myositis-specific autoantibodies in adult dermatomyositis. b Cuticular overgrowth and hemorrhage in an anti-Mi-2 patient. c Alopecia in an anti-MDA-5 patient. d Calcinosis cutis in an anti-MDA-5 patient. e Peripheral edema in an anti-NXP-2 patient. Photo credit to Andy Mammen, MD. f V-neck sign in an anti-TIF-1γ patient. g Red-on-white in an anti-TIF-1γ patient. h Mechanics hands in an anti-Jo-1 patient
Anti-Mi-2 Autoantibodies
Mi-2 antigen is part of the nucleosome-remodeling deacetylase complex (Table 1), and anti-Mi-2 autoantibodies were first reported as a MSA in DM by Targoff et al. in 1985 [5]. The prevalence ranges from 2 to 38% within adult DM populations and 4 to 10% within juvenile dermatomyositis (JDM) populations [6,7,8,9,10,11,12,13,14]. The highest prevalence in adults was reported in India and the lowest in Japan, while a study of JDM patients suggests a higher prevalence among Hispanic patients [6, 7, 13]. Various HLA associations have identified DRB1*0302 as the primary allelic risk factor among African Americans and DRB1*0701/DQA1*0201 as the primary allelic risk factors among European Americans [15,16,17,18]. DRB1*0302 and DRB1*0701 have a conserved amino acid sequence that may be contributing to the preferential binding of the Mi-2 peptide [16]. A large cohort in the USA identified a positive association between UV radiation and the expression of anti-Mi-2 autoantibodies within women (OR 17.3, CI 1.8–162.4, p = 0.012), and more recently, a Brazilian cohort found a positive association between anti-Mi-2 and photosensitivity [11, 19]. However, a recent study from Mexico found a large difference between the percentage of DM patients with anti-Mi-2 autoantibodies in two different cities of similar latitude, suggesting that UV irradiation is not the only risk factor for developing anti-Mi-2 autoantibodies [20].
Anti-Mi-2-positive DM patients have significantly decreased risk of interstitial lung disease (ILD) compared to anti-Mi-2-negative patients and have low rates of malignancy [6, 13, 21]. Higher creatine kinase (CK) and lactate dehydrogenase levels have been observed among both juvenile and adult DM patients [7, 20]. Initial work investigating muscle disease pathogenesis in these patients demonstrated Mi-2 overexpression in regenerating human DM myofibers and accelerated myoblast differentiation in vitro after suppression of Mi-2 expression, leading to the hypothesis that Mi-2 inhibits myoblast differentiation through muscle repair [22]. More recently, muscle biopsies from anti-Mi-2-positive DM patients have been shown to have significantly more inflammation compared to other autoantibody subgroups, and those from JDM patients reflect more severe disease [23•, 24•].
The cutaneous manifestations commonly found in anti-Mi-2 DM include Gottron’s sign (violaceous erythematous patches and thin scaly plaques over the knee and elbow extensor surfaces), heliotrope rash, V-neck sign, Shawl sign, and cuticular overgrowth (Fig. 1b) [14, 20]. An increased prevalence of punctate perionychium hemorrhages has been described in Japanese anti-Mi-2-positive DM patients compared to those with anti-MDA-5 and anti-TIF-1γ autoantibodies [13]. In JDM Gottron’s papules, heliotrope rash and malar rash are frequently seen [7].
Anti-Mi-2 patients have a favorable prognosis with good response to treatment, but may have significant risk of recurrence [13, 14, 25]. Most recently, Rituximab has emerged as a favorable treatment option for anti-Mi-2 autoantibody-positive patients with data collected through the Rituximab in Myositis (RIM) trial demonstrating significant improvement in interferon chemokine scores and clinical disease [26, 27].
Anti-MDA-5 Autoantibodies
MDA-5, which encodes a cytosolic double-stranded RNA sensor, is one of the three retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) (Table 1). As a group, the RLRs have a critical role in innate immunity and recognition of viral RNAs [28]. Autoantibodies to MDA-5 are more prevalent in Asian (11–57%) than Caucasian populations (0–13%) [13, 28,29,30,31,32,33]. A recent study found an even higher prevalence in Chinese (57%) vs. Japanese (24%) patients [30]. Anti-MDA-5 autoantibodies are associated with DRB1*04:01 and DRB1*12:02 alleles in Japanese patients [34]. A similar genetic analysis study looking at Chinese patients found a higher frequency of the DRB1*12:01 allele in anti-MDA-5-positive patients and identified DRB1*09:01 as a poor prognostic factor [35]. In the JDM population, the reported prevalence is 7–12% [23•, 36].
Muscle biopsies from adult anti-MDA-5-positive patients often lack classic DM findings of perifascicular fiber atrophy, major histocompatibility complex class I expression, capillary loss, and tubuloreticular inclusions, but, uniquely, have nitric oxide synthase 2-positive muscle fibers [37]. In the JDM population, muscle biopsies are less likely to have the destructive histological changes observed in biopsies from non-anti-MDA-5-positive JDM patients and have less overall histopathologic severity compared to other autoantibody groups [23•, 36]. Neutrophil extracellular traps (NETs) are associated with a worse overall outcome in ILD among DM patients and serum cfDNA can be used as a proxy for NET formation [38,39,40]. Research investigating the role of NETs in anti-MDA-5-positive DM patients with ILD found that serum cfDNA levels were elevated in anti-MDA-5 autoantibody-positive patients and even higher in those with rapidly progressive ILD (RP-ILD) [38]. Finally, autoantibodies against splicing factor proline/glutamine-rich protein (SFPQ), which has a role in the innate immune system, have been exclusively identified in the anti-MDA-5-positive subset of DM patients [41]. Differences in detection timing of the anti-SFPQ autoantibody suggest a seasonal pattern of disease with 77% of those in the early-detection group diagnosed between August and October and 57% of those in the delayed-detection group diagnosed between January and March [41].
The use of anti-MDA-5 titers in disease monitoring and prognosis is an ongoing area of research. Significantly lower baseline titers in survivors vs. non-survivors and a meaningful decrease in post-treatment titers are reported by some, but not all, authors [42•, 43,44,45]. One study proposed using duration of elapsed time before therapy or anti-MDA-5 isotype as a more accurate predictor of outcome [46]. Elevated serum ferritin levels are uniquely associated with this autoantibody group, and their elevation in patients with RP-ILD may be a useful alternative to anti-MDA-5 titer levels when evaluating treatment response and pulmonary disease status [28, 45,46,47].
The rapidly progressive subtype of ILD that is associated with high mortality is particularly predominant in Japanese anti-MDA-5-positive populations; however, there have also been reports of anti-MDA-5-positive DM patients presenting with aggressive ILD in the USA, China, and Europe [13, 28, 29, 48, 49]. A recent meta-analysis, including both Asian and Caucasian patients, demonstrated a significant association between the anti-MDA-5 autoantibody and ILD with an OR of 16.47 (95% CI 10.16–26.70, p < 0.001) [50]. Mortality in anti-MDA-5-positive patients is largely attributed to ILD with mortality rates as high as 38% in pooled studies, and 5-year survival rates as low as 56% [13, 48]. Studies in Asian cohorts report improved ILD outcomes in patients treated early with an aggressive regimen of corticosteroids, cyclophosphamide, and calcineurin inhibitors [51,52,53].
