
Abstract
The fundamental mechanisms that drive the pathogenesis of systemic sclerosis (SSc) remain elusive, despite over 50 years of investigation. Here, we review recent progress in the understanding of the immunopathogenesis of SSc. In particular, we consider interleukin-13 (IL13), and its upstream and downstream pathways, as an example of an immune system-derived mediator involved in fibrotic and vascular pathology. Emerging results linking pattern-recognition receptors and interferon pathways to SSc are also stressed. We discuss genetic data linking the immune system to SSc risk and efforts to apply animal models to subsets of patients recently resolved by gene expression profiling. These developments will help build a context for better understanding of previous observations and design of the next generation of studies that may eventually lead to effective treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The fundamental mechanisms of initiation of SSc and of its progression remain elusive, despite over 50 years of investigation. One of the challenges in this regard is that SSc involves aspects of inflammation, vascular biology, and growth factor-propagated fibrosis. Thus, the pathologic mechanisms involved invoke the full complexity of each of these. Despite the co-occurrence of these seemingly disparate forms of pathology in SSc, accumulating evidence, for example the discovery of a strong association of human leukocyte antigens (HLA) with disease risk and direct observation of histologic and molecular immunopathology, increasingly suggest that immune dysregulation is a proximal cause of SSc. From this perspective, immune dysregulation is poised to emerge as a unifying explanation for the fibrotic, vasculopathic, and inflammatory manifestations of SSc.
Here, we review recent progress in the understanding the immunopathogenesis of SSc, focusing on pathways that link inflammation with pro-fibrotic signaling and, potentially, the vasculopathy observed in SSc. We consider interleukin-13 (IL13) and its upstream inducer IL33 in detail, as examples of immune system-derived mediators involved in fibrotic and vascular pathology. We also discuss recent genetic data linking the immune system to SSc risk, efforts to apply animal models to specific SSc patient subsets, and new data regarding pattern-recognition receptors and interferon pathways in SSc. In particular, patient stratification efforts are emphasized on the basis that they provide an explicit molecular demonstration of the long-standing clinical observation that SSc is a heterogeneous disorder. This heterogeneity in turn suggests that consideration of context, in terms of the clinical, molecular, and genetic background of the patients and animal model systems under investigation, will be critical to interpreting the results of individual studies and ultimately generating the insight necessary to develop successful new treatment.
Recent Genetic Evidence Implicating the Immune System in SSc
Despite its classification as an autoimmune disease, the primary involvement of the immune system in the pathogenesis of SSc remains a topic of debate. The uniform observation of increased extracellular matrix production and early vascular phenomena, for example Raynaud’s, has led to the suggestion that this disease is a primary disorder of either fibroblast dysfunction or endothelial cell injury [1, 2]. Although autoantibody production characterizes most SSc patients, some have argued this is an epiphenomenon as a consequence of chronic tissue damage [3]. Furthermore, classic immunosuppressive medication is of limited benefit [4], in contrast with other autoimmune conditions.
Recent genetic evidence that implicates immune system dysfunction as a primary driver of SSc pathogenesis have affected this debate. Genome wide association studies (GWAS) of single nucleotide polymorphisms (SNPs) enable high-resolution screening for genetic risk factors of complex diseases. To date, three GWAS studies have been performed on SSc patients. Each of these identified strong associations within the major histocompatibility locus, which most probably indicates that adaptive antigen-driven immunity contributes to SSc pathogenesis [5, 6••, 7•]. In these and follow-up studies, associations of SSc risk or SSc clinical sub-phenotypes were identified in genes encoding six proteins implicated in the immune response:
- 1.
-
2.
STAT4 and IRF5, transcription factors mediating immune responses [6••, 7•, 8];
-
3.
IL12RB2, a subunit of the IL12 receptor complex [9];
-
4.
CD226, an immunoglobulin superfamily member involved in NK cell and lymphocyte activation [10]; and
-
5.
TNFSF4 (OXL40), a co-stimulatory molecule expressed on antigen presenting cells [11].
