
Abstract
T regulatory cells (Tregs) represent a phenotypically and functionally heterogeneous group of lymphocytes that exert immunosuppressive activities on effector immune responses. Tregs play a key role in maintaining immune tolerance and homeostasis through diverse mechanisms which involve interactions with components of both the innate and adaptive immune systems. As in many autoimmune diseases, Tregs have been proposed to play a relevant role in the pathogenesis of systemic lupus erythematosus (SLE), an autoimmune disease characterized by a progressive breakdown of tolerance to self-antigens and the presence of concomitant hyperactive immune responses. Here, we review how Tregs dysfunction in SLE has been manipulated experimentally and preclinically in the attempt to restore, at last in part, the immune disturbances in the disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease that associates with a progressive deterioration of the mechanisms of immune tolerance. An impaired ability to discriminate between foreign and self-antigens results in autoimmune attacks in which both the innate and adaptive arms of the immune system take part. The prevalent pathologic manifestations of the disease result from aberrant production of autoantibodies and the release of proinflammatory mediators that promote and/or exacerbate tissue damage. Ultimately, these events can occur in multiple organs and can cause a prolonged local inflammatory response that can lead to compromised organ function.
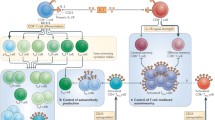
Given that the severity and symptoms of SLE are often heterogeneous among patients, the investigations on the molecular mechanisms of the disease pathogenesis have often resulted in complex pictures that have included multiple components. Among them, dysregulated numbers and/or function of T regulatory cells (Tregs) have been reported by many groups (reviewed in [1]), as well as impaired mechanisms of Tregs activities on their target cells (Fig. 1).

Schematic mechanisms of Treg-mediated suppression on Teffs, B cells, and APCs (DCs). The figure shows the Treg-mediated paracrine effects through cytokines, cytotoxicity, apoptosis, suppression of antibody production, contact-dependent induction of anergy, inhibition of costimulatory signals (needed for cell activation)
Since the initial discovery of the Tregs, a dysregulation of this cell subset has been identified in multiple autoimmune diseases including type 1 diabetes, multiple sclerosis, inflammatory bowel disease, rheumatoid arthritis, and SLE [2].
The critical role of Tregs in immune homeostasis is best exemplified by the syndrome named immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX), a rare and often fatal autoimmune disease caused by a loss-of-function mutation in the FOXP3 gene (a transcription factor of the forkhead box P family that is the master regulator for Tregs). The degree of disease severity resulting from the mutation of this single gene offers insights into how fundamental FOXP3 is in inducing immune tolerance [3].
Phenotypic Features of Tregs
Tregs can be classified according to their origin and then further divided according to their phenotype and function. The broad categories include tTregs (derived from the thymus), pTregs (induced in the periphery), and iTregs (or in vitro-induced Tregs) [4]. However, there are no clear-cut phenotypic markers that can distinguish one subset from another, which is a problematic aspect for discerning effectiveness of Tregs as suppressor cells (not last, for possible use in immunotherapy). While it has been challenging to find a phenotypic marker that can be unique to Tregs, two main Treg markers have long remained important to infer a rather reliable phenotype: the IL-2 receptor-α (CD25) and FOXP3. However, these markers can also be transiently expressed in activated T effector cells (Teffs) [5] and may not be present in IL-10-producing Tr1 Tregs [6].
Other molecules associated with Treg suppressive function include CTLA-4, glucocorticoid-induced tumor necrosis factor receptor-related protein (GITR), LAG-3, OX40, CD73, and CD39 but, again, these markers are also not exclusive to Tregs [7]. While there have been several molecules purported to be tTreg-specific, such as Helios in murine and human tTregs and neuropilin-1 in murine tTregs, it has subsequently been shown that these molecules may also be expressed on Teffs [7]. A good marker for suppressor Tregs is GITR [8], and another one for tTregs is the presence of the Treg-specific demethylated region (TSDR) [9], which strongly correlates with FOXP3 expression [10]. Nonetheless, the fact that TSDR and FOXP3 are intracellular makes the sorting of live Tregs using those markers problematic.
Given this scenario and the lack of markers exclusive to Tregs, the most practical and common way to differentiate Tregs generally uses a rather simplistic combination of markers—CD4+CD25+Foxp3+ for mice and CD4+CD25highCD127-FoxP3+ for humans [5].
Physiopathology of Tregs in SLE
Data on Tregs in SLE have been at times controversial in regard to the numbers and function of these cells in lupus patients. Although most studies indicate a decreased frequency of Tregs in SLE [11, 12], with possibly greater decrease during active disease [13], some studies have not confirmed such correlations [14].