Numerous studies have reported a high prevalence of inflammatory arthritis among anti-MDA-5-positive patients with 69, 82, and 86% of anti-MDA5 patients being affected in Japanese adult, US adult, and European JDM cohorts, respectively [28, 32, 36]. Anti-MDA-5-positive DM patients have been misdiagnosed with psoriatic arthritis and often have a similar clinical presentation to rheumatoid arthritis [32, 54]. Although there is a large subset of anti-MDA-5-positive patients with myositis, both Japanese and US studies have consistently identified a significantly higher number of anti-MDA-5 patients with clinically amyopathic disease (50–77%) compared to other autoantibodies [13, 32, 33, 55].
Other less common clinical associations with anti-MDA5 autoantibodies have recently been described. A significant number of anti-MDA-5-positive patients (35–74%) experience fever compared to anti-MDA-5 negative patients, and anti-MDA-5 DM has presented as fever of unknown origin [13, 32, 47, 56]. Severe myocardial dysfunction leading to third degree heart block has also been observed in anti-MDA-5 patients in the absence of RP-ILD or pre-existing cardiovascular disease [57]. Finally, 7% of patients who developed chronic graft-vs.-host disease following allogenic stem cell transplantation (AHSCT) tested positive for anti-MDA-5 autoantibodies [58]. A majority developed ILD and half developed DM cutaneous manifestations, suggesting that post-AHSCT patients who develop either pulmonary or atypical cutaneous symptoms be tested for anti-MDA-5 autoantibodies [58].
The cutaneous phenotype is often particularly severe in anti-MDA-5 patients. Cutaneous manifestations include alopecia (Fig. 1c), Gottron’s sign and papules, heliotrope rash, and mechanics hands, as well as cutaneous ulcers (hyperkeratotic pulp lesions, lateral nailfold ulcers, ulceration of Gottron’s papules, and ulcerations over elbow and knee extensor surfaces) in both the adult and juvenile DM populations [13, 32, 33, 36, 48]. Skin findings also include painful, erythematous papules and macules on the palmar surfaces of the metacarpophalangeal and interphalangeal joints, and a higher prevalence of oral erosions compared to anti-MDA5 autoantibody-negative DM patients [33, 48]. Calcinosis cutis (Fig. 1d) has been associated with fingertip ulcers in some patients and may be driven by vascular injury [59,60,61]. A recent study suggests that clinical remission of skin disease in adults is less likely in this autoantibody group [62]. In the JDM population, the data on a skin disease course is conflicting—one study suggested that these patients have a prolonged disease course, while another indicated that anti-MDA-5-positive patients are more likely to be in remission at 2 years than their negative counterparts [23•, 36].
Anti-NXP-2 Autoantibodies
The nuclear matrix protein (NXP-2 or MORC3) autoantibody was initially identified as anti-MJ in the JDM population and has a role in p53 regulation (Table 1) [63, 64]. Its prevalence within DM varies and appears to be higher in USA (14–25%) than Japanese (2–5%) populations [65,66,67,68, 69•]. In JDM patients, the reported prevalence is 20–25% [7, 70]. The variability in prevalence among adults may be attributed to differences among study populations and/or antibody assays.
With respect to disease pathology, recent investigations have demonstrated that there is decreased inflammation in anti-NXP-2-positive muscle biopsies compared to those from anti-NXP-2-negative patients (0 vs. 28%, p = 0.01), and increased clinical muscle atrophy (29 vs. 11%, p = 0.02) [24•, 69•].
Recent studies have associated anti-NXP-2 autoantibodies with internal malignancy. In a combined cohort study from East and West Coast tertiary referral centers in the USA, 14% of patients had an internal malignancy and a majority of patients with malignancy had either anti-NXP-2 or anti-TIF-1γ autoantibodies [68]. One study found that anti-NXP-2 autoantibody-positive patients had a 3.68-fold increased risk of cancer (95% CI 1.2–8.6) compared to the same age and gender patients in the general population, while the other found that the trend for increased prevalence of malignancy in anti-NXP-2 autoantibody-positive patients did not reach statistical significance [67, 69•].
In multiple cohorts, anti-NXP-2 patients have been shown to have frequent and severe muscle weakness, myalgias, and dysphagia [65,66,67]. This autoantibody has also been associated with distal weakness that is atypical for DM [67, 69•]. Data from two US cohorts demonstrated an increased prevalence of peripheral edema (Fig. 1e) in anti-NXP-2-positive vs. anti-NXP-2-negative patients (35–36 vs. 11–19%) [67, 69•]. ILD is uncommon in anti-NXP-2 DM compared to the non-NXP-2 population [65, 66, 69•]. In JDM patients, the anti-NXP-2 phenotype was defined earlier with contractures and atrophy, (proximal) muscle weakness, and increased muscle cramps [7, 70, 71]. JDM patients with anti-NXP-2 autoantibodies tend to be sicker with both frequent hospitalizations and poor functional status over a 2-year follow-up period [7, 70]. Calcinosis cutis is a distinguishing feature of anti-NXP-2 in both adult and JDM patients, and multiple studies have showed an increased prevalence in this population [61, 67, 69•, 71,72,73].
In JDM, Oddis et al. initially described the association between intestinal vasculitis and anti-NXP-2 and later studies have also demonstrated an increased frequency of gastrointestinal bleeding and ulcers compared to other autoantibody groups [7, 63]. It is likely that a similar association exists in adults, as we have seen a similar association in several patients.
Anti-TIF-1γ Autoantibodies
In 2006, anti-155/140 autoantibodies were discovered in two independent groups of adult DM patients and these autoantibodies were ultimately shown to be part of the TIF1 family of homologous proteins of which TIF-1γ (TRIM33) is most commonly targeted in DM patients (Table 1) [74]. Depending on the model system, TIF-1γ has been shown to serve as a regulator of transcription, a tumor suppressor, a mediator of DNA damage repair, and an E3 ligase that modulates TGF-ß signaling [75, 76]. Anti-TIF-1γ autoantibodies are particularly prevalent in Caucasian (38–41%) compared to Japanese patients (7–14%), and in the JDM literature, 80% of patients were identified as Caucasian with an overall prevalence of 20–32% [7, 13, 23•, 74, 77, 78]. Using immunoprecipitation assays, we have identified a group of patients who are positive for both anti-Mi-2 and anti-TIF-1γ autoantibodies [77]. Whether patients truly develop two unique autoantibodies or if the double-positive results are the consequence of anti-Mi-2 autoantibodies weakly binding TIF-1γ, as demonstrated in a recent Japanese study, has not yet been fully explained [79].
The relationship between malignancy and anti-TIF-1γ autoantibodies has been well established, with a prevalence ranging from 38 to 71% in Japanese seropositive patients [13, 74, 80]. The prevalence of cancer in the US anti-TIF-1γ population is comparatively reduced although there is still a trend for increased malignancy in this patient population [77, 78]. Recent efforts to understand the pathogenesis of anti-TIF-1γ-positive DM and the relationship with malignancy suggest that the association between malignancy and anti-TIF-1γ autoantibodies may be explained by the presence of somatic mutations in tumor TIF1 family genes [81••].