Interestingly, some of these risk alleles are shared with other autoimmune diseases, suggesting that a common pathway leading to loss of tolerance may underlie phenotypically different diseases. As the genetics of SSc are a rapidly developing field, the authors encourage the reader to seek dedicated reviews on this topic [12, 13]. Although neither the mechanistic effects of the genes implicated in SSc pathogenesis nor an integrated picture of how these risk alleles interact with one another has emerged, these data indicate the immune system is involved in the development and manifestations of this disease.
Molecular Stratification of SSc Patients
SSc is a heterogeneous disorder with a wide range of clinical manifestations and variations in severity [14–16]. Major obstacles to elucidation of the involvement of the immune system in the pathogenesis of SSc include a lack of insight into molecular aspects of different manifestations of the disease and poor understanding of which subsets of patients might be best represented by a given animal model of this disease. Given the relative accessibility of the skin to tissue harvesting, recent efforts to classify patients have focused on transcriptional profiling of skin biopsies [15, 17, 18••]. Whitfield and colleagues performed unsupervised clustering of skin gene expression profiles from patients with diffuse SSc (dSSc), limited SSc (lSSc), and morphea, and identified an inflammatory subset of patients with highly enriched expression of chemokines, chemokine receptors, interferon response genes, and transcripts characteristic of immune cells [17, 18••]. Interestingly, according to these two reports the inflammatory subset cuts across traditional clinical stratifications of SSc, encompassing an estimated 17 % to 36 % of patients with dSSc, 28 % with lSSc, and 100 % with localized scleroderma. In contrast, for a second fibroproliferative subset, largely composed of patients with clinical diffuse disease, upregulation of genes involved in cell proliferation predominates [17, 18••]. Accordingly, higher numbers of T-cells and Ki-67 positive proliferating cells are observed for patients in the inflammatory and fibroproliferative subsets, respectively [17]. A recent longitudinal analysis demonstrates that these expression profiles are stable, suggesting that these subsets represent different disease entities as opposed to different time points along the course of a single disorder [18••]. These findings suggest that stratifying patients according to these molecularly defined subsets may reduce a major source of heterogeneity in studies of SSc and, accordingly, increase the robustness of future clinical studies. Furthermore, they provide a basis for understanding the different animal models of SSc and a means of converting pathogenic pathways identified in these models to personalized treatment for patients.
The Sclerodermatous Graft Versus Host Disease Model and its Relationship to the Inflammatory Subset of SSc
Investigation of the causes of SSc relies heavily on the use of mouse models, and systematic consideration of these models has been the topic of recent reviews [19, 20]. The sclerodermatous graft versus host disease model (SclGVHD) is based on the observation that clinical manifestations for patients with chronic GVHD overlap those for patients with SSc, including skin sclerosis and myofibroblast accumulation [21, 22]. This model is generated by transferring splenocytes into irradiated or immunodeficient mouse strains allogenic at multiple minor histocompatibility loci [23]. Notably, the specific tissue manifestations of the disease vary depending on the MHC haplotype shared between the graft and host. When the model is run on donor–host pairs that share the H-2k or H-2b MHC haplotype instead of the “native” BALB/c H-2d haplotype, skin manifestations are less prominent and, instead, mice develop constitutional symptoms and diarrhea [24]. This implies that shaping of the thymic repertoire by specific MHC alleles may be an important determinant in the range of tissues affected in individual SSc patients.
The version of SclGVHD using Rag2 −/− immunodeficient hosts on the BALB/c background is particularly well characterized [23, 25••]. These mice develop skin thickening and fibrosis characterized by dermal infiltration of graft CD4+ T-cells. There is also a resident dermal macrophage population that expands over the first few weeks of the disease. Fibrosis is also observed in the renal interstitium and intestinal lamina propria and Scl-70 autoantibodies, a characteristic serologic marker of human SSc, are produced. Interestingly, this clearly immune-driven model includes some of the vasculopathic manifestations of SSc, with luminal narrowing in the capillaries of the skin and kidney, associated with increased expression of the vasoconstrictor endothelin-1 [23]. Human chronic GVHD seen after BMT, however, lacks the vascular rarefaction seen in SSc skin [22]. These data suggest that murine SclGVHD may differ from human chronic GVHD in its vascular manifestations, with the former more accurately modeling SSc. Further investigation is needed to understand this potential difference between human chronic GVHD and murine SclGVHD.
