
Opinion statement
Purpose of Review In dermatomyositis (DM), antibodies have been shown to closely correlate with clinical phenotypes. The focus of this review is to describe the known clinical associations of the different antibodies related to DM.
Recent Findings The DM-specific antibodies include anti-Mi-2, anti-NXP2, anti-TIF1-gamma, anti-MDA5, and anti-SAE. They present with varying levels of skin, muscle, and other target organ involvement. The anti-synthetase antibodies can present as DM, but define a distinct subset displaying other features known as the anti-synthetase syndrome. Anti-PM/Scl, anti-Ro, anti-RNP, and anti-Ku are myositis-associated antibodies that can present as DM as well as other overlap syndromes.
Summary More homogenous subgroups are created by viewing DM through the filter of antibodies. The demonstration of one of these antibodies in a patient suspected of having DM is valuable for informing the diagnosis, prognosis, and treatment of the disease. As antibody testing becomes more widely available, we expect even better characterization of disease and treatment response based on antibody groups to emerge in the coming years.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The idiopathic inflammatory myopathies (IIMs) are a heterogeneous group of immune-mediated conditions that affect the muscles and various other target organs including the skin, lung, and joints. The manifestations are varied but inflammation of the target organs leading to organ dysfunction is seen. On the basis of differences in clinical presentation, histopathology, serology, and response to treatment, four distinct subgroups are generally recognized: dermatomyositis (DM), polymyositis (PM), immune-mediated necrotizing myopathy (IMNM), and sporadic inclusion body myositis (IBM) [1].
Dermatomyositis is characterized by microvascular injury affecting both the skin and muscle, resulting in the development of proximal muscle weakness and a polymorphous rash [1]. It can manifest with a skin rash and muscle weakness, skin rash alone (amyopathic), or only muscle weakness (DM sine dermatitis). Organ involvement can span the setting of mild skin disease to severe cases of profound weakness, lung disease, and calcinosis.
Diagnosing dermatomyositis
Patients can be categorized as DM on the basis of clinical presentation and presence of a rash, muscle or skin biopsy findings, and/or serology. The diagnostic criteria proposed by Bohan and Peter remain the most widely used to date [2, 3]. However, in this classification scheme published in 1975, only two forms of IIMs were recognized—PM and DM. Criteria used to define PM and DM included the presence of symmetrical proximal muscle weakness, muscle histopathology showing evidence of inflammation, elevation of serum muscle enzymes, electromyographic changes of an irritable myopathy, and characteristic dermatologic changes in DM. The point of differentiation for these two entities was the presence of a rash, leading to a diagnosis of DM. Muscle biopsy findings were not used to differentiate between the two groups.
With further understanding of these diseases and better characterization of histopathology, distinct differences on muscle biopsy were noted between PM and DM and also led to the recognition of two other distinct groups, IBM and IMNM [4]. In patients with DM, inflammatory infiltrates are mainly composed of CD4+ T cells, macrophages, and B cells and are found in the perimysium and perivascular areas. Perifascicular muscle fiber atrophy as well as deposition of the C5b-9 complement membrane attack complex (MAC) on small blood vessels is also seen and noted to be characteristic [5]. Therefore, by the European Neuromuscular Centre (ENMC) 2003 criteria, a diagnosis of definite DM was proposed as the fulfillment of all clinical criteria (subacute onset of weakness, proximal > distal/neck flexor > neck extensor weakness, presence of typical rashes of DM) with muscle biopsy findings which included the presence of perifascicular atrophy [4]. Probable DM could be diagnosed when all clinical criteria were seen, with other muscle biopsy features or alternative laboratory criteria. Laboratory criteria included elevated muscle enzymes, compatible EMG changes, MRI findings of muscle edema, or the presence of myositis-specific antibodies. Muscle biopsy changes for this category included that of MAC deposition on small blood vessels, MHC-class I expression on perifascicular fibers, or perivascular/perimysial inflammatory cell infiltrates. As pathognomonic features on muscle biopsy were noted for DM, the ENMC criteria also made provision for cases of dermatomyositis sine dermatitis where a typical rash was not seen but muscle changes were noted. It also allowed for the distinction of amyopathic dermatomyositis where skin changes were noted, but without discernable muscle involvement.
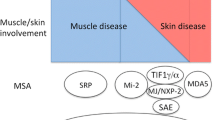
The discovery of several myositis-specific and myositis-associated antibodies (MAA) has been an important advancement in the study of IIMs, where autoimmunity plays a key role. Myositis-specific autoantibodies occur in approximately 70% of DM patients and are remarkably specific for DM and IMNM while myositis-associated autoantibodies (MAA) are generally detected in overlap syndromes (OS) and other connective tissue diseases (Table 1) [6, 7•, 8, 9]. These MSAs and MAAs target diverse intracellular components. In dermatomyositis, the MSAs have been found to be tightly linked to specific phenotypes which can be very useful in aiding diagnosis, treatment, and prognosis [7•, 10••, 11]. Although most of the widely accepted classification criteria for myositis do not specifically incorporate antibody data, it is clear that the presence of these antibodies is very useful to create more homogenous patient subgroups within DM and the rest of the IIMs. Further, while the MSAs can coexist, they are usually mutually exclusive, allowing for even better delineation of clinical phenotypes [12, 13].