Assays evaluating Treg function in SLE patients vs. healthy controls have also given varying results, although most studies have reported a decreased suppressive function of Tregs in SLE [15]. This has led to the complementary and non-mutually exclusive possibility that a reduction in Treg activity in SLE might in part also depend on the fact that Teffs can acquire resistance to suppression by Tregs, at least under certain circumstances [16]. In any case, the multifaceted immune derangements in SLE seem to convincingly include Tregs as major contributors to the pathologic events in the disease.
Tregs Effects on Humoral Immunity
Scurfy (Sf) mice carry a missense mutation in Foxp3 [3] that leads to the absence of tTregs and renders these mice incapable of producing iTregs. This indicates a requirement of Foxp3 for the generation and survival of Tregs [17]. Sf share features with lupus phenotypes including lymphopenia, anemia, pneumonitis, arthritis, and nephritis, although their multiorgan inflammation also involves systems that are usually spared in SLE [17]. Notably, Sf mice have autoantibodies that are also seen in SLE (anti-nuclear antibody (ANA), anti-Smith, and anti-dsDNA) [17], suggesting that the absence of Tregs can favor B cell dysfunction. This possibility has more directly been demonstrated by studies that showed Treg-mediated suppression of autoantibody-producing lupus B cells through mechanisms including apoptosis of B cells and contact-dependent cytotoxicity mechanisms mediated by perforin and granzyme [18]. Those mechanisms could be complementary to the paracrine function of Tregs in the modulation of the immune response that involves the release of the anti-inflammatory cytokines TGF-β, IL-10, and IL-35 (e.g., TGF-β is essential to the peripheral induction of Tregs, and its serum levels are decreased in lupus patients) [19].
Treg Effects on Cell-Mediated Immunity
Tregs from lupus patients also induce anergy in CD4+ Teffs that help lupus B cells to produce autoantibodies [20]. CD8+ Tregs can also mediate apoptosis of CD4+ Teffs [21], and the frequency of these Foxp3-expressing cells increases following tolerization protocols that prevent the development of SLE manifestations [21]. CD8+ Tregs may facilitate induction of CD4+ Tregs in SLE [22], and their frequency increases in renal tissue after therapy with i.v. methylprednisolone [23] and in the peripheral blood after autologous HSCT [24].
Treg Effects on APCs
Antigen-presenting cells (APCs) mediate sensitization and activation of Teffs and represent therefore important players in immune regulation. One mechanism by which Tregs modulate APC function is through CTLA-4, an inhibitory molecule expressed on Tregs that can prevent the upregulation or downregulate the expression of CD80 and CD86 on dendritic cells (DCs). CD80/CD86 can bind to CTLA-4 and serve as costimulatory signals that are essential to the activation of Teffs [25]. The importance of this pathway is highlighted by the observation that genetic deficiency in CTLA-4 in mice causes systemic autoinflammatory disease, and that CTLA-4-deficient Tregs have reduced suppressive function both in vivo and in vitro [26].
Additional mediators of APC-mediated regulation include IL-10 and neuropilin-1. The latter is expressed on Foxp3+ cells and promotes long interactions with APCs that could possibly give Tregs an advantage in competing for antigen with Teffs, particularly under conditions of low antigen availability [2].
Effects of Proinflammatory Cytokines on Treg Function
There is evidence that a proinflammatory cytokine milieu can attenuate Treg function [27]. Serum levels of IL-6 are elevated in SLE [28], and this cytokine renders Th17 cells more resistant to the suppressive effects mediated by Tregs [29]. The conversion of Tregs into Th17 in the presence of IL-6 is a finding of particular interest, considering the abnormally increased levels of IL-17 levels in lupus patients [30]. Interestingly, naturally and in vitro-induced Tregs respond to IL-6 differently, since iTregs can resist conversion under defined settings [29].
IFN-α is another proinflammatory cytokine hyperexpressed in SLE that reduces the tolerogenic effects of Tregs on DCs and weakens Treg-suppressor activity [31]. Increased levels of TNF-α have also been associated by some authors with an attenuated Treg-suppressive function as well as a downregulation of Foxp3 [32, 33].
More recently, a Treg population operating in a proinflammatory environment has been described, the so-called effector Tregs (eTregs). The broad, dichotomous classification of Tregs into central Tregs (cTregs) and eTregs is based on localization and function. cTregs regulate T cell priming in secondary lymphoid tissue while eTregs, which express high levels of molecules involved in cellular activation such as ICOS, GITR, and CD69, typically home to non-lymphoid tissue [34]. eTregs are also comparatively much more proliferative and short-lived than cTregs are and express lower levels of the antiapoptotic molecule Bcl-2 [35]. Additionally, eTregs have the capacity to vary the scale of their effector response in reaction to inflammation, using the transcription factor IRF-4 to promote their own survival through the upregulation of autophagy genes [36•, 37].