Anti-TIF-1γ autoantibodies are negatively associated with Raynaud’s phenomenon (RP), arthralgias, and ILD [7, 74, 77]. Although the majority of anti-TIF-1γ-positive patients have muscle disease, they also have lower mean levels of both aldolase and CK compared to patients without this autoantibody [7, 77, 82]. Muscle biopsies from DM patients express TIF-1γ at low levels in the peripheral nuclei of morphologically normal muscle fibers and high levels in the myonuclei/perifascicular muscle cells, and skin biopsies from DM patients suggest that TIF-1γ expression correlates with cellular stress [83•, 84]. Together these results indicate that persistently high levels of autoantigens in regenerating or stressed tissue may be providing an ongoing autoantigen source to drive the autoimmune response [83•, 84]. Examination of muscle biopsies has also revealed cytochrome oxidase-deficient fibers indicating an association of mitochondrial dysfunction with anti-TIF-1γ autoantibodies [24•].
Cutaneous manifestations in anti-TIF-1γ autoantibody patients typically follow the classic pattern, which includes V-neck sign (Fig. 1f), and are often severe and chronic in both adult and juvenile DM populations [7, 74, 77]. Despite the severity of cutaneous disease activity in these patients, there is a decreased risk of calcinosis cutis, at least in adults [7, 74, 77]. In addition to the classic phenotype, a number of unique yet characteristic skin lesions have been recently identified within this autoantibody group: psoriasis-like lesions, hyperkeratotic small papules on the palmar and digital flexor surfaces, red-on-white lesions (Fig. 1g), and an ovoid palatal patch in adults [77, 85]. The ovoid palatal patch demonstrates interface dermatitis and may indicate an increased risk of malignancy [85]. In the JDM population, cuticular overgrowth is also predominant [7].
Anti-SAE1/2 Autoantibodies
Anti-SAE1/2 autoantibodies target the A and B subunits of small ubiquitin-like modifier 1 (SUMO-1) activating enzyme (Table 1) and were identified as a novel MSA in DM patients in 2007 [86]. The presence of anti-SAE1/2 autoantibodies is extremely uncommon in the JDM population and characteristic clinical features among adults with this autoantibody have not yet emerged. Studies have identified a variable prevalence of anti-SAE1/2 autoantibodies ranging from 1 to 3% in Asian populations and 5–10% in European populations [31, 87,88,89,90,91].
Data from relatively small studies are conflicting regarding the clinical phenotype of anti-SAE1/2-positive patients. There is a subset of patients who initially present with cutaneous disease and progress to develop myositis, while another subset present with skin and muscle disease simultaneously [31, 87, 88, 90, 91]. Dysphagia is a frequent finding among patients with muscle disease and was significantly more common in a Chinese study comparing anti-SAE1/2 autoantibody-positive vs. anti-SAE1/2 autoantibody-negative patients [88,89,90]. Cutaneous manifestations in DM include the classic findings of heliotrope rash, Gottron’s sign, and papules, as well as a newly described dark red/violaceous rash presenting in subsets of patients from China, Italy, and Hungary [31, 87, 89,90,91]. Persistent cutaneous ulcers were also observed in a group of patients and a study from the UK identified a significantly higher frequency of periungual changes in anti-SAE1/2-positive vs. anti-SAE1/2-negative patients [31, 89, 90, 92].
Interstitial lung disease in anti-SAE1/2-positive patients is usually mild, with few patients experiencing respiratory symptoms despite having evidence of ILD with imaging [87,88,89,90]. Recently, studies in both China and Japan have identified patients with anti-SAE1/2 autoantibodies and pulmonary arterial hypertension [87, 89]. The etiology of pulmonary arterial hypertension in these patients continues to be investigated as it cannot be sufficiently explained by the severity of ILD. While recent investigations have found the frequency of malignancy to be significantly higher in anti-SAE1/2-positive patients, these findings are limited to small studies from Japan and have not been confirmed in non-Asian populations [87, 93].
Antisynthetase Autoantibodies
The antisynthetase autoantibodies (ASAs) target different tRNA synthetases, a family of cytoplasmic enzymes that generate aminoacyl tRNAs (Table 1). There are eight ASAs that are associated with the antisynthetase syndrome (ASSD) and have been identified in myositis: anti-Jo-1 (histidyl-tRNA synthetase), anti-PL-12 (alanyl-tRNA synthetase), anti-PL-7 (threonyl-tRNA synthetase), anti-EJ (glycyl-tRNA synthetase), anti-OJ (isoleucyl-tRNA synthetase), anti-KS (asparaginyl-tRNA synthetase), anti-Zo (phenylalanyl-tRNA synthetase), and anti-Ha/YRS (tyrosyl-tRNA synthetase). The prevalence of ASAs varies between populations, and anti-Jo-1 is the most common with 5–20% of DM patients testing positive for this autoantibody [6, 12, 94]. A meta-analysis of 27 studies identified anti-PL-7 autoantibodies in 2% of patients, anti-PL-12 in 3%, anti-KS in 1%, anti-OJ in 1%, and anti-EJ in 1% [94]. Only 4% of JDM patients have been reported to have ASAs [7].
Patients with ASSD have heterogeneous clinical presentations and there is significant variety within each autoantibody group [95,96,97,98]. A meta-analysis revealed a significantly increased risk for myositis (RR 1.6; CI 1.38–1.85), arthralgias (RR 1.52; CI 1.32–1.76), and mechanics hands (RR 1.47; CI 1.11–1.94) in anti-Jo-1-positive patients compared to patients with other ASA autoantibodies [94]. Additional studies found that while ILD is prevalent among patients with a range of ASAs, it is even more prevalent and severe among the anti-Jo-1 subgroup [6, 96, 97, 99]. Finally, a recent muscle biopsy analysis revealed that anti-Jo-1-positive patients uniquely develop a necrotizing perifascicular myositis vs. the perifascicular atrophy myositis associated with DM [100•].
The classic ASSD triad includes ILD, myositis, and arthritis [101]. Accompanying symptoms of RP and fever are frequently identified, and cutaneous findings include dry, hyperkeratotic, and cracked skin on the hands (mechanics hands, Fig. 1h) and plantar surfaces of the feet (“hiker’s feet”) [102, 103]. Other than these, the most “classic” associated cutaneous findings of DM (e.g., V-neck sign, shawl sign, calcinosis) tend to be found at a lower frequency in this subgroup among JDM patients [7]. A recent large cohort of combined European and US patients identified 165 patients with only 1–2 of the classic ASSD triad of findings at the disease onset and found that 58% went on to develop an additional manifestation of the triad within 15 (IQR 9–51) months. The odds of developing new ILD, myositis, or arthritis were significantly increased among patients who also developed accompanying symptoms of RP, mechanics hands, or fever during follow-up [101].