Although versions of the SclGVHD model using irradiated versus Rag2 −/− immunodeficient hosts have not been directly compared, one notable difference is that only the irradiated version develops significant lung fibrosis [23, 26]. One possible explanation is that inflammatory “danger” signals, for example those elicited by radiation injury, are important for the induction of a fibroinflammatory program in the SclGVHD lung. Interestingly, lung involvement in SSc patients is also variable, and studies suggest it may be linked to gastroesophageal reflux, leading to acid aspiration and epithelial injury [27]. Taken together, these findings suggest a “second hit” hypothesis to explain the variable lung involvement among SclGVHD models and SSc patients whereby immune system activation must be accompanied by tissue injury to incite this organ-specific manifestation.
Given the complexity and heterogeneity of SSc, an important part of the evaluation of any relevant animal model is determination of whether it accurately reflects the human disease, and, if so, which subset of disease it models best. Although these judgments have typically occurred at the level of gross and histologic assessment of the disease, the advent of molecular stratification of SSc subsets has enabled a more systematic molecular approach to this question. In particular, SclGVHD seems to best approximate the inflammatory subset of patients [25••]. Thus, the SclGVHD model is an example of immunopathology resulting in fibrotic and vasculopathic manifestations similar to those seen in SSc, and at the molecular level best represents patients with the inflammatory skin gene expression profile.
IL13 as a Pro-Fibrotic Mediator in SSc: Background
Several pro-fibrotic factors with a peripheral relationship to the immune system are under investigation in SSc, including TGFβ, CTGF, PDGF, and endothelin; these are reviewed elsewhere [28–31]. In addition to these, IL13 has emerged as a putative immune system-derived mediator of fibrotic disease in SSc. IL13 is a 15-kDa secreted cytokine encoded at chromosome 5q31. IL13 was discovered, with IL4, as a predominant product of Th2-skewed T cells [32]. However, in addition to T cells, other cell types produce IL13, including mast cells, macrophages, and dendritic cells [25••, 33–35]. Another potential source of IL13 is a recently described cell population called the “nuocyte” [36], so named because of their production of IL13, with “Nu” being the 13th letter of the Greek alphabet. Nuocytes are induced by IL33 and IL25, and are grouped within the growing category of innate lymphoid cells (ILC) [37•, 38•, 39•], a term derived from their responsiveness to IL-7, a prototypic lymphoid growth factor, but persistence in mice lacking adaptive immunity.
IL13 has effects that partially overlap those of IL4, and this is reflected in the mechanisms of IL13 signaling. IL4 signals through two receptor complexes, a type I receptor comprised of IL4RA and the common γ-chain, and a type II receptor comprised of IL4RA and IL13RA1 [40]. IL13 only utilizes the type II receptor, which activates the STAT6 transcription through TYK2 and JAK2 [41]. In addition to IL13RA1, there is a related receptor, IL13RA2, which does not partner with IL4RA. IL13RA2 has two variants: a secreted form and a transmembrane form with a short cytoplasmic tail devoid of known signaling motifs [42]. Early studies on IL13RA2 did not find that it conferred responsiveness to IL13, which led to speculation that it functions as a decoy receptor [43–45]. Accordingly, increased levels of IL13-driven disease are observed for IL13RA2 knockouts [46, 47], and soluble IL13RA2-Fc fusion protein therapeutically blocks IL13 [48]. In contrast, IL13RA2 promotes fibrosis in some TGFβ-dependent models, suggesting it may have signaling capacity [49, 50]. Further study of the mechanisms of regulation of IL13RA2 and the pathways it engages is needed to generate a clear picture of its biological relevance to IL13 signaling.