Tregs and Teffs
Under normal circumstances, the balance between Tregs and Teffs is finely regulated, and the plasticity of T cells allows Teffs—or at least certain populations of them—to acquire Treg functions, at least temporarily [38]. Investigations have shown that the activation of self-antigen-specific CD4+ Teffs results in an anergic response characterized by upregulation of CD73 and FR4 in the presence of Tregs [39, 40]. These cells can become precursors of pTregs lacking CD25 expression and with TSDR hypomethylation profiles in genes related to Treg function: Ctla4, Foxp3, Ikzf4, and Tnfrsf18 [40–42].
Also of note, cell metabolism seems to differ between Tregs and Teffs. Comparative proteomic analyses of Teffs and Tregs showed that both types of cells use predominantly glycolytic pathways in vivo, with Tregs more metabolically active than Teffs are at least under certain circumstances [43]. Of interest, metabolic intervention depressing FoxP3 expression reduced Treg-suppressive function through a mammalian target of rapamycin (mTOR)-dependent process [44]. Indeed, mTOR is highly expressed in proliferating Tregs and is instrumental in coupling TCR activation and IL-2 signaling for the suppressive function of Tregs [45•]. In this sense, the balance between Tregs and Teffs is highly influenced by the Teffs’ production of IL-2 required for Treg differentiation and survival [46]. This links Teffs and Tregs in that a greater Teff activity results in increased release of IL-2, which in turn promotes Treg activity. Thus, under physiologic conditions, a rise in immunogenic activity of Teffs is balanced by the activity of Tregs.
Tregs-Based Therapies in SLE
Most Treg-based therapeutic approaches build upon the premise that a reduced number and/or suppressive function of Tregs can favor SLE pathology. Below, we summarize the attempts designed thus far to increase the numbers and/or function of Tregs in SLE (Table 1).
Adoptive Transfer of Ex Vivo-Expanded Tregs
Human Tregs expanded in vitro can display an enhanced immunoregulatory activity that has been observed when using both autologous polyclonal and antigen-specific Tregs [47,48]. The success of those experiments, as well as that of adoptive transfers of Tregs in lupus mice [49], facilitated the implementation of phase I and II clinical trials in other autoimmune diseases. Adoptive transfers of Tregs in patients with type 1 diabetes (T1D) and graft-versus-host disease (GVHD) showcased the feasibility of generating therapeutic quantities of sufficiently pure iTreg through polyclonal expansion. [50•, 51, 52, 53•]. Unfortunately, there was no evidence of clinical improvement after adoptive Treg transfer in T1D, as measured by C-peptide levels and HbA1C over a follow-up period of 1–2 years [50•]. However, a small clinical trial using Tregs derived from cord blood showed significant improvement in patients with acute GVHD [53•]. These studies exposed a critical aspect of adoptive Treg immunotherapy: the limited survival of Tregs in vivo. Treg levels experience a dose-independent dramatic decline by 14 days after treatment. Despite the short life span of Tregs in vivo, up to 25 % of the transferred Tregs survived for over 1 year after transplant in some T1D patients [50•], leaving hope for future research. However, for SLE, there are no available data at present on the use of adoptively transferred iTregs in patients.
Hematopoietic and Mesenchymal Stem Cell Transplant
Immune reconstitution through hematopoietic and/or mesenchymal stem cell transplant (HSCT and MSCT, respectively) after chemotherapy has been a therapeutic option for patients with severe autoimmune disease refractory to standard management. HSCT has been shown to be effective in inducing long-term remission in SLE, and if disease returned, it tended to be milder [54].
Mesenchymal stem cells (MSCs) have potent immunosuppressive function and do not require MHC restriction to operate, making them good therapeutic candidate agents [55].
MSCT/HSCTs have been successful in inducing remissions in lupus patients refractory to treatment or with organ damage [56]. The therapeutic benefits deriving from those approaches seem to involve Treg modulation [24]. A clinical trial employing autologous HSCT in 15 patients with refractory SLE found that the treatment increased the frequency of CD4+CD25highFoxP3+ cells to levels comparable to those seen in healthy patients [24]. It also induced a population of CD8+FoxP3+ Treg that was absent in patients with active disease and that exerted powerful suppression through contact-independent, TGF-β-mediated mechanisms that involved antigen-specific and non-specific responses [24]. In contrast, a smaller trial of autologous HSCT in 12 patients with various refractory autoimmune diseases (SLE, rheumatoid arthritis, mixed connective tissue disease, systemic sclerosis) found no changes in Tregs levels after transplant, although only three SLE patients took part in that trial [57].
For MSCT in SLE, a 4-year follow-up on 87 patients showed a 28 % remission rate at 12 months posttransplant [58]. However, the effects of MSCT on Tregs were not assessed. Instead, a case report of a refractory SLE patient receiving autologous HSCT followed by MSCT did not show a significant uptrend in Treg numbers but did demonstrate persistently increased Tregs/Teffs ratios, peaking at 6 months [59].