Therefore, when considering DM, we see an evolution of diagnostic criteria initially based on clinical presentation and the demonstration of a rash, to a heavy reliance on histopathology, to the current era where antibody testing is not only diagnostic, but also useful to define the disease syndrome (Table 1).
Treatment
Dermatomyositis-specific autoantibodies and clinical phenotypes
Anti-Mi-2
The anti-Mi-2 antibody targeting DNA helicase was first detected in 1984 [14]. Phenotypically, it is associated with the development of the classic cutaneous features of DM. Patients can present with the heliotrope rash, Gottron’s papules (Fig. 1), V-sign (Fig. 2), arm erythema (Fig. 3), shawl sign, photosensitive poikiloderma, and periungual and cuticular overgrowth (Fig. 1). Additional cutaneous manifestations recently described include facial dermatosis and flagellate erythema [15•]. The pattern of muscle involvement follows the typical DM distribution with proximal muscle weakness and notably elevated muscle enzymes. There is usually sparing of the lung and joints and a remarkable response to steroids, resulting in a good overall prognosis with a 5-year survival rate of > 90% [16]. On muscle histopathology, anti-Mi-2 patients have been reported to have more primary inflammation compared to those with other DM-specific antibodies [17]. In terms of cancer risk, anti-Mi-2 is not typically associated with a paraneoplastic syndrome; however, a European cohort reported an elevated malignancy risk, but only in anti-Mi-2 Ab-positive patients possessing the N-terminal fragment of the Mi-2 antigen [18,19,20].

Multiple erythematous to pinkish, slightly hyperkeratotic papules on the skin overlying the distal interphalangeal and proximal interphalangeal joints on the hand known as “Gottron’s papules” associated with periungual erythema and cuticular overgrowth.

Deep red, coalescent macules, and patches on the neck and chest, known as the “V-sign”.

Erythematous to violaceous patches spanning the lateral aspect of the upper extremity.
Anti-p140 or anti-NXP-2
First described in juvenile DM in 1997, anti-NXP-2 autoantibodies target a protein involved in various nuclear processes known as nuclear matrix protein 2. As with other MSAs, there has been significant ethnogeographic variation in prevalence, ranging from < 2% in a Japanese cohort to > 15% in their Italian counterparts [21, 22]. In juvenile DM, a more severe phenotype has been described which includes severe weakness, muscle atrophy, polyarthritis, contractures, and intestinal vasculitis [23, 24]. Calcinosis cutis is a feature of both the juvenile and adult form [22,23,24,25,26]. In adults, more severe systemic disease can be seen with myalgia and dysphagia but relatively milder skin disease [27]. Distal weakness as well as subcutaneous edema has also been described [28]. In contrast to anti-Mi-2 antibodies, biopsy of affected muscle of patients with anti-NXP-2 has shown less primary inflammation than those without the antibody [17]. The relationship with malignancy is conflicting using different cohorts, but possible malignancy has been described only in adults [22,23,24,25,26, 29]. In a study of patients at our institution, we noted a 3.68-fold increased risk of malignancy compared to the general population.
Anti-MDA5 or anti-CADM-140
The anti-clinically amyopathic DM (CADM) 140 antibody was noted by Sato et al. in 2005 to be expressed in over half of Japanese patients with amyopathic DM but none in classic DM or PM [30]. It targets a type of RNA helicase integral to the innate immune system response to RNA viruses called the melanoma differentiation-associated gene 5 protein. Clinically, it is associated with skin-limited disease, minimal muscle involvement, and varying severities of interstitial lung disease (ILD). The cutaneous manifestations transcend those of classic DM and include characteristic skin ulcerations (Fig. 4), palmar papules, and diffuse hair thinning [15•, 31]. Asian patients appear to express anti-CADM-140 at a higher frequency and are particularly prone to the rapidly progressive form of ILD (RPILD), a finding that was not duplicated in earlier studies on Europeans and Americans [32]. However, a recent study on a cohort in the USA showed equal frequency of anti-MDA5 in both CADM and classic DM. This suggests not only that the occurrence of either CADM or classic DM in patients with anti-MDA5 may vary among ethnic groups, but also that in certain populations, anti-MDA5 may not be specific to CADM, but rather to ILD and RPILD [33]. Compared to classic DM, patients with MDA5-positive CADM have a relatively lower risk of associated malignancy [29, 30, 34].

Distinct ulcers surrounding and overlying Gottron’s papules characteristic of MDA5-associated DM.