Overall, anti-ARS-positive patients have a favorable prognosis compared to anti-ARS-negative patients with a 10-year survival rate of 92 vs. 59% (p = 0.02) [104]. The 5-year survival rate is significantly higher in anti-ARS-positive patients with ILD compared to both non-ARS patients with ILD and anti-MDA-5-positive patients with ILD [105]. Malignancy has not classically been associated with this autoantibody group, but recent research has identified malignancy in as many as 12% of anti-ARS patients, and thus, additional investigation into this relationship is required [98].
MSA Detection and Future Directions
Myositis-specific autoantibodies hold tremendous promise with respect to their use as adjunct tools for diagnosis, sub-classification, and risk stratification of patients with dermatomyositis. They may also provide an important foothold into understanding the mechanism of disease pathogenesis in DM. The latter will require a careful investigation of antigen structure and/or expression in diseased tissues and investigation of how autoimmune effector pathways serve to propagate the immune response in a tissue-specific manner [106]. As described in the previous sections, there are numerous features associated with specific MSAs, but these phenotypes are typically indistinct, overlapping, and incompletely penetrant in a given patient. At present, there is a lack of data on how autoantibodies can be used to make specific decisions regarding clinical care. This can be at least partially attributed to the fact that available data comes from relatively small studies with different patient populations and varying methodologies for identifying autoantibodies. It is likely that clinical phenotypes are the product of an interaction between patient factors (i.e., genetics, environment) and autoimmunity. Thus, it is conceivable that instead of universal “antibody phenotypes,” there will be phenotypes particular to carefully defined clinical strata. Adopting a universal “platform” for MSA detection and continuing to acquire data from larger prospective and diverse patient cohorts will increase our ability to interpret the significance of serologic data in DM.
Conclusions
MSAs provide a powerful tool with which we can differentiate patients with DM. Although there are limitations in this technique, including lack of cross-validation of assays, autoantibody phenotype overlap, and incomplete autoantibody penetrance, MSAs are important adjunct tools for diagnosis and sub-classification of patients that will guide increasingly specific clinical care in the future. MSAs are associated with characteristic combinations of systemic and cutaneous phenotypes, and even in the absence of a common autoantibody testing platform, recognition of these patterns and combinations can help clinicians make a diagnosis of dermatomyositis. Accurate sub-classification of DM patients is critical to guiding patient care with respect to follow-up, treatment selection, and anticipation of disease sequelae.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292(7):344–7. https://doi.org/10.1056/NEJM197502132920706.
Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975;292(8):403–7. https://doi.org/10.1056/NEJM197502202920807.
Lundberg IE, Tjarnlund A, Bottai M, Werth VP, Pilkington C, Visser M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. 2017;76(12):1955–64. https://doi.org/10.1136/annrheumdis-2017-211468.
Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57(5):375–81. https://doi.org/10.4103/0019-5154.100486.
Targoff IN, Reichlin M. The association between Mi-2 antibodies and dermatomyositis. Arthritis Rheum. 1985;28(7):796–803.
Srivastava P, Dwivedi S, Misra R. Myositis-specific and myositis-associated autoantibodies in Indian patients with inflammatory myositis. Rheumatol Int. 2016;36(7):935–43. https://doi.org/10.1007/s00296-016-3494-3.
Rider LG, Shah M, Mamyrova G, Huber AM, Rice MM, Targoff IN, et al. The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore). 2013;92(4):223–43. https://doi.org/10.1097/MD.0b013e31829d08f9.
Betteridge Z, McHugh N. Myositis-specific autoantibodies: an important tool to support diagnosis of myositis. J Intern Med. 2016;280(1):8–23. https://doi.org/10.1111/joim.12451.
Shah M, Mamyrova G, Targoff IN, Huber AM, Malley JD, Rice MM, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore). 2013;92(1):25–41. https://doi.org/10.1097/MD.0b013e31827f264d.
Brouwer R, Hengstman GJ, Vree Egberts W, Ehrfeld H, Bozic B, Ghirardello A, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis. 2001;60(2):116–23.
Cruellas MG, Viana Vdos S, Levy-Neto M, Souza FH, Shinjo SK. Myositis-specific and myositis-associated autoantibody profiles and their clinical associations in a large series of patients with polymyositis and dermatomyositis. Clinics (Sao Paulo). 2013;68(7):909–14. https://doi.org/10.6061/clinics/2013(07)04.
Selva-O'Callaghan A, Labrador-Horrillo M, Solans-Laque R, Simeon-Aznar CP, Martinez-Gomez X, Vilardell-Tarres M. Myositis-specific and myositis-associated antibodies in a series of eighty-eight Mediterranean patients with idiopathic inflammatory myopathy. Arthritis Rheum. 2006;55(5):791–8. https://doi.org/10.1002/art.22237.
Hamaguchi Y, Kuwana M, Hoshino K, Hasegawa M, Kaji K, Matsushita T, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol. 2011;147(4):391–8. https://doi.org/10.1001/archdermatol.2011.52.
Love LA, Leff RL, Fraser DD, Targoff IN, Dalakas M, Plotz PH, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore). 1991;70(6):360–74.
O'Hanlon TP, Carrick DM, Targoff IN, Arnett FC, Reveille JD, Carrington M, et al. Immunogenetic risk and protective factors for the idiopathic inflammatory myopathies: distinct HLA-A, -B, -Cw, -DRB1, and -DQA1 allelic profiles distinguish European American patients with different myositis autoantibodies. Medicine (Baltimore). 2006;85(2):111–27. https://doi.org/10.1097/01.md.0000217525.82287.eb.
O'Hanlon TP, Rider LG, Mamyrova G, Targoff IN, Arnett FC, Reveille JD, et al. HLA polymorphisms in African Americans with idiopathic inflammatory myopathy: allelic profiles distinguish patients with different clinical phenotypes and myositis autoantibodies. Arthritis Rheum. 2006;54(11):3670–81. https://doi.org/10.1002/art.22205.
Mierau R, Dick T, Bartz-Bazzanella P, Keller E, Albert ED, Genth E. Strong association of dermatomyositis-specific Mi-2 autoantibodies with a tryptophan at position 9 of the HLA-DR beta chain. Arthritis Rheum. 1996;39(5):868–76.
Shamim EA, Rider LG, Pandey JP, O'Hanlon TP, Jara LJ, Samayoa EA, et al. Differences in idiopathic inflammatory myopathy phenotypes and genotypes between Mesoamerican Mestizos and North American Caucasians: ethnogeographic influences in the genetics and clinical expression of myositis. Arthritis Rheum. 2002;46(7):1885–93. https://doi.org/10.1002/art.10358.
Love LA, Weinberg CR, McConnaughey DR, Oddis CV, Medsger TA Jr, Reveille JD, et al. Ultraviolet radiation intensity predicts the relative distribution of dermatomyositis and anti-Mi-2 autoantibodies in women. Arthritis Rheum. 2009;60(8):2499–504. https://doi.org/10.1002/art.24702.
Petri MH, Satoh M, Martin-Marquez BT, Vargas-Ramirez R, Jara LJ, Saavedra MA, et al. Implications in the difference of anti-Mi-2 and -p155/140 autoantibody prevalence in two dermatomyositis cohorts from Mexico City and Guadalajara. Arthritis Res Ther. 2013;15(2):R48. https://doi.org/10.1186/ar4207.