IL-2
As mentioned before, IL-2 has a crucial role in immune homeostasis, promoting the differentiation and survival of Tregs [60]. IL-2 levels are reduced in SLE, and administration of low-dose IL-2 in SLE patients associated with disease improvement as manifested by reduced need for glucocorticoids, normalized numbers of blood cells and platelets, and significant reductions in SLEDAI score [61]. An expansion of CD25highCD127low Tregs and a decrease in Teffs/Tregs ratios were also observed [62, 63]. Also encouraging were the data of low-dose IL-2 in GVHD, where significant increases in numbers of CD4+CD25+FoxP3+ cells were observed [64].
Retinoids
Data from lupus mice and clinical studies suggest that all-trans retinoic acid (atRA) can improve symptoms and laboratory indices of SLE [65]. However, the benefits of this therapy are inconsistent, and the small sample sizes used in clinical studies does not allow conclusions on the use of retinoids in SLE [66]. A combination of atRA and prednisolone enhanced survival and improved proteinuria in lupus mice as compared to prednisolone treatment alone [65], and two lupus nephritis patients showed improvement of urinary protein and anti-dsDNA titers after 6 month treatment with atRA [67].
Tolerogenic Peptides
Multiple tolerogenic peptides have been developed for possible therapeutic use in SLE. The concept behind this approach is that a dysregulated immune system can modify a pre-established response to a self-antigen through the induction of tolerogenic responses to the same self-antigen. One requirement for a successful outcome using this approach requires the maintenance of effective immune responses to unrelated exogenous antigens, i.e., lack of generalized immune suppression. The peptides hCDR1, pCons, P140, and nucleosomal peptides H471-94 are based on amino acid sequences from various self-antigens that are known to be targets of autoimmune attack in SLE. The tolerogenic activities of these peptides are manifested by an expansion of Tregs and the suppression of the production of proinflammatory cytokines and effector immune cells [20].
Low-dose injections of nucleosomal histone-derived peptide induced tolerance against this self-antigen, primarily via splenic DCs which upregulated TGF-β expression and stimulated the differentiation of antigen-specific CD4+ and CD8+ Tregs [68]. Other studies showed that the induction of CD8+ Tregs anticipated that of CD4+ Tregs [22].
Encouraging data in mouse models of SLE [20, 68, 69] also led to clinical trials using two peptides, hCDR1and P140. Edratide (hCDR1) underwent phase II clinical trials that did not meet endpoints, while Lupuzor (P140) is undergoing a phase III clinical trial after the phase IIb clinical trial indicated efficacy of 3 month therapy, with an improvement rate of 84.2 % in peptide-treated patients vs. 45.8 % in the placebo group [70•].
Statins
Increased numbers of Tregs have been linked to therapies using statins, which are drugs used in the management of atherosclerosis (and SLE is a known risk factor for atherosclerosis). Statins have modulatory effects on Tregs and Th17 cells [71], and a trial on three patients with refractory lupus found that 80 mg simvastatin o.d. for 8 days associated with a significant improvement in proteinuria and downregulation of the immune activation marker CD69 [72]. Larger, mechanistic studies are required to draw possible conclusions on the impact of statins in SLE.
Effect of Common Lupus Therapies on Tregs
There are also studies investigating the effects of lupus therapies on Tregs. High-dose methylprednisolone augmented eTreg numbers, at least for the first 8 days after treatment [73]. An increase in Tregs at day 2 associated with the absence of acute disease for at least 1 year, suggesting that the body’s capacity to generate Tregs after induction therapy in SLE may be a key factor in maintaining immune homeostasis, as confirmed by the finding of significant increases in pTregs after cyclophosphamide pulse therapy [74]. These observations also raise the possibility of using Tregs for monitoring disease activity in SLE, since changes in Treg numbers can reflect changes of disease status.
Interestingly, transient B cell depletion using anti-CD20 monoclonal antibody (rituximab) was found to enhance the numbers and function of Tregs [75, 76] and FoxP3 expression that persisted in patients with clinical remission [77].
Challenges in Using Tregs-Based Therapies for SLE
While preliminary data on the use of Treg-based immunotherapies show promise and some potential in several autoimmune diseases, for SLE, we have less information on the possibility of whether beneficial effects can ensue from this approach. This can be due to multiple factors. For example, Tregs types include—in addition to CD25+FoxP3+ T cells—Tr1, Th3, CD8+ and CD4+CD25lowCD127lowGITR+ Tregs, just to name the best known subtypes [78], and one should evaluate which subset(s) could have better potential in suppressing SLE in vivo. Importantly, since SLE is a systemic disease in which multiple self-antigens are targeted by the autoimmune process, it may be more difficult to promote antigen-specific inhibitory responses resulting in beneficial clinical outcomes. Additionally, it is not well known where Tregs distribute once transferred and how they can preserve their phenotype and regulatory function once exposed to the proinflammatory cytokine environment in SLE. It is also not known how long they can survive and maintain an immunosuppressive phenotype in vivo under the pressure of hyperactive immune responsiveness. To further complicate these aspects, tissue-resident Tregs’ functions may associate with site-dependent differences in the local immune response (see as an example the eTregs).