Anti-p155/140 or anti-TIF1 antibodies
Initially discovered in 2006, anti-TIF1 antibodies were found to target nuclear transcription factors in the human transcriptional intermediatory factor family [35]. Across various ethnogeographic subsets, anti-p155/140 or anti-TIF1 antibodies have been strongly linked to malignancy in DM and have been found in up to 100% of adult patients with cancer-associated myositis in several cohorts [35,36,37]. In juvenile DM, however, it phenotypically manifests as calcinosis [38]. While it carries a reduced risk of ILD, the skin manifestations are typically severe and present as diffuse photoerythema and facial dermatitis often described as a “dusky red” face [15•, 31]. Muscle involvement occurs in the majority of patients. In a report by Fujimoto on Japanese patients, 68% had classic DM while the rest had clinically amyopathic DM [28]. On biopsies of affected muscle, patients who were TIF1-positive appeared to have more mitochondrial dysfunction than their TIF1-negative counterparts [17]. The lung is relatively spared, with only 4% developing ILD in a Japanese cohort [28, 39].
Anti-SAE
Discovered in the sera of DM patients in 2007, anti-SAE antibodies seek out the small ubiquitin-like modifier (SUMO)-activating enzyme involved in post-translational modification [40]. Clinically, anti-SAE positivity has been linked to a CADM-like phenotype featuring prominent cutaneous disease that can precede onset of myositis [7•, 12]. While dysphagia and other systemic features are typical, a high prevalence of ILD was found to be unique to Asian patients only [7•, 12, 41]. The risk of cancer is not clearly established and has ranged from 0 to 57% in various cohorts [40,41,42,43,44,45]. Overall, the prognosis of patients with anti-SAE positivity is considered good [44].
Anti-synthethase antibodies
First discovered in the 1980s, the anti-synthetase antibodies form a group of autoantibodies that target various forms of the aminoacyl-tRNA synthetase enzyme. Clinically, they can present as either dermatomyositis or polymyositis and are therefore not DM-specific but constitute a discrete subgroup. They occur in varying frequencies (approximately 20% for anti-Jo-1 and < 5% for all the rest) and are likewise associated with the development of distinct phenotypes collectively referred to as the anti-synthetase syndrome (ASyS) [37, 46, 47]. In general, ASyS is characterized by the classic triad of myositis, ILD, and polyarthritis occurring alongside other findings such as mechanic’s hands, Raynaud’s phenomenon, or fever [48,49,50]. The classic triad is eventually noted in the majority of ASyS patients especially those expressing anti-Jo-1 positivity but the component symptoms may initially manifest at different points in time [51]. The presence of anti-Jo-1 antibodies, which are detected most frequently in ASyS, portend more severe muscle involvement, whereas anti-PL-7 and anti-PL-12 denote more severe interstitial lung disease as well as increased frequency of gastroesophageal reflux [48, 50, 52]. Incidentally, the Black race has been reported to be an independent predictor of severity of pulmonary involvement [50].
Myositis-associated autoantibodies
DM and PM can occur together with various autoimmune and connective tissue diseases such as systemic sclerosis (SSc), systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), Sjogren’s syndrome, or mixed connective tissue disease. These overlap syndromes are also characterized by the presence of autoantibodies, namely, anti-Ro/SSA, anti-PM/Scl, anti-Ku, and anti-U1-RNP that also have considerable diagnostic and prognostic value. The most common OS involving myositis is PM-Scl, also known as scleromyositis, which makes up 40% of myositis-related overlap syndromes [53].
Anti-Ro/SSA (Anti-Ro52 in particular) is frequently seen in patients with anti-Jo-1-positive ASyS and is associated with earlier onset of arthritis, mechanic’s hands, and similarly appearing, hyperkeratotic, fissured lesions on the feet dubbed “Hiker’s feet” [50, 54, 55]. It has also been linked to more severe myositis and arthritis with worse prognosis [56]. Several studies showed that anti-Ro positivity in the context of ASyS was associated with a higher risk of malignancy whereas others did not yield similar findings [50, 56, 57].
Anti-PM/Scl, also referred to as anti-exosome, is seen in up to 40% of patients with scleromyositis but can also occur in isolated SSc, DM, or PM. It clinically manifests as myositis, arthritis, and Raynaud’s phenomenon with or without concomitant esophageal dysmotility, ILD, pulmonary hypertension, or renal dysfunction [58]. It can mimic the anti-synthetase syndrome. Some reports have found an association with mechanic’s hands and plantar hyperkeratosis [59, 60]. In general, anti-PM/Scl antibodies portend a good prognosis in that skin involvement tends to be limited and responsive to steroids [61].
Anti-Ku antibodies directed against the subunit of a protein kinase involved in DNA repair and recombination were first detected in the sera of Japanese patients with scleromyositis in the 1980s and have been linked to a wide array of clinical findings including arthralgias, myositis, Raynaud’s phenomenon, skin thickening, GERD, and even primary pulmonary hypertension [62,63,64]. As with other autoantibodies, significant differences have been noted across ethnogeographic groups not just in terms of presentation but also with regard to prevalence and disease associations [63,64,65,66].