IL13 as a Pro-Fibrotic Mediator in SSc: Translational Studies
Keeping in mind the caveats discussed in this review regarding the heterogeneity of SSc, if IL13 is involved in the pathogenesis of SSc it should be detectable in patients at levels above those in controls. In the serum of both dSSc and lSSc patients, levels of both IL4 and IL13 were increased [51]. In the skin of SSc patients, the total number of IL13-expressing cells, as judged by immunohistochemical staining, was elevated, and some of the positive cells also stained for a macrophage marker, suggesting that resident dermal macrophages are a source of IL13 in the skin of SSc patients [25••]. In another study, IL13 expression was increased in peripheral blood CD8+ T-cells in patients with SSc [52]. IL13 levels correlated with the biomarkers CRP and ESR, suggesting that IL13 may be linked to inflammatory mechanisms. In a follow-up study, CD8+ T-Cells from SSc patients were found to upregulate the Th2 transcription factor GATA3, which promotes IL13 expression and inhibits differentiation toward IFNγ-producing Th1 cells [53]. The proportion of GATA3-expressing CD8+ T-cells correlated with measures of disease severity, for example modified Rodnan skin scores (mRSS) and interstitial lung disease. In addition to IL13 itself, mRNA levels of the genes encoding the IL13 receptor complex (IL13RA1 or IL4RA) and an IL13 regulated gene, CCL2, were shown to correlate with both the presence of SSc and disease severity as measured by the mRSS [25••]. Last, an IL13 responsive gene profile derived from treating fibroblasts in vitro with IL13 was significantly enriched in the inflammatory subset [25••], suggesting that IL13 pathway activation may be most relevant to this group of patients.
Pulmonary arterial hypertension (PAH) is a complication of SSc resulting in particularly high morbidity and mortality [54]. PAH is most commonly associated with lSSc, although it can be observed in dSSc also. Among lSSc patients, IL13 levels were increased in patients with PAH, and expression of the IL13 target gene MRC1 in circulating monocytes correlated with pulmonary arterial pressures [55]. Serum IL13 levels also correlated with abnormalities in the capillary loops visible through the nails of SSc patients, an early feature of SSc that may precede other manifestations [56, 57]. Thus, an association between IL13 expression and the presence of SSc has been observed in several studies, and several studies suggest that IL13 levels correlate with vasculopathic manifestations of scleroderma, for example PAH and nailfold capillary abnormalities.
Studies of the genetics of scleroderma have yielded conflicting results regarding the effect of polymorphisms in the IL13 pathway. Granel et al. showed that polymorphisms in IL13 or IL13RA2 contribute to genetic risk of SSc [58, 59]. However, none of the SSc GWAS studies identified signals within loci relevant to the IL13 pathway and a recent study targeting polymorphisms in IL4, IL13, and the genes encoding the type II IL4 receptor (IL13RA1, IL4RA) failed to find an association with SSc or clinical subtype [60]. These conflicting studies await further evaluation using independent cohorts.
IL13 as a Pro-Fibrotic Mediator in SSc: Mouse Models
Perhaps one of the strongest demonstrations of the potent pro-fibrotic activity of IL13 is the observation that transgenic overexpression of IL13 in the skin and lungs of mice is sufficient to drive chronic inflammation and fibrosis [61, 62]. With regard to more specific SSc models, both IL13 and a receptor IL13RA2, are elevated in skin treated with the pro-fibrotic agent, bleomycin [63]. Furthermore, bleomycin-induced skin sclerosis and lung fibrosis are attenuated by genetic deletion of Il13 or therapeutic neutralization, respectively [50, 64, 65].