This is in addition to considerations on practical aspects such as optimization protocols to obtain sufficiently pure Tregs in therapeutic quantities and in a cost-effective manner. In all, these knowledge gaps and considerations have hampered so far the implementation of therapeutic uses of Tregs in SLE that have instead been faster for organ-specific autoimmune disease.
Conclusions
Tregs utilize multiple mechanisms to maintain peripheral tolerance, including the suppression of multiple immune cell types via cell contact-dependent and/or independent mechanisms. Most studies have indicated quantitative and/or qualitative deficits in Tregs in SLE patients, yet the immunotherapeutic use of Tregs in SLE is still at its beginnings, despite the promise carried by successful preclinical studies in lupus mice. Considerations should be made on whether adoptive transfers of ex vivo-expanded Tregs could sort better outcomes than protocols expanding iTregs in vivo, and which approaches could favor Treg stability under unfavorable inflammatory conditions. In any case, advantages that could be envisioned from the use of Tregs in the immunotherapy of SLE—most likely usable in combination with other therapies—would be a lack of generalized immunosuppression associated with the current conventional therapies. Secondarily to this, one might anticipate improved immune homeostasis and lower risks of off-target effects.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
La Cava A. T-regulatory cells in systemic lupus erythematosus. Lupus. 2008;17:421–5. doi:10.1177/0961203308090028.
Shevach EM. Mechanisms of Foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–45. doi:10.1016/j.immuni.2009.04.010.
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61. doi:10.1126/science.1079490.
Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev. 2014;259:88–102. doi:10.1111/imr.12160.
Lourenço EV, La Cava A. Natural regulatory T cells in autoimmunity. Autoimmunity. 2011;44:33–42. doi:10.3109/08916931003782155.
Roncarolo MG, Gregori S, Bacchetta R, Battaglia M. Tr1 cells and the counter-regulation of immunity: natural mechanisms and therapeutic applications. Curr Top Microbiol Immunol. 2014;380:39–68. doi:10.1007/978-3-662-43492-5_3.
Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–64. doi:10.1146/annurev.immunol.25.022106.141623.
McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, et al. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–23. doi:10.1016/S1074-7613(02)00280-7.
Schreiber L, Pietzsch B, Floess S, Farah C, Jänsch L, Schmitz I, et al. The Treg-specific demethylated region stabilizes Foxp3 expression independently of NF-kB signaling. PLoS One. 2014;9:e88318. doi:10.1371/journal.pone.0088318.
Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol. 2008;38:1654–63. doi:10.1002/eji.200838105.
Miyara M, Amoura Z, Parizot C, Badoual C, Dorgham K, Trad S, et al. Global natural regulatory T cell depletion in active systemic lupus erythematosus. J Immunol. 2005;175:8392–400. doi:10.4049/jimmunol.175.12.8392.
Crispin JC, Martinez A, Alcocer-Varela J. Quantification of regulatory T cells in patients with systemic lupus erythematosus. J Autoimmun. 2003;21:273–6. doi:10.1016/S0896-8411(03)00121-5.
Valencia X, Yarboro C, Illei G, Lipsky PE. Deficient CD4+CD25high T regulatory cell function in patients with active systemic lupus erythematosus. J Immunol. 2007;178:2579–88. doi:10.4049/jimmunol.178.4.2579.
Suarez A, Lopez P, Gomez J, Gutierrez C. Enrichment of CD4+CD25high T cell population in patients with systemic lupus erythematosus treated with glucocorticoids. Ann Rheum Dis. 2006;65:1512–7. doi:10.1136/ard.2005.049924.
Bonelli M, Savitskaya A, von Dalwigk K, Steiner CW, Aletaha D, Smolen JS, et al. Quantitative and qualitative deficiencies of regulatory T cells in patients with systemic lupus erythematosus (SLE). Int Immunol. 2008;20:861–8. doi:10.1093/intimm/dxn044.
Yu Y, Liu Y, Shi FD, Zou H, Hahn BH, La Cava A. Tolerance induced by anti-DNA Ig peptide in (NZB × NZW)F1 lupus mice impinges on the resistance of effector T cells to suppression by regulatory T cells. Clin Immunol. 2012;142:291–5. doi:10.1016/j.clim.2011.11.004.
Hadaschik EN, Wei X, Leiss H, Heckmann B, Niederreiter B, Steiner G, et al. Regulatory T cell-deficient scurfy mice develop systemic autoimmune features resembling lupus-like disease. Arthritis Res Ther. 2015;17:35. doi:10.1186/s13075-015-0538-0.
Iikuni N, Lourenço EV, Hahn BH, La Cava A. Cutting edge: regulatory T cells directly suppress B cells in systemic lupus erythematosus. J Immunol. 2009;183:1518–22. doi:10.4049/jimmunol.0901163.