Anti-U1-RNP occurs in high titers in mixed connective tissue disease but can also be seen in lower titers in SLE and SSc. It has been reported to occur with anti-Jo-1 in severe forms of myositis and with anti-CCP in erosive arthritis [67, 68].
Serologic testing for autoantibodies
Although knowledge of antibody status is very useful for myositis, clinical testing is not as widely available, with a variety of methodologies leading to issues of standardization and validation. Immunoprecipitation (IP) or double immunodiffusion (DID) assays have historically been used to detect autoantibodies in the sera of myositis patients; however, they are cumbersome to perform and are not widely available [69]. To fill the need for improved access to testing by clinicians and researchers alike, commercial kits utilizing enzyme-linked immunosorbent assays (ELISA) or immunoblot assays have been developed and show good performance [32, 37, 70, 71•]. The advent of these high-precision commercial kits that allow for more accessible testing will further enhance the clinical utility of autoantibodies in evaluating DM and other IIMs.
Whether or not MSA and/or MAA titers can be used to monitor disease activity or progression remains to be fully investigated. Two recent studies revealed a correlation between anti-Jo-1 titers and various measures of myositis disease activity including creatinine kinase (CK) levels, while two others found that levels of anti-CADM-140 were either significantly lower or virtually undetectable in patients who were considered treatment-responsive or in clinical remission [30, 72,73,74]. Additionally, anti-TIF1-γ and anti-Mi-2 titers were also shown to correlate with improvement in myositis disease activity indices [74]. Thus, the possibility that certain autoantibodies can serve as biomarkers for monitoring disease activity and treatment response further expands its clinical utility in the management of IIMs.
Caveats to the clinical utility of autoantibodies in DM and other IIMs
Since the first myositis-specific autoantibodies were discovered in the 1980s, there has been a surge in interest in elucidating their prevalence and disease associations. Cohort studies of different sizes involving subjects of varying ethnicities from a wide range of countries have contributed meaningfully to the wealth of information that is now available. As was evident throughout this review, the findings are not always consistent and may even vary considerably across populations. Whether this is attributed to (1) inherent immunogenetic differences partly determined by race, (2) degree or nature of exposure to different exogenous (e.g., viral or ultraviolet [UV]) stimuli depending on geographic location, or (3) non-standardized methods of measuring antibodies and the lack of a uniform set of cut-offs is still uncertain [47]. However, as more and more studies are performed and as testing becomes more accessible, it is reasonable to expect that more definitive trends and patterns will become apparent.
It is also important to keep in mind that other factors independent of autoantibody profile can influence the severity and specific symptomatology of the different DM and IIM subsets. For instance, a recent cohort study in the USA that looked at both Black and Caucasian patients found that the Black race was an independent predictor of interstitial lung disease severity in anti-synthetase syndrome (ASyS) that was unrelated to increased anti-PL-12 expression in Black patients. Further, while amyopathic DM has been traditionally ascribed to the presence of anti-CADM-140 antibodies based on large studies on Asian populations, results gleaned from a US-based cohort found anti-CADM positivity to occur just as frequently in patients with classic DM as those with CADM [33, 75, 76]. Thus, while there may be a mechanistic link between race and autoantibody expression, other racially determined immunogenetic factors independent of autoantibody status may contribute to the observed phenotypic differences.
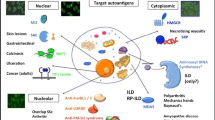
Finally, the exact nature of the relationship between expression of these autoantibodies and their disease associations remains unclear. Whether or not they play a direct role in pathogenesis or merely represent an epiphenomenon is yet to be elucidated [7•, 77,78,79,80]. Some evidence to support a pathogenetic role in genetically susceptible individuals lies in the discovery that myotoxic proteins induced by type 1 interferons (IFN) are upregulated in large numbers in the muscle tissue of patients with DM and that certain autoantibodies such as anti-Jo-1 are capable of inducing type 1 IFN production, leading to a sustained inflammatory response [79, 81, 82]. As further proof, animal studies have shown that the anti-Mi-2 antibody associated with the classic cutaneous manifestations of DM targets a keratinocyte-derived protein whose production is stimulated by UV exposure [83]. The inhibition of this UV-protective response by anti-Mi-2 autoantibodies may thus contribute to development of the photosensitive, polymorphous rashes of DM. As such, a deeper understanding of the function of not just the autoantibodies themselves but of their target antigens is crucial to determining their precise roles, if any, in disease pathogenesis.
Conclusions
Myositis-specific and myositis-associated autoantibodies play an ever-increasing role in the diagnosis, management, and prognostication of DM and the other idiopathic inflammatory myopathies. With the introduction of more easily performable quantification methods such as ELISA and the push to establish standardized cut-offs, their usefulness may eventually extend to disease activity and treatment response monitoring. However, variations in the frequency of autoantibody positivity and their corresponding phenotypes across different cohorts that are likely driven by a complex interplay of racially, genetically, and environmentally determined factors independent of autoantibody status serve as limitations to its usefulness. As more and more data from studies on diverse ethnogeographic groups becomes available, it is likely that more definitive and reliable population-specific correlations in autoantibody and phenotypic expression will emerge. Regardless, it falls on the clinician to adopt an individualized approach to each patient and incorporate all the other available clinical, histopathologic, serologic, and radiographic tools to arrive at the correct diagnosis and determine the appropriate treatment strategy.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971–82.
Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344–7.
Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:403–7.
Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10–12 October 2003, Naarden, The Netherlands. Neuromuscul Disord. 2004;14:337–45.
Kissel JT, Mendell JR, Rammohan KW. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med. 1986;314:329–34.
Brouwer R, Hengstman GJ, Vree Egberts W, Ehrfeld H, Bozic B, Ghirardello A, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis. 2001;60:116–23.
• Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology. 2009;48:607–12. This review highlights the clinical utility and pathogenic significance of myositis-specific antibodies in disease expression.
Hamaguchi Y, Kuwana M, Hoshino K, Hasegawa M, Kaji K, Matsushita T, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol. 2011;147:391–8.
Merlo G, Clapasson A, Cozzani E, Sanna L, Pesce G, Bagnasco M, et al. Specific autoantibodies in dermatomyositis: a helpful tool to classify different clinical subsets. Arch Dermatol Res. 2017;309:87–95.
•• Betteridge Z, McHugh N. Myositis-specific autoantibodies: an important tool to support diagnosis of myositis. J Intern Med. 2016;280:8–23. This review highlights the clinical utility of antibodies as both diagnostic and prognostic markers of disease.
Tansley SL, Betteridge ZE, McHugh NJ. The diagnostic utility of autoantibodies in adult and juvenile myositis. Curr Opin Rheumatol. 2013;25:772–7.
Ghirardello A, Borella E, Beggio M, Franceschini F, Fredi M, Doria A. Myositis autoantibodies and clinical phenotypes. Auto- Immun Highlights. 2014;5:69–75.
Yoshifuji H. Biomarkers and autoantibodies of interstitial lung disease with idiopathic inflammatory myopathies. Clin Med Insights Circ Respir Pulm Med. 2016;9:141–6.
Targoff IN, Reichlin M. The association between Mi-2 antibodies and dermatomyositis. Arthritis Rheum. 1985;28:796–803.
• Daly ML, Gordon PA, Creamer D. Cutaneous features of dermatomyositis associated with myositis-specific antibodies. Br J Dermatol. 2016;176:1662–5. This paper described the associations between specific dermatologic manifestations and dermatomyositis antibodies.
Aggarwal R, Cassidy E, Fertig N, Koontz DC, Lucas M, Ascherman DP, et al. Patients with non-Jo-1 anti-tRNA-synthetase autoantibodies have worse survival than Jo-1 positive patients. Ann Rheum Dis. 2014;73:227–32.
Pinal-Fernandez I, Casciola-Rosen LA, Christopher-Stine L, Corse AM, Mammen AL. The prevalence of individual histopathologic features varies according to autoantibody status in muscle biopsies from patients with dermatomyositis. J Rheumatol. 2015;42:1448–54.
Ghirardello A, Zampieri S, Iaccarino L, Tarricone E, Bendo R, Gambari PF, et al. Anti-Mi-2 antibodies. Autoimmunity. 2005;38:79–83.
Komura K, Fujimoto M, Matsushita T, Kaji K, Kondo M, Hirano T, et al. Prevalence and clinical characteristics of anti-Mi-2 antibodies in Japanese patients with dermatomyositis. J Dermatol Sci. 2005;40:215–7.
Madan V, Chinoy H, Griffiths CEM, Cooper RG. Defining cancer-risk, and assessing diagnostic usefulness of myositis serology, in dermatomyositis—Part 2. Clin Exp Dermatol. 2009;34:561–5.
Ichimura Y, Matsushita T, Hamaguchi Y, Kaji K, Hasegawa M, Tanino Y, et al. Anti-NXP2 autoantibodies in adult patients with idiopathic inflammatory myopathies: possible association with malignancy. Ann Rheum Dis. 2012;71:710–3.
Ceribelli A, Fredi M, Taraborelli M, Cavazzana I, Franceschini F, Quinzanini M, et al. Anti-MJ/NXP-2 autoantibody specificity in a cohort of adult Italian patients with polymyositis/dermatomyositis. Arthritis Res Ther. 2012;14:R97.
Oddis CV, Fertig N, Goel A, Espada G, Confalone Gregorian M, Maldonado Cocco JA. Clinical and serological characterization of the anti-MJ antibody in childhood myositis. Arthritis Rheum. 1997;40:S139.
Espada G, Maldonado Cocco JA, Fertig N, Oddis CV. Clinical and serologic characterization of an Argentine pediatric myositis cohort: identification of a novel autoantibody (anti-MJ) to a 142-kDa protein. J Rheumatol. 2009;36:2547–51.