Komura K, Fujimoto M, Matsushita T, Kaji K, Kondo M, Hirano T, et al. Prevalence and clinical characteristics of anti-Mi-2 antibodies in Japanese patients with dermatomyositis. J Dermatol Sci. 2005;40(3):215–7. https://doi.org/10.1016/j.jdermsci.2005.09.004.
Mammen AL, Casciola-Rosen LA, Hall JC, Christopher-Stine L, Corse AM, Rosen A. Expression of the dermatomyositis autoantigen Mi-2 in regenerating muscle. Arthritis Rheum. 2009;60(12):3784–93. https://doi.org/10.1002/art.24977.
• Deakin CT, Yasin SA, Simou S, Arnold KA, Tansley SL, Betteridge ZE, et al. Muscle biopsy findings in combination with myositis-specific autoantibodies aid prediction of outcomes in juvenile dermatomyositis. Arthritis Rheumatol. 2016;68(11):2806–16. https://doi.org/10.1002/art.39753. First study to show that degree of severity of muscle biopsy, in combination with MSA subtype, is predictive of increased treatment duration in JDM.
• Pinal-Fernandez I, Casciola-Rosen LA, Christopher-Stine L, Corse AM, Mammen AL. The prevalence of individual histopathologic features varies according to autoantibody status in muscle biopsies from patients with dermatomyositis. J Rheumatol. 2015;42(8):1448–54. First paper to systematically analyze muscle biopsies in adult DM and identify histology features associated with specific MSAs.
Hengstman GJ, Vree Egberts WT, Seelig HP, Lundberg IE, Moutsopoulos HM, Doria A, et al. Clinical characteristics of patients with myositis and autoantibodies to different fragments of the Mi-2 beta antigen. Ann Rheum Dis. 2006;65(2):242–5. https://doi.org/10.1136/ard.2005.040717.
Reed AM, Crowson CS, Hein M, de Padilla CL, Olazagasti JM, Aggarwal R, et al. Biologic predictors of clinical improvement in rituximab-treated refractory myositis. BMC Musculoskelet Disord. 2015;16:257. https://doi.org/10.1186/s12891-015-0710-3.
Aggarwal R, Bandos A, Reed AM, Ascherman DP, Barohn RJ, Feldman BM, et al. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheumatol. 2014;66(3):740–9. https://doi.org/10.1002/art.38270.
Nakashima R, Imura Y, Kobayashi S, Yukawa N, Yoshifuji H, Nojima T, et al. The RIG-I-like receptor IFIH1/MDA5 is a dermatomyositis-specific autoantigen identified by the anti-CADM-140 antibody. Rheumatology (Oxford). 2010;49(3):433–40. https://doi.org/10.1093/rheumatology/kep375.
Cao H, Pan M, Kang Y, Xia Q, Li X, Zhao X, et al. Clinical manifestations of dermatomyositis and clinically amyopathic dermatomyositis patients with positive expression of anti-melanoma differentiation-associated gene 5 antibody. Arthritis Care Res (Hoboken). 2012;64(10):1602–10. https://doi.org/10.1002/acr.21728.
Chen Z, Hu W, Wang Y, Guo Z, Sun L, Kuwana M. Distinct profiles of myositis-specific autoantibodies in Chinese and Japanese patients with polymyositis/dermatomyositis. Clin Rheumatol. 2015;34(9):1627–31. https://doi.org/10.1007/s10067-015-2935-9.
Bodoki L, Nagy-Vincze M, Griger Z, Betteridge Z, Szollosi L, Danko K. Four dermatomyositis-specific autoantibodies-anti-TIF1gamma, anti-NXP2, anti-SAE and anti-MDA5-in adult and juvenile patients with idiopathic inflammatory myopathies in a Hungarian cohort. Autoimmun Rev. 2014;13(12):1211–9. https://doi.org/10.1016/j.autrev.2014.08.011.
Hall JC, Casciola-Rosen L, Samedy LA, Werner J, Owoyemi K, Danoff SK, et al. Anti-melanoma differentiation-associated protein 5-associated dermatomyositis: expanding the clinical spectrum. Arthritis Care Res (Hoboken). 2013;65(8):1307–15. https://doi.org/10.1002/acr.21992.
Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola-Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65(1):25–34. https://doi.org/10.1016/j.jaad.2010.09.016.
Chen Z, Wang Y, Kuwana M, Xu X, Hu W, Feng X, et al. HLA-DRB1 alleles as genetic risk factors for the development of anti-MDA5 antibodies in patients with dermatomyositis. J Rheumatol. 2017;44(9):1389–93. https://doi.org/10.3899/jrheum.170165.
Lin JM, Zhang YB, Peng QL, Yang HB, Shi JL, Gu ML, et al. Genetic association of HLA-DRB1 multiple polymorphisms with dermatomyositis in Chinese population. HLA. 2017;90(6):354–9. https://doi.org/10.1111/tan.13171.
Tansley SL, Betteridge ZE, Gunawardena H, Jacques TS, Owens CM, Pilkington C, et al. Anti-MDA5 autoantibodies in juvenile dermatomyositis identify a distinct clinical phenotype: a prospective cohort study. Arthritis Res Ther. 2014;16(4):R138. https://doi.org/10.1186/ar4600.
Allenbach Y, Leroux G, Suarez-Calvet X, Preusse C, Gallardo E, Hervier B, et al. Dermatomyositis with or without anti-melanoma differentiation-associated gene 5 antibodies: common interferon signature but distinct NOS2 expression. Am J Pathol. 2016;186(3):691–700. https://doi.org/10.1016/j.ajpath.2015.11.010.
Peng Y, Zhang S, Zhao Y, Liu Y, Yan B. Neutrophil extracellular traps may contribute to interstitial lung disease associated with anti-MDA5 autoantibody positive dermatomyositis. Clin Rheumatol. 2017;37:107–15. https://doi.org/10.1007/s10067-017-3799-y.
Marie I, Hachulla E, Cherin P, Dominique S, Hatron PY, Hellot MF, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum. 2002;47(6):614–22. https://doi.org/10.1002/art.10794.
Schnabel A, Reuter M, Biederer J, Richter C, Gross WL. Interstitial lung disease in polymyositis and dermatomyositis: clinical course and response to treatment. Semin Arthritis Rheum. 2003;32(5):273–84. https://doi.org/10.1053/sarh.2002.50012.
Hosono Y, Nakashima R, Serada S, Murakami K, Imura Y, Yoshifuji H, et al. Splicing factor proline/glutamine-rich is a novel autoantigen of dermatomyositis and associated with anti-melanoma differentiation-associated gene 5 antibody. J Autoimmun. 2017;77:116–22. https://doi.org/10.1016/j.jaut.2016.11.006.
• Matsushita T, Mizumaki K, Kano M, Yagi N, Tennichi M, Takeuchi A, et al. Antimelanoma differentiation-associated protein 5 antibody level is a novel tool for monitoring disease activity in rapidly progressive interstitial lung disease with dermatomyositis. Br J Dermatol. 2017;176(2):395–402. https://doi.org/10.1111/bjd.14882. Novel finding suggesting that an increase in anti-MDA-5 titers is associated with relapse of interstitial lung disease.