Banchereau J, Pascual V, O’Garra A. From IL-2 to IL-37: the expanding spectrum of anti-inflammatory cytokines. Nat Immunol. 2012;13:925–31. doi:10.1038/ni.2406.
La Cava A, Ebling FM, Hahn BH. Ig-reactive CD4+CD25+ T cells from tolerized (New Zealand Black x New Zealand White)F1 mice suppress in vitro production of antibodies to DNA. J Immunol. 2004;173:3542–8. doi:10.4049/jimmunol.173.5.3542.
Singh RP, La Cava A, Hahn BH. pConsensus peptide induces tolerogenic CD8+ T cells in lupus-prone (NZB x NZW)F1 mice by differentially regulating Foxp3 and PD1 molecules. J Immunol. 2008;180:2069–80. doi:10.4049/jimmunol.180.4.2069.
Sharabi A, Mozes E. The suppression of murine lupus by a tolerogenic peptide involves Foxp3-expressing CD8 cells that are required for the optimal induction and function of foxp3-expressing CD4 cells. J Immunol. 2008;181:3243–51.
Tsai YG, Lee CY, Lin TY, Lin CY. CD8+ Treg cells associated with decreasing disease activity after intravenous methylprednisolone pulse therapy in lupus nephritis with heavy proteinuria. PLoS One. 2014;9:e81344. doi:10.1371/journal.pone.0081344.
Zhang L, Bertucci AM, Ramsey-Goldman R, Burt RK, Datta SK. Regulatory T cell (Treg) subsets return in patients with refractory lupus following stem cell transplantation, and TGF-β-producing CD8+ Treg cells are associated with immunological remission of lupus. J Immunol. 2009;183:6346–58. doi:10.4049/jimmunol.0901773.
Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci U S A. 2008;105:10113–8. doi:10.1073/pnas.0711106105.
Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–5. doi:10.1126/science.1160062.
La Cava A. Tregs are regulated by cytokines: implications for autoimmunity. Autoimmun Rev. 2008;8:83–7. doi:10.1016/j.autrev.2008.08.002.
Linker-Israeli M, Deans RJ, Wallace DJ, Prehn J, Ozeri-Chen T, Klinenberg JR. Elevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesis. J Immunol. 1991;147:117–23.
Zheng SG, Wang J, Horwitz DA. Cutting edge: Foxp3+CD4+CD25+ regulatory T cells induced by IL-2 and TGF-β are resistant to Th17 conversion by IL-6. J Immunol. 2008;180:7112–6. doi:10.4049/jimmunol.180.11.7112.
Wong CK, Lit LC, Tam LS, Li EK, Wong PT, Lam CW. Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: implications for Th17-mediated inflammation in autoimmunity. Clin Immunol. 2008;127:385–93. doi:10.1016/j.clim.2008.01.019.
Mao C, Wang S, Xiao Y, Xu J, Jiang Q, Jin M, et al. Impairment of regulatory capacity of CD4+CD25+ regulatory T cells mediated by dendritic cell polarization and hyperthyroidism in Graves’ disease. J Immunol. 2011;186:4734–43. doi:10.4049/jimmunol.0904135.
Bjarnadottir U, Lemarquis AL, Halldorsdottir S, Freysdottir J, Ludviksson BR. The suppressive function of human CD8+ iTregs is inhibited by IL-1β and TNFα. Scand J Immunol. 2014;80:313–22. doi:10.1111/sji.12212.
Gómez J, Prado C, López P, Suárez A, Gutiérrez C. Conserved anti-proliferative effect and poor inhibition of TNFα secretion by regulatoryCD4+CD25+ T cells in patients with systemic lupus erythematosus. Clin Immunol. 2009;132:385–92. doi:10.1016/j.clim.2009.05.012.
Yamaguchi T, Wing JB, Sakaguchi S. Two modes of immune suppression by Foxp3+ regulatory T cells under inflammatory or non-inflammatory conditions. Semin Immunol. 2011;23:424–30. doi:10.1016/j.smim.2011.10.002.
Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, et al. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med. 2014;211:121–36. doi:10.1084/jem.20131142.
Chandrasekaran U, Yi W, Gupta S, Weng CH, Giannopoulou E, Chinenov Y, et al. Regulation of effector Treg cells in murine lupus. Arthritis Rheum. 2016;68:1454–66. doi:10.1002/art.39599. Unveiled new modalities by which Teffs can survive in hostile SLE inflammatory settings.
La Cava A. Survive to fight: effector Treg cells in systemic lupus erythematosus. Arthritis Rheum. 2016;68:1327–9. doi:10.1002/art.39616.
Horwitz DA, Zheng SG, Gray JD. Natural and TGF-β-induced Foxp3+CD4+CD25+ regulatory T cells are not mirror images of each other. Trends Immunol. 2008;29:429–35. doi:10.1016/j.it.2008.06.005.