Tansley SL, Betteridge ZE, Shaddick G, Gunawardena H, Arnold K, Wedderburn LR, et al. Calcinosis in juvenile dermatomyositis is influenced by both anti-NXP2 autoantibody status and age at disease onset. Rheumatol Oxf Engl. 2014;53:2204–8.
Albayda J, Pinal-Fernandez I, Huang W, Parks C, Paik J, Casciola-Rosen L, et al. Dermatomyositis patients with anti-nuclear matrix protein-2 autoantibodies have more edema, more severe muscle disease, and increased malignancy risk. Arthritis Care Res. 2017 [cited 2017 May 6]. doi: https://doi.org/10.1002/acr.23188/full.
Rogers A, Chung L, Li S, Casciola-Rosen L, Fiorentino DF. The cutaneous and systemic findings associated with nuclear matrix protein-2 antibodies in adult dermatomyositis patients. Arthritis Care Res. 2017; https://doi.org/10.1002/acr.23210.
Fujimoto M, Hamaguchi Y, Kaji K, Matsushita T, Ichimura Y, Kodera M, et al. Myositis-specific anti-155/140 autoantibodies target transcription intermediary factor 1 family proteins. Arthritis Rheum. 2012;64:513–22.
Fiorentino DF, Chung LS, Christopher-Stine L, Zaba L, Li S, Mammen AL, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1γ. Arthritis Rheum. 2013;65:2954–62.
Sato S, Hirakata M, Kuwana M, Suwa A, Inada S, Mimori T, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum. 2005;52:1571–6.
Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allergy Immunol. 2016;51:293–302.
Nakashima R, Hosono Y, Mimori T. Clinical significance and new detection system of autoantibodies in myositis with interstitial lung disease. Lupus. 2016;25:925–33.
Moghadam-Kia S, Oddis CV, Sato S, Kuwana M, Aggarwal R. Antimelanoma differentiation-associated gene 5 antibody: expanding the clinical spectrum in North American patients with dermatomyositis. J Rheumatol. 2017;44:319–25.
Hoshino K, Muro Y, Sugiura K, Tomita Y, Nakashima R, Mimori T. Anti-MDA5 and anti-TIF1-γ antibodies have clinical significance for patients with dermatomyositis. Rheumatology. 2010;49:1726–33.
Targoff IN, Mamyrova G, Trieu EP, Perurena O, Koneru B, O’Hanlon TP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. 2006;54:3682–9.
Fujikawa K, Kawakami A, Kaji K, Fujimoto M, Kawashiri S, Iwamoto N, et al. Association of distinct clinical subsets with myositis-specific autoantibodies towards anti-155/140-kDa polypeptides, anti-140-kDa polypeptides, and anti-aminoacyl tRNA synthetases in Japanese patients with dermatomyositis: a single-centre, cross-sectional study. Scand J Rheumatol. 2009;38:263–7.
Satoh M, Tanaka S, Ceribelli A, Calise SJ, Chan EKL. A comprehensive overview on myositis-specific antibodies: new and old biomarkers in idiopathic inflammatory myopathy. Clin Rev Allergy Immunol. 2017;52:1–19.
Gunawardena H, Wedderburn LR, Chinoy H, Betteridge ZE, North J, Ollier WER, et al. Autoantibodies to a 140-kd protein in juvenile dermatomyositis are associated with calcinosis. Arthritis Rheum. 2009;60:1807–14.
Fiorentino DF, Kuo K, Chung L, Zaba L, Li S, Casciola-Rosen L. Distinctive cutaneous and systemic features associated with antitranscriptional intermediary factor-1γ antibodies in adults with dermatomyositis. J Am Acad Dermatol. 2015;72:449–55.
Betteridge ZE, Gunawardena H, Chinoy H, North J, Ollier WER, Cooper RG, et al. Clinical and human leucocyte antigen class II haplotype associations of autoantibodies to small ubiquitin-like modifier enzyme, a dermatomyositis-specific autoantigen target, in UK Caucasian adult-onset myositis. Ann Rheum Dis. 2009;68:1621–5.
Fujimoto M, Matsushita T, Hamaguchi Y, Kaji K, Asano Y, Ogawa F, et al. Autoantibodies to small ubiquitin-like modifier activating enzymes in Japanese patients with dermatomyositis: comparison with a UK Caucasian cohort. Ann Rheum Dis. 2013;72:151–3.
Muro Y, Sugiura K, Nara M, Sakamoto I, Suzuki N, Akiyama M. High incidence of cancer in anti-small ubiquitin-like modifier activating enzyme antibody-positive dermatomyositis. Rheumatology. 2015;54:1745–7.
Ge Y, Lu X, Shu X, Peng Q, Wang G. Clinical characteristics of anti-SAE antibodies in Chinese patients with dermatomyositis in comparison with different patient cohorts. Sci Rep. 2017;7:188.
Tarricone E, Ghirardello A, Rampudda M, Bassi N, Punzi L, Doria A. Anti-SAE antibodies in autoimmune myositis: identification by unlabelled protein immunoprecipitation in an Italian patient cohort. J Immunol Methods. 2012;384:128–34.