Abe Y, Matsushita M, Tada K, Yamaji K, Takasaki Y, Tamura N. Clinical characteristics and change in the antibody titres of patients with anti-MDA5 antibody-positive inflammatory myositis. Rheumatology (Oxford). 2017;56(9):1492–7. https://doi.org/10.1093/rheumatology/kex188.
Sato S, Kuwana M, Fujita T, Suzuki Y. Anti-CADM-140/MDA5 autoantibody titer correlates with disease activity and predicts disease outcome in patients with dermatomyositis and rapidly progressive interstitial lung disease. Mod Rheumatol. 2013;23(3):496–502. https://doi.org/10.1007/s10165-012-0663-4.
Gono T, Sato S, Kawaguchi Y, Kuwana M, Hanaoka M, Katsumata Y, et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology (Oxford). 2012;51(9):1563–70. https://doi.org/10.1093/rheumatology/kes102.
Muro Y, Sugiura K, Akiyama M. Limitations of a single-point evaluation of anti-MDA5 antibody, ferritin, and IL-18 in predicting the prognosis of interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Clin Rheumatol. 2013;32(3):395–8. https://doi.org/10.1007/s10067-012-2142-x.
Lee LW, Narang NS, Postolova A, Seminara N, Kantor MA. Anti-MDA5-positive dermatomyositis presenting as fever of unknown origin. J Gen Intern Med. 2016;31(12):1530–6. https://doi.org/10.1007/s11606-016-3769-0.
Chaisson NF, Paik J, Orbai AM, Casciola-Rosen L, Fiorentino D, Danoff S, et al. A novel dermato-pulmonary syndrome associated with MDA-5 antibodies: report of 2 cases and review of the literature. Medicine (Baltimore). 2012;91(4):220–8. https://doi.org/10.1097/MD.0b013e3182606f0b.
Labrador-Horrillo M, Martinez MA, Selva-O'Callaghan A, Trallero-Araguas E, Balada E, Vilardell-Tarres M, et al. Anti-MDA5 antibodies in a large Mediterranean population of adults with dermatomyositis. J Immunol Res. 2014;2014:290797. https://doi.org/10.1155/2014/290797.
Li L, Wang Q, Wen X, Liu C, Wu C, Yang F, et al. Assessment of anti-MDA5 antibody as a diagnostic biomarker in patients with dermatomyositis-associated interstitial lung disease or rapidly progressive interstitial lung disease. Oncotarget. 2017;8(44):76129–40. https://doi.org/10.18632/oncotarget.19050.
Kawasumi H, Gono T, Kawaguchi Y, Yamanaka H. Recent treatment of interstitial lung disease with idiopathic inflammatory myopathies. Clin Med Insights Circ Respir Pulm Med. 2015;9(Suppl 1):9–17. https://doi.org/10.4137/CCRPM.S23313.
Zou J, Guo Q, Chi J, Wu H, Bao C. HRCT score and serum ferritin level are factors associated to the 1-year mortality of acute interstitial lung disease in clinically amyopathic dermatomyositis patients. Clin Rheumatol. 2015;34(4):707–14. https://doi.org/10.1007/s10067-015-2866-5.
Kotani T, Takeuchi T, Makino S, Hata K, Yoshida S, Nagai K, et al. Combination with corticosteroids and cyclosporin-A improves pulmonary function test results and chest HRCT findings in dermatomyositis patients with acute/subacute interstitial pneumonia. Clin Rheumatol. 2011;30(8):1021–8. https://doi.org/10.1007/s10067-011-1713-6.
Cabezas-Rodriguez I, Morante-Bolado I, Brandy-Garcia A, Queiro-Silva R, Mozo L, Ballina-Garcia FJ. Anti-MDA5 dermatomyositis mimicking psoriatic arthritis. Reumatol Clin. 2016; https://doi.org/10.1016/j.reuma.2016.10.010.
Gono T, Kawaguchi Y, Satoh T, Kuwana M, Katsumata Y, Takagi K, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49(9):1713–9. https://doi.org/10.1093/rheumatology/keq149.
Chen Z, Cao M, Plana MN, Liang J, Cai H, Kuwana M, et al. Utility of anti-melanoma differentiation-associated gene 5 antibody measurement in identifying patients with dermatomyositis and a high risk for developing rapidly progressive interstitial lung disease: a review of the literature and a meta-analysis. Arthritis Care Res (Hoboken). 2013;65(8):1316–24. https://doi.org/10.1002/acr.21985.
Pau-Charles I, Moreno PJ, Ortiz-Ibanez K, Lucero MC, Garcia-Herrera A, Espinosa G, et al. Anti-MDA5 positive clinically amyopathic dermatomyositis presenting with severe cardiomyopathy. J Eur Acad Dermatol Venereol. 2014;28(8):1097–102. https://doi.org/10.1111/jdv.12300.
Lepelletier C, Bengoufa D, Lyes Z, de Masson A, Chasset F, Jachiet M, et al. Dermatopulmonary syndrome associated with anti-MDA5 antibodies after allogeneic hematopoietic stem cell transplantation. JAMA Dermatol. 2017;2016 https://doi.org/10.1001/jamadermatol.2016.3976.
Avouac J, Guerini H, Wipff J, Assous N, Chevrot A, Kahan A, et al. Radiological hand involvement in systemic sclerosis. Ann Rheum Dis. 2006;65(8):1088–92. https://doi.org/10.1136/ard.2005.044602.
Avouac J, Mogavero G, Guerini H, Drape JL, Mathieu A, Kahan A, et al. Predictive factors of hand radiographic lesions in systemic sclerosis: a prospective study. Ann Rheum Dis. 2011;70(4):630–3. https://doi.org/10.1136/ard.2010.134304.
Valenzuela A, Chung L, Casciola-Rosen L, Fiorentino D. Identification of clinical features and autoantibodies associated with calcinosis in dermatomyositis. JAMA Dermatol. 2014;150(7):724–9. https://doi.org/10.1001/jamadermatol.2013.10416.
Wolstencroft PW, Chung L, Li S, Casciola-Rosen L, Fiorentino DF. Factors associated with clinical remission of skin disease in dermatomyositis. JAMA Dermatol. 2018;2017 https://doi.org/10.1001/jamadermatol.2017.3758.
Oddis CV, Fertig N, Goel A, Espada G, Gregorian MC, Cocco JAM et al. Clinical and serological characterization of the anti-MJ antibody in childhood myositis. Arthritis Rheum-Us. 1997;40(9):652-.
Takahashi K, Yoshida N, Murakami N, Kawata K, Ishizaki H, Tanaka-Okamoto M, et al. Dynamic regulation of p53 subnuclear localization and senescence by MORC3. Mol Biol Cell. 2007;18(5):1701–9. https://doi.org/10.1091/mbc.E06-08-0747.
Ichimura Y, Matsushita T, Hamaguchi Y, Kaji K, Hasegawa M, Tanino Y, et al. Anti-NXP2 autoantibodies in adult patients with idiopathic inflammatory myopathies: possible association with malignancy. Ann Rheum Dis. 2012;71(5):710–3. https://doi.org/10.1136/annrheumdis-2011-200697.