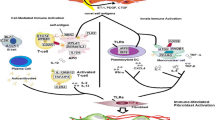
In the SclGVHD model, dermal infiltrating CD4+ T-cells and a dermal macrophage population were identified as sources of IL13 (Fig. 1) [25••]. Accordingly, hosts lacking the common IL4 and IL13 receptor subunit, IL4RA, were completely protected from disease. Hosts lacking IL13 were partially protected, which likely reflects continued production of IL13 by graft CD4+ T-cells. Moreover, the IL13 response profile enriched in inflammatory patients is also enriched in SclGVHD mice but not the IL13 unresponsive IL4ra −/− hosts. IL13 seems to act indirectly via host cells to shape the responses of the graft-derived lymphocytes, because regulatory T cells and total CD4+ T-cell IL-10 expression were increased in IL4RA-deficient hosts. Furthermore, a mixed phenotype of classical and alternative activation markers [66], which may be driven by IL13, were observed in macrophages in the skin of SclGVHD [25••]. Interestingly, similar phenotypes have been observed with tumor-associated macrophages, suggesting a parallel between the fibrotic stromal reaction seen with some tumors and the fibrotic disease seen in SSc [67]. To further identify the mediators downstream of IL13, the overlap between genes upregulated in the skin of SclGVHD mice, the human inflammatory subset of SSc, and IL-13-treated fibroblasts was considered [25••]. The chemokine CCL2 emerged as the only gene present in all three data sets, consistent with the action of CCL2 as a secondary mediator of disease in the IL-13 transgenic lung model [68]. Accordingly, CCL2 is upregulated in both macrophages and stromal cells by an IL4RA-dependent pathway and blockade of CCL2 prevents SclGVHD [25••]. Taken together, IL13 derived from dermal macrophages and infiltrating lymphocytes promotes SclGVHD by inducing CCL2 expression, affecting macrophage polarization and indirectly inhibiting immunoregulatory T-cell expansion (Fig. 1).

IL13 in the skin of inflammatory SSc patients. This figure depicts the possible cellular sources, targets, and effects of IL13 in the skin of inflammatory SSc patients, as determined in animal models and SSc patient samples
IL33: An Emerging Profibrotic Cytokine Upstream of IL13
As evidence gathers that IL13 has a variety of pro-fibrotic functions that may be relevant to the pathogenesis of SSc, it becomes of great interest to understand how IL13 expression is induced. Recent evidence implicates IL33 as a broad upstream inducer of IL13 in the immune system. IL33 is an IL1 family member constitutively expressed in cell nuclei [69, 70]. Necrotic cell death initiates IL33 release, leading to the suggestion that IL33 acts as an innate “danger signal” driving the pro-inflammatory effects of necrosis. IL33 signals through a dimeric receptor composed of an IL33-specific receptor chain (ST2) and a subunit shared by several IL-1 family members (IL-1RAcP) [71]. Whereas the precursor forms of other IL1 family members are activated by caspase-1/inflammasome-dependent cleavage, cleavage of pro-IL33 by caspase-1 is inactivating [72]. Instead, other proteases, for example neutrophil elastase or cathepsin G, may mediate maturation of pro-IL33 to the bioactive form [73].
IL33 expression has been demonstrated in a range of cells implicated in the pathogenesis of SSc—endothelial and epithelial cells, and fibroblasts and smooth muscle cells [74, 75]. This suggests that vasculopathic insults, which could be an initial event in SSc, may lead to IL33 release via ischemia. Alternatively, IL33 could be released via inflammation or toxin-induced tissue damage. Ultimately, IL33 would induce a fibroinflammatory response via IL13 and other downstream factors. Thus, IL33 release activated by different upstream events could explain the observation that seemingly disparate stimuli (e.g. bleomycin, SclGVHD, ischemia) lead to fibrotic pathology. With regard to human translational studies, depletion of nuclear IL-33 has been observed in skin endothelial cells and keratinocytes of early SSc patients despite similar or increased levels of IL33 transcripts, leading to speculation that microenvironmental stressors in SSc skin result in release of IL33 [76]. Moreover, expression of both the IL33-specific receptor ST2 and levels of circulating IL33 are increased in the serum of SSc patients [76–78]. In an Italian cohort, IL33 levels were higher in patients with early disease, and in those with active nailfold vascular changes [78], an observation that emphasizes the possible link between IL13 and SSc vasculopathy [55–57]. In a Japanese cohort, IL33 levels correlated with skin scores and inversely with forced vital capacity, a measure of lung fibrosis [77].