Bodoki L, Nagy-Vincze M, Griger Z, Betteridge Z, Szöllősi L, Dankó K. Four dermatomyositis-specific autoantibodies-anti-TIF1γ, anti-NXP2, anti-SAE and anti-MDA5-in adult and juvenile patients with idiopathic inflammatory myopathies in a Hungarian cohort. Autoimmun Rev. 2014;13:1211–9.
Nishikai M, Reichlin M. Heterogeneity of precipitating antibodies in polymyositis and dermatomyositis. Characterization of the Jo-1 antibody system. Arthritis Rheum. 1980;23:881–8.
Mimori T. Clinical significance of anti-Ku autoantibodies—a serologic marker of overlap syndrome? Intern Med Tokyo Jpn. 2002;41:1096–8.
Hervier B, Benveniste O. Clinical heterogeneity and outcomes of antisynthetase syndrome. Curr Rheumatol Rep. 2013;15:349.
Mirrakhimov AE. Antisynthetase syndrome: a review of etiopathogenesis, diagnosis and management. Curr Med Chem. 2015;22:1963–75.
Pinal-Fernandez I, Casal-Dominguez M, Huapaya JA, Albayda J, Paik JJ, Johnson C, et al. A longitudinal cohort study of the anti-synthetase syndrome: increased severity of interstitial lung disease in black patients and patients with anti-PL7 and anti-PL12 autoantibodies. Rheumatol Oxf Engl. 2017;56:999–1007.
Bartoloni E, Gonzalez-Gay MA, Scirè C, Castaneda S, Gerli R, Lopez-Longo FJ, et al. Clinical follow-up predictors of disease pattern change in anti-Jo1 positive anti-synthetase syndrome: results from a multicenter, international and retrospective study. Autoimmun Rev. 2017;16:253–7.
Marie I, Josse S, Decaux O, Dominique S, Diot E, Landron C, et al. Comparison of long-term outcome between anti-Jo1- and anti-PL7/PL12 positive patients with antisynthetase syndrome. Autoimmun Rev. 2012;11:739–45.
Troyanov Y, Targoff IN, Tremblay J-L, Goulet J-R, Raymond Y, Senécal J-L. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore). 2005;84:231–49.
Cruellas MGP, dos Santos Trindade Viana V, Levy-Neto M, de Souza FHC, Shinjo SK. Myositis-specific and myositis-associated autoantibody profiles and their clinical associations in a large series of patients with polymyositis and dermatomyositis. Clinics. 2013;68:909–14.
Cox JT, Gullotti DM, Mecoli CA, Lahouti AH, Albayda J, Paik J, et al. “Hiker’s feet”: a novel cutaneous finding in the inflammatory myopathies. Clin Rheumatol. 2017;36:1683–6.
Marie I, Hatron PY, Dominique S, Cherin P, Mouthon L, Menard J-F, et al. Short-term and long-term outcome of anti-Jo1-positive patients with anti-Ro52 antibody. Semin Arthritis Rheum. 2012;41:890–9.
Chinoy H, Fertig N, Oddis CV, Ollier WER, Cooper RG. The diagnostic utility of myositis autoantibody testing for predicting the risk of cancer-associated myositis. Ann Rheum Dis. 2007;66:1345–9.
Mahler M, Raijmakers R. Novel aspects of autoantibodies to the PM/Scl complex: clinical, genetic and diagnostic insights. Autoimmun Rev. 2007;6:432–7.
Marie I, Lahaxe L, Benveniste O, Delavigne K, Adoue D, Mouthon L, et al. Long-term outcome of patients with polymyositis/dermatomyositis and anti-PM-Scl antibody. Br J Dermatol. 2010;162:337–44.
Muro Y, Hosono Y, Sugiura K, Ogawa Y, Mimori T, Akiyama M. Anti-PM/Scl antibodies are found in Japanese patients with various systemic autoimmune conditions besides myositis and scleroderma. Arthritis Res Ther. 2015;17:57.
Vandergheynst F, Ocmant A, Sordet C, Humbel RL, Goetz J, Roufosse F, et al. Anti-pm/scl antibodies in connective tissue disease: clinical and biological assessment of 14 patients. Clin Exp Rheumatol. 2006;24:129–33.
Isern RA, Yaneva M, Weiner E, Parke A, Rothfield N, Dantzker D, et al. Autoantibodies in patients with primary pulmonary hypertension: association with anti-Ku. Am J Med. 1992;93:307–12.
Cooley HM, Melny BJ, Gleeson R, Greco T, Kay TW. Clinical and serological associations of anti-Ku antibody. J Rheumatol. 1999;26:563–7.
Franceschini F, Cavazzana I, Generali D, Quinzanini M, Viardi L, Ghirardello A, et al. Anti-Ku antibodies in connective tissue diseases: clinical and serological evaluation of 14 patients. J Rheumatol. 2002;29:1393–7.