Martinez RJ, Zhang N, Thomas SR, Nandiwada SL, Jenkins MK, Binstadt BA, et al. Arthritogenic self-reactive CD4+ T cells acquire a FR4hi CD73hi anergic state in the presence of Foxp3+ T regulatory cells. J Immunol. 2012;188:170–81. doi:10.4049/jimmunol.1101311.
Kalekar LA, Schmiel SE, Nandiwada SL, Lam WY, Barsness LO, Zhang N, et al. CD4+ T cell anergy prevents autoimmunity and generates regulatory T cell precursors. Nat Immunol. 2016;17:304–14. doi:10.1038/ni.3331.
Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity. 2012;37:785–99. doi:10.1016/j.immuni.2012.09.010.
Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity. 2013;38:414–23. doi:10.1016/j.immuni.2013.03.002.
Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, et al. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity. 2016;44:406–21. doi:10.1016/j.immuni.2016.01.028.
Procaccini C, De Rosa V, Galgani M, Abanni L, Cali G, Porcellini A, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. 2010;33:929–41. doi:10.1016/j.immuni.2010.11.024.
Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish Treg-cell function. Nature. 2013;499:485–90. doi:10.1038/nature12297. Link of mTOR to metabolism-driven regulation of Treg-suppressive activity.
Maloy KJ, Powrie F. Fueling regulation: IL-2 keeps CD4+ Treg cells fit. Nat Immunol. 2005;6:1071–2. doi:10.1038/ni1105-1071.
Cao T, Wenzel SE, Faubion WA, Harriman G, Li L. Enhanced suppressive function of regulatory T cells from patients with immune-mediated diseases following successful ex vivo expansion. Clin Immunol. 2010;136:329–37. doi:10.1016/j.clim.2010.04.014.
Hahn BH, Anderson M, Le E, La Cava A. Anti-DNA Ig peptides promote Treg cell activity in systemic lupus erythematosus patients. Arthritis Rheum. 2008;58:2488–97. doi:10.1002/art.23609.
Scalapino KJ, Tang Q, Bluestone JA, Bonyhadi ML, Daikh DI. Suppression of disease in New Zealand Black/New Zealand White lupus-prone mice by adoptive transfer of ex vivo expanded regulatory T cells. J Immunol. 2006;177:1451–9.
Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. 2015;7:315ra189. doi:10.1126/scitranslmed.aad4134. Feasibility of preparing purified Tregs for immunotherapy in autoimmune disease (lifespan of transferred Tregs was >1 year).
Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juścińska J, et al. Therapy of type 1 diabetes with CD4+CD25highCD127-regulatory T cells prolongs survival of pancreatic islets—results of one year follow-up. Clin Immunol. 2014;153:23–30. doi:10.1016/j.clim.2014.03.016.
Theil A, Tuve S, Oelschlagel U, Maiwald A, Dohler D, Ossmann D, et al. Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft-versus-host disease. Cytotherapy. 2015;17:473–86. doi:10.1016/j.jcyt.2014.11.005.
Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood. 2016;127:1044–51. doi:10.1182/blood-2015-06-653667. Successful large-scale expansion of Tregs and efficacy in GVHD.
Hügle T, Daikeler T. Stem cell transplantation for autoimmune diseases. Haematologica. 2010;95:185–8. doi:10.3324/haematol.2009.017038.
Figueroa FE, Cuenca Moreno J, La Cava A. Novel approaches to lupus drug discovery using stem cell therapy. Role of mesenchymal-stem-cell-secreted factors. Expert Opin Drug Discovery. 2014;9:555–66. doi:10.1517/17460441.2014.89769.
Burt RK, Traynor A, Statkute L, Barr WG, Rosa R, Schroeder J, et al. Nonmyeloablative hematopoietic stem cell transplantation for systemic lupus erythematosus. JAMA. 2006;295:527–35. doi:10.1001/jama.295.5.527.
Szodoray P, Varoczy L, Papp G, Barath S, Nakken B, Szegedi G, et al. Immunological reconstitution after autologous stem cell transplantation in patients with refractory systemic autoimmune diseases. Scand J Rheumatol. 2012;41:110–5. doi:10.3109/03009742.2011.606788.
Wang D, Zhang H, Liang J, Li X, Feng X, Wang H, et al. Allogeneic mesenchymal stem cell transplantation in severe and refractory systemic lupus erythematosus: 4 years of experience. Cell Transplant. 2013;22:2267–77. doi:10.3727/096368911x582769.
Wang Q, Qian S, Li J, Che N, Gu L, Wang Q, et al. Combined transplantation of autologous hematopoietic stem cells and allogenic mesenchymal stem cells increases T regulatory cells in systemic lupus erythematosus with refractory lupus nephritis and leukopenia. Lupus. 2015;24:1221–6. doi:10.1177/0961203315583541.1.
Goudy K, Aydin D, Barzaghi F, Gambineri E, Vignoli M, Ciullini Mannurita S, et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol. 2013;146:248–61. doi:10.1016/j.clim.2013.01.004.