Since the discovery of IL33 as the ligand utilizing the ST2 receptor, IL33 stimulation has been linked to the induction of Th2 cytokines, including IL13 [70]. Although best described as activating IL13 expression in Th2 cells [70], B-cells [79], and a subset of ILCs, termed nuocytes [37•, 39•], IL-33 also stimulates IL-13 production by innate immune cells, for example mast cells [80] and basophils [81]. Consistent with the known pro-fibrotic effects of IL13, subcutaneous injection of IL-33 is sufficient to drive skin fibrosis, and this response is attenuated in IL13-deficient mice [82]. Thus, these studies suggest a hierarchy among pro-fibrotic cytokines whereby tissue injury activates IL-33 release, which in turn induces the effector cytokine IL-13 in a variety of target cells. Although the above studies suggest that IL33 is involved in the development of SSc, the involvement of this cytokine in pathogenesis must be investigated further.
Insights From Chemical-Induced Fibrosis Models
Several animal models of SSc rely on the administration of compounds with known pro-fibrotic toxicity. With comparison with other complimentary models and correlation with human SSc to provide context, chemical models provide unique insights. In addition to the widely studied agent bleomycin [83], other chemical agents have been used as a model of fibrosis. In particular, building on observations that a type I interferon gene expression signature is elevated in patients with SSc [84, 85], and that the TLR3 and RIG1 ligand poly I:C is a potent inducer of type I interferons [86], subcutaneous infusion of poly I:C was found to induce fibrotic and inflammatory dermal changes [87••]. These changes included induction of the vasculopathic mediator endothelin-1, and parallel studies revealed a concordant increase in endothelin-1 mRNA expression in the skin of SSc patients [88]. Additionally, poly I:C was also found to induce TGFβ target genes, suggesting that TLR3 or other dsRNA sensors are a point of interaction between inflammatory signaling and pro-fibrotic growth factors. Although it is currently unclear how TLR3 is involved in the pathogenesis of SSc, these results reveal a potential pathway whereby tissue damage or viruses may contribute to SSc skin disease [89].
Conclusions
Although it has long been suspected that SSc is an immune-mediated disease, only in recent years is evidence of this starting to emerge. Perhaps the reason for this slow progress is not that the volume or quality of studies has been lacking, but rather that appropriate contexts for combining the disparate observations made in this heterogeneous systemic disorder has previously been lacking. These various contexts encompass the genetic, molecular, and clinical framework that places each individual study in perspective. In that respect, GWAS studies have implicated polymorphisms in the HLA locus and other immune-associated genes as risk factors for the development of SSc, providing strong evidence that the immune system is a central mediator of SSc [5, 6••, 7•]. Similarly, use of molecular data to allocate SSc patients to subsets revealed significant heterogeneity of these patients and enabled identification a particular inflammatory subset with evidence of immune activation [15]. The results from these subset studies correlate with those from animal models revealing that specific cytokines, for example IL13, are critical mediators of fibroinflammatory pathology in models of SSc; these findings may be most relevant for this inflammatory subset of patients [25••]. All of these studies providing evidence for the immune-mediated pathogenesis of SSc build upon many years of observation of mixed lymphoid infiltrates present in the skin and lungs of SSc patients [90, 91].
An important achievement leading to understanding of SSc is the placing of the observations of the past 30 years in the context of these newly defined patient subsets. For instance, the number of cytokines and other mediators observed to be elevated in SSc is so large that constructing a single coherent model that incorporates each of these is challenging. From this perspective, subdividing SSc patients may deconvolute this complexity to the point where the contributions and mechanistic interrelations of the pathways involved for individual patients can be resolved. Similarly, molecular stratification of patients may help build a more unified picture of the genetic risk for SSc, as the power of previously published studies was probably diluted by inclusion of patients that varied in their underlying mechanism of disease.