Ishikawa A, Muro Y, Sugiura K, Akiyama M. Development of an ELISA for detection of autoantibodies to nuclear matrix protein 2. Rheumatology (Oxford). 2012;51(7):1181–7. https://doi.org/10.1093/rheumatology/kes033.
Rogers A, Chung L, Li S, Casciola-Rosen L, Fiorentino DF. The cutaneous and systemic findings associated with nuclear matrix protein-2 antibodies in adult dermatomyositis patients. Arthritis Care Res (Hoboken). 2017. doi:https://doi.org/10.1002/acr.23210, 69, 1909, 1914.
Fiorentino DF, Chung LS, Christopher-Stine L, Zaba L, Li SF, Mammen AL, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1 gamma. Arthritis Rheum-Us. 2013;65(11):2954–62. https://doi.org/10.1002/art.38093.
• Albayda J, Pinal-Fernandez I, Huang W, Parks C, Paik J, Casciola-Rosen L et al. Dermatomyositis patients with anti-nuclear matrix protein-2 autoantibodies have more edema, more severe muscle disease, and increased malignancy risk. Arthritis Care Res (Hoboken). 2017. doi:https://doi.org/10.1002/acr.23188. First paper to comprehensively describe the unique clinical phenotype of anti-NXP-2-positive DM patients.
Espada G, Maldonado Cocco JA, Fertig N, Oddis CV. Clinical and serologic characterization of an Argentine pediatric myositis cohort: identification of a novel autoantibody (anti-MJ) to a 142-kDa protein. J Rheumatol. 2009;36(11):2547–51. https://doi.org/10.3899/jrheum.090461.
Tansley SL, Betteridge ZE, Shaddick G, Gunawardena H, Arnold K, Wedderburn LR, et al. Calcinosis in juvenile dermatomyositis is influenced by both anti-NXP2 autoantibody status and age at disease onset. Rheumatology (Oxford). 2014;53(12):2204–8. https://doi.org/10.1093/rheumatology/keu259.
Ceribelli A, Fredi M, Taraborelli M, Cavazzana I, Franceschini F, Quinzanini M, et al. Anti-MJ/NXP-2 autoantibody specificity in a cohort of adult Italian patients with polymyositis/dermatomyositis. Arthritis Res Ther. 2012;14(2):R97. https://doi.org/10.1186/ar3822.
Gunawardena H, Wedderburn LR, Chinoy H, Betteridge ZE, North J, Ollier WE, et al. Autoantibodies to a 140-kd protein in juvenile dermatomyositis are associated with calcinosis. Arthritis Rheum. 2009;60(6):1807–14. https://doi.org/10.1002/art.24547.
Kaji K, Fujimoto M, Hasegawa M, Kondo M, Saito Y, Komura K, et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy. Rheumatology (Oxford). 2007;46(1):25–8. https://doi.org/10.1093/rheumatology/kel161.
Agricola E, Randall RA, Gaarenstroom T, Dupont S, Hill CS. Recruitment of TIF1gamma to chromatin via its PHD finger-bromodomain activates its ubiquitin ligase and transcriptional repressor activities. Mol Cell. 2011;43(1):85–96. https://doi.org/10.1016/j.molcel.2011.05.020.
Kulkarni A, Oza J, Yao M, Sohail H, Ginjala V, Tomas-Loba A, et al. Tripartite motif-containing 33 (TRIM33) protein functions in the poly(ADP-ribose) polymerase (PARP)-dependent DNA damage response through interaction with amplified in liver cancer 1 (ALC1) protein. J Biol Chem. 2013;288(45):32357–69. https://doi.org/10.1074/jbc.M113.459164.
Fiorentino DF, Kuo K, Chung L, Zaba L, Li S, Casciola-Rosen L. Distinctive cutaneous and systemic features associated with antitranscriptional intermediary factor-1gamma antibodies in adults with dermatomyositis. J Am Acad Dermatol. 2015;72(3):449–55. https://doi.org/10.1016/j.jaad.2014.12.009.
Fiorentino DF, Chung LS, Christopher-Stine L, Zaba L, Li S, Mammen AL, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1gamma. Arthritis Rheum. 2013;65(11):2954–62. https://doi.org/10.1002/art.38093.
Fujimoto M, Murakami A, Kurei S, Okiyama N, Kawakami A, Mishima M, et al. Enzyme-linked immunosorbent assays for detection of anti-transcriptional intermediary factor-1 gamma and anti-Mi-2 autoantibodies in dermatomyositis. J Dermatol Sci. 2016;84(3):272–81. https://doi.org/10.1016/j.jdermsci.2016.09.013.
Targoff IN, Mamyrova G, Trieu EP, Perurena O, Koneru B, O'Hanlon TP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. 2006;54(11):3682–9. https://doi.org/10.1002/art.22164.
•• Pinal-Fernandez I, Ferrer-Fabregas B, Trallero-Araguas E, Balada E, Martinez MA, Milisenda JC et al. Tumour TIF1 mutations and loss of heterozygosity related to cancer-associated myositis. Rheumatology (Oxford). 2017. doi:https://doi.org/10.1093/rheumatology/kex413. First study to investigate genetic changes in the TIF1 genes of tumors from patients with paraneoplastic anti-TIF-1γ autoantibody-positive DM.
Fujimoto M, Watanabe R, Ishitsuka Y, Okiyama N. Recent advances in dermatomyositis-specific autoantibodies. Curr Opin Rheumatol. 2016;28(6):636–44. https://doi.org/10.1097/BOR.0000000000000329.
• Mohassel P, Rosen P, Casciola-Rosen L, Pak K, Mammen AL. Expression of the dermatomyositis autoantigen transcription intermediary factor 1gamma in regenerating muscle. Arthritis Rheumatol. 2015;67(1):266–72. https://doi.org/10.1002/art.38863. Illustrates a role for TIF-1γ in muscle regeneration and demonstrates that high expression of TIF-1γ within the muscle may be driving the myositis autoimmune response within DM.
Scholtissek B, Ferring-Schmitt S, Maier J, Wenzel J. Expression of the autoantigen TRIM33/TIF1gamma in skin and muscle of patients with dermatomyositis is upregulated, together with markers of cellular stress. Clin Exp Dermatol. 2017;42(6):659–62. https://doi.org/10.1111/ced.13180.
Bernet LL, Lewis MA, Rieger KE, Casciola-Rosen L, Fiorentino DF. Ovoid palatal patch in dermatomyositis: a novel finding associated with anti-TIF1gamma (p155) antibodies. JAMA Dermatol. 2016;152(9):1049–51. https://doi.org/10.1001/jamadermatol.2016.1429.
Betteridge Z, Gunawardena H, North J, Slinn J, McHugh N. Identification of a novel autoantibody directed against small ubiquitin-like modifier activating enzyme in dermatomyositis. Arthritis Rheum. 2007;56(9):3132–7. https://doi.org/10.1002/art.22862.