Reeves WH. Use of monoclonal antibodies for the characterization of novel DNA-binding proteins recognized by human autoimmune sera. J Exp Med. 1985;161:18–39.
Váncsa A, Gergely L, Ponyi A, Lakos G, Németh J, Szodoray P, et al. Myositis-specific and myositis-associated antibodies in overlap myositis in comparison to primary dermatopolymyositis: relevance for clinical classification: retrospective study of 169 patients. Joint Bone Spine. 2010;77:125–30.
Feist E, Schneider M, Brychcy M, Dorner T, Burmester G, Hiepe F. A unique autoantibody pattern of positive anti-Jo-1, anti-U1RNP, and antiproteasome antibodies in autoimmune myositis as a diagnostic challenge. Ann Rheum Dis. 2003;62:370–1.
Szodoray P, Hajas A, Kardos L, Dezso B, Soos G, Zold E, et al. Distinct phenotypes in mixed connective tissue disease: subgroups and survival. Lupus. 2012;21:1412–22.
Cavazzana I, Fredi M, Ceribelli A, Mordenti C, Ferrari F, Carabellese N, et al. Testing for myositis specific autoantibodies: comparison between line blot and immunoprecipitation assays in 57 myositis sera. J Immunol Methods. 2016;433:1–5.
Aggarwal R, Oddis CV, Goudeau D, Fertig N, Metes I, Stephens C, et al. Anti-transcription intermediary factor 1-gamma autoantibody ELISA development and validation. Rheumatology. 2014;53:433–7.
• Fujimoto M, Murakami A, Kurei S, Okiyama N, Kawakami A, Mishima M, et al. Enzyme-linked immunosorbent assays for detection of anti-transcriptional intermediary factor-1 gamma and anti-Mi-2 autoantibodies in dermatomyositis. J Dermatol Sci. 2016;84:272–81. This study demonstrates the usefulness of newly established ELISAs to detect particular MSAs such as TIF1-gamma and anti-Mi-2, which can lead to more routine testing.
Stone KB, Oddis CV, Fertig N, Katsumata Y, Lucas M, Vogt M, et al. Anti-Jo-1 antibody levels correlate with disease activity in idiopathic inflammatory myopathy. Arthritis Rheum. 2007;56:3125–31.
Muro Y, Sugiura K, Hoshino K, Akiyama M. Disappearance of anti-MDA-5 autoantibodies in clinically amyopathic DM/interstitial lung disease during disease remission. Rheumatol Oxf Engl. 2012;51:800–4.
Aggarwal R, Oddis CV, Goudeau D, Koontz D, Qi Z, Reed AM, et al. Autoantibody levels in myositis patients correlate with clinical response during B cell depletion with rituximab. Rheumatol Oxf Engl. 2016;55:991–9.
Sato S, Kuwana M, Fujita T, Suzuki Y. Anti-CADM-140/MDA5 autoantibody titer correlates with disease activity and predicts disease outcome in patients with dermatomyositis and rapidly progressive interstitial lung disease. Mod Rheumatol. 2013;23:496–502.
Fujisawa T, Hozumi H, Kono M, Enomoto N, Hashimoto D, Nakamura Y, et al. Prognostic factors for myositis-associated interstitial lung disease. PLoS One. 2014;9:e98824.
Arad-Dann H, Isenberg D, Ovadia E, Shoenfeld Y, Sperling J, Sperling R. Autoantibodies against a nuclear 56 kDa protein: a marker for inflammatory muscle disease. J Autoimmun. 1989;2:877–88.
Stuhlmüller B, Jerez R, Hausdorf G, Barthel H-R, Meurer M, Genth E, et al. Novel autoantibodies against muscle-cell membrane proteins in patients with myositis. Arthritis Rheum. 1996;39:1860–8.
Lundberg IE, Dastmalchi M. Possible pathogenic mechanisms in inflammatory myopathies. Rheum Dis Clin N Am. 2002;28:799–822.
Katirji B, Kaminski HJ, Ruff RL. Neuromuscular disorders in clinical practice. New York: Springer; 2013.
Greenberg SA. Inflammatory myopathies: disease mechanisms. Curr Opin Neurol. 2009;22:516–23.
Day J, Otto S, Proudman S, Hayball JD, Limaye V. Dysregulated innate immune function in the aetiopathogenesis of idiopathic inflammatory myopathies. Autoimmun Rev. 2017;16:87–95.
Burd CJ, Kinyamu HK, Miller FW, Archer TK. UV radiation regulates Mi-2 through protein translation and stability. J Biol Chem. 2008;283:34976–82.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Albayda declares that she has no conflict of interest. Dr. Castillo declares that she has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Other CTD: Inflammatory Myopathies and Sjogren’s
Rights and permissions
About this article

Cite this article
Castillo, R., Albayda, J. Dermatomyositis: Autoantibodies and Their Corresponding Phenotypes. Curr Treat Options in Rheum 3, 254–266 (2017). https://doi.org/10.1007/s40674-017-0078-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40674-017-0078-7