Definition of context is also critical in ongoing investigations into the mechanistic contribution of fibroinflammatory mediators to animal models and use of this understanding to improve patient care. Given the complexity of the disease and the large number of potential mediators, we suggest specific priorities to enrich our collective ability to relate and compare parallel studies. First, wherever possible, consistency among investigators in terms of methods and models examined should be encouraged. Second, although it is desirable to develop new SSc models, replication of findings across existing models should be highly valued. Third, further attempts should be made to develop the relationships and hierarchies among individual mediators and cellular participants to determine which are upstream, downstream, and orthogonal to one another. Fourth, establishing the relationship of the different animal models within the heterogeneous nature of SSc is critical. Thus, future studies of the immunopathogenesis of SSc should, ideally, use models with rigorously defined relationships to SSc patient subsets. Last, priority should be given to corroborating findings from animal models by use of patient-derived samples.
In summary the most notable advances in our understanding the immunopathogenesis of SSc in recent years are an improved contextual structure comprising genetic data, molecular patient stratification, and an emerging understanding of how SSc animal models relate to the human disease. These resources will orient the future study of SSc, enabling investigators to build momentum toward understanding this complex disorder.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Strehlow D, Korn JH. Biology of the scleroderma fibroblast. Curr Opin Rheumatol. 1998;10(6):572–8.
Sgonc R. The vascular perspective of systemic sclerosis: of chickens, mice and men. Int Arch Allergy Immunol. 1999;120(3):169–76.
Stone OJ. Autoimmunity as a secondary phenomenon in scleroderma (and so-called human adjuvant disease). Med Hypotheses. 1991;34(2):127–30.
Manno R, Boin F. Immunotherapy of systemic sclerosis. Immunotherapy. 2010;2(6):863–78.
Zhou X, Lee JE, Arnett FC, et al. HLA-DPB1 and DPB2 are genetic loci for systemic sclerosis: a genome-wide association study in Koreans with replication in North Americans. Arthritis Rheum. 2009;60(12):3807–14.
•• Radstake TR, Gorlova O, Rueda B, et al. Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat Genet. 2010;42(5):426–9. This paper reports the findings of a large GWAS study of SSc, identifying the CD247 locus as involved and confirming previous observations regarding the MHC locus, IRF5, and STAT4.
• Allanore Y, Saad M, Dieude P, et al. Genome-wide scan identifies TNIP1, PSORS1C1, and RHOB as novel risk loci for systemic sclerosis. PLoS Genet. 2011;7(7):e1002091. This GWAS study of SSc confirms the linkage with MHC and identifies the TNIP1, RHOB, and PSORS1C1 loci as involved.
Sharif R, Mayes MD, Tan FK, et al. IRF5 polymorphism predicts prognosis in patients with systemic sclerosis. Ann Rheum Dis. 2012;71(7):1197–202.
Bossini-Castillo L, Martin JE, Broen J, et al. A GWAS follow-up study reveals the association of the IL12RB2 gene with systemic sclerosis in Caucasian populations. Hum Mol Genet. 2012;21(4):926–33.
Bossini-Castillo L, Simeon CP, Beretta L, et al. A multicenter study confirms CD226 gene association with systemic sclerosis-related pulmonary fibrosis. Arthritis Res Ther. 2012;14(2):R85.
Bossini-Castillo L, Broen JC, Simeon CP, et al. A replication study confirms the association of TNFSF4 (OX40L) polymorphisms with systemic sclerosis in a large European cohort. Ann Rheum Dis. 2011;70(4):638–41.
Broen JC, Coenen MJ, Radstake TR. Genetics of systemic sclerosis: an update. Curr Rheumatol Rep. 2012;14(1):11–21.
Romano E, Manetti M, Guiducci S, et al. The genetics of systemic sclerosis: an update. Clin Exp Rheumatol. 2011;29(2 Suppl 65):S75–86.
Beyer C, Schett G, Distler O, Distler JH. Animal models of systemic sclerosis: prospects and limitations. Arthritis Rheum. 2010;62(10):2831–44.
Sargent JL, Whitfield ML