He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-dose interleukin-2 treatment selectively modulates CD4+ T cell subsets in patients with systemic lupus erythematosus. Nat Med. 2016. doi:10.1038/nm.4148.
von Spee-Mayer C, Siegert E, Abdirama D, Rose A, Klaus A, Alexander T, et al. Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann Rheum Dis. 2016;75:1407–15. doi:10.1136/annrheumdis-2015-207776.
Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med. 2011;365:2067–77. doi:10.1056/NEJMoa1105143.
Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea 3rd EP, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. 2011;365:2055–66. doi:10.1056/NEJMoa1108188.
Nozaki Y, Yamagata T, Yoo BS, Sugiyama M, Ikoma S, Kinoshita K, et al. The beneficial effects of treatment with all-trans-retinoic acid plus corticosteroid on autoimmune nephritis in NZB/W mice. Clin Exp Immunol. 2005;139:74–83. doi:10.1111/j.1365-2249.2005.02654.x.
Miyabe Y, Miyabe C, Nanki T. Could retinoids be a potential treatment for rheumatic diseases? Rheumatol Int. 2015;35:35–41. doi:10.1007/s00296-014-3067-2.
Kinoshita K, Kishimoto K, Shimazu H, Nozaki Y, Sugiyama M, Ikoma S, et al. Successful treatment with retinoids in patients with lupus nephritis. Am J Kidney Dis. 2010;55:344–7. doi:10.1053/j.ajkd.2009.06.012.
Kang HK, Michaels MA, Berner BR, Datta SK. Very low-dose tolerance with nucleosomal peptides controls lupus and induces potent regulatory T cell subsets. J Immunol. 2005;174:3247–55. doi:10.4049/jimmunol.174.6.3247.
Eilat E, Dayan M, Zinger H, Mozes E. The mechanism by which a peptide based on complementarity-determining region-1 of a pathogenic anti-DNA auto-Ab ameliorates experimental systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2001;98:1148–53. doi:10.1073/pnas.98.3.1148.
Zimmer R, Scherbarth HR, Rillo OL, Gomez-Reino JJ, Muller S. Lupuzor/P140 peptide in patients with systemic lupus erythematosus: a randomised, double-blind, placebo-controlled phase IIb clinical trial. Ann Rheum Dis. 2013;72:1830–5. doi:10.1136/annrheumdis-2012-202460. Evidence of efficacy and safety of a peptide-based therapy in SLE.
Ulivieri C, Baldari CT. Statins: from cholesterol-lowering drugs to novel immunomodulators for the treatment of Th17-mediated autoimmune diseases. Pharmacol Res. 2014;88:41–52. doi:10.1016/j.phrs.2014.03.001.
Abud-Mendoza C, de la Fuente H, Cuevas-Orta E, Baranda L, Cruz-Rizo J, González-Amaro R. Therapy with statins in patients with refractory rheumatic diseases: a preliminary study. Lupus. 2003;12:607–11.
Mathian A, Jouenne R, Chader D, Cohen-Aubart F, Haroche J, Fadlallah J, et al. Regulatory T cell responses to high-dose methylprednisolone in active systemic lupus erythematosus. PLoS One. 2015;10:e0143689. doi:10.1371/journal.pone.0143689.
Tselios K, Sarantopoulos A, Gkougkourelas I, Papagianni A, Boura P. Increase of peripheral T regulatory cells during remission induction with cyclophosphamide in active systemic lupus erythematosus. Int J Rheum Dis. 2014;17:790–5. doi:10.1111/1756-185x.12500.
Liossis SN, Sfikakis PP. Rituximab-induced B cell depletion in autoimmune diseases: potential effects on T cells. Clin Immunol. 2008;127:280–5. doi:10.1016/j.clim.2008.01.011.
Vallerskog T, Gunnarsson I, Widhe M, Risselada A, Klareskog L, van Vollenhoven R, et al. Treatment with rituximab affects both the cellular and the humoral arm of the immune system in patients with SLE. Clin Immunol. 2007;122:62–74.
Sfikakis PP, Souliotis VL, Fragiadaki KG, Moutsopoulos HM, Boletis JN, Theofilopoulos AN. Increased expression of the FoxP3 functional marker of regulatory T cells following B cell depletion with rituximab in patients with lupus nephritis. Clin Immunol. 2007;123:66–73.
Jonuleit H, Schmitt E. The regulatory T cell family: distinct subsets and their interrelations. J Immunol. 2003;171:6323–7. doi:10.4049/jimmunol.171.12.6323.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
This article is part of Topical Collection on Systemic Lupus Erythematosus
Rights and permissions
About this article

Cite this article
Giang, S., La Cava, A. Regulatory T Cells in SLE: Biology and Use in Treatment. Curr Rheumatol Rep 18, 67 (2016). https://doi.org/10.1007/s11926-016-0616-6
Published:
DOI: https://doi.org/10.1007/s11926-016-0616-6