Muro Y, Sugiura K, Akiyama M. Low prevalence of anti-small ubiquitin-like modifier activating enzyme antibodies in dermatomyositis patients. Autoimmunity. 2013;46(4):279–84. https://doi.org/10.3109/08916934.2012.755958.
Fujimoto M, Matsushita T, Hamaguchi Y, Kaji K, Asano Y, Ogawa F, et al. Autoantibodies to small ubiquitin-like modifier activating enzymes in Japanese patients with dermatomyositis: comparison with a UK Caucasian cohort. Ann Rheum Dis. 2013;72(1):151–3. https://doi.org/10.1136/annrheumdis-2012-201736.
Ge Y, Lu X, Shu X, Peng Q, Wang G. Clinical characteristics of anti-SAE antibodies in Chinese patients with dermatomyositis in comparison with different patient cohorts. Sci Rep. 2017;7(1):188. https://doi.org/10.1038/s41598-017-00240-6.
Betteridge ZE, Gunawardena H, Chinoy H, North J, Ollier WE, Cooper RG, et al. Clinical and human leucocyte antigen class II haplotype associations of autoantibodies to small ubiquitin-like modifier enzyme, a dermatomyositis-specific autoantigen target, in UK Caucasian adult-onset myositis. Ann Rheum Dis. 2009;68(10):1621–5. https://doi.org/10.1136/ard.2008.097162.
Tarricone E, Ghirardello A, Rampudda M, Bassi N, Punzi L, Doria A. Anti-SAE antibodies in autoimmune myositis: identification by unlabelled protein immunoprecipitation in an Italian patient cohort. J Immunol Methods. 2012;384(1–2):128–34. https://doi.org/10.1016/j.jim.2012.07.019.
Lee S, Findeisen J, McLean C, Stavrakoglou A. Recalcitrant ulcers associated with anti-small ubiquitin-like modifier activating enzyme-positive dermatomyositis treated with surgery followed by intravenous immunoglobulin. Australas J Dermatol. 2017;59:e76–8. https://doi.org/10.1111/ajd.12659.
Muro Y, Sugiura K, Nara M, Sakamoto I, Suzuki N, Akiyama M. High incidence of cancer in anti-small ubiquitin-like modifier activating enzyme antibody-positive dermatomyositis. Rheumatology (Oxford). 2015;54(9):1745–7. https://doi.org/10.1093/rheumatology/kev247.
Lega JC, Fabien N, Reynaud Q, Durieu I, Durupt S, Dutertre M, et al. The clinical phenotype associated with myositis-specific and associated autoantibodies: a meta-analysis revisiting the so-called antisynthetase syndrome. Autoimmun Rev. 2014;13(9):883–91. https://doi.org/10.1016/j.autrev.2014.03.004.
Kalluri M, Sahn SA, Oddis CV, Gharib SL, Christopher-Stine L, Danoff SK, et al. Clinical profile of anti-PL-12 autoantibody. Cohort study and review of the literature. Chest. 2009;135(6):1550–6. https://doi.org/10.1378/chest.08-2233.
Ang CC, Anyanwu CO, Robinson E, Okawa J, Feng R, Fujimoto M, et al. Clinical signs associated with an increased risk of interstitial lung disease: a retrospective study of 101 patients with dermatomyositis. Br J Dermatol. 2017;176(1):231–3. https://doi.org/10.1111/bjd.14801.
Labirua-Iturburu A, Selva-O'Callaghan A, Vincze M, Danko K, Vencovsky J, Fisher B, et al. Anti-PL-7 (anti-threonyl-tRNA synthetase) antisynthetase syndrome: clinical manifestations in a series of patients from a European multicenter study (EUMYONET) and review of the literature. Medicine (Baltimore). 2012;91(4):206–11. https://doi.org/10.1097/MD.0b013e318260977c.
Hamaguchi Y, Fujimoto M, Matsushita T, Kaji K, Komura K, Hasegawa M, et al. Common and distinct clinical features in adult patients with anti-aminoacyl-tRNA synthetase antibodies: heterogeneity within the syndrome. PLoS One. 2013;8(4):e60442. https://doi.org/10.1371/journal.pone.0060442.
Johnson C, Connors GR, Oaks J, Han S, Truong A, Richardson B, et al. Clinical and pathologic differences in interstitial lung disease based on antisynthetase antibody type. Respir Med. 2014;108(10):1542–8. https://doi.org/10.1016/j.rmed.2014.09.003.
• Mescam-Mancini L, Allenbach Y, Hervier B, Devilliers H, Mariampillay K, Dubourg O, et al. Anti-Jo-1 antibody-positive patients show a characteristic necrotizing perifascicular myositis. Brain. 2015;138(Pt 9):2485–92. https://doi.org/10.1093/brain/awv192. Results indicate that perifascicular necrosis is a unique phenotype of anti-Jo-1-positive DM.
Bartoloni E, Gonzalez-Gay MA, Scire C, Castaneda S, Gerli R, Lopez-Longo FJ, et al. Clinical follow-up predictors of disease pattern change in anti-Jo1 positive anti-synthetase syndrome: results from a multicenter, international and retrospective study. Autoimmun Rev. 2017;16(3):253–7. https://doi.org/10.1016/j.autrev.2017.01.008.
Cox JT, Gullotti DM, Mecoli CA, Lahouti AH, Albayda J, Paik J, et al. “Hiker’s feet”: a novel cutaneous finding in the inflammatory myopathies. Clin Rheumatol. 2017;36(7):1683–6. https://doi.org/10.1007/s10067-017-3598-5.
Bachmeyer C, Tillie-Leblond I, Lacert A, Cadranel J, Aractingi S. “Mechanic’s hands”: a misleading cutaneous sign of the antisynthetase syndrome. Br J Dermatol. 2007;156(1):192–4. https://doi.org/10.1111/j.1365-2133.2006.07593.x.
Hozumi H, Enomoto N, Kono M, Fujisawa T, Inui N, Nakamura Y, et al. Prognostic significance of anti-aminoacyl-tRNA synthetase antibodies in polymyositis/dermatomyositis-associated interstitial lung disease: a retrospective case control study. PLoS One. 2015;10(3):e0120313. https://doi.org/10.1371/journal.pone.0120313.
Hozumi H, Fujisawa T, Nakashima R, Johkoh T, Sumikawa H, Murakami A, et al. Comprehensive assessment of myositis-specific autoantibodies in polymyositis/dermatomyositis-associated interstitial lung disease. Respir Med. 2016;121:91–9. https://doi.org/10.1016/j.rmed.2016.10.019.
Rosen A, Casciola-Rosen L. Autoantigens as partners in initiation and propagation of autoimmune rheumatic diseases. Annu Rev Immunol. 2016;34:395–420. https://doi.org/10.1146/annurev-immunol-032414-112205.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
This article is part of the Topical Collection on Inflammatory Muscle Disease
Rights and permissions
About this article

Cite this article
Wolstencroft, P.W., Fiorentino, D.F. Dermatomyositis Clinical and Pathological Phenotypes Associated with Myositis-Specific Autoantibodies. Curr Rheumatol Rep 20, 28 (2018). https://doi.org/10.1007/s11926-018-0733-5
Published:
DOI: https://doi.org/10.1007/s11926-018-0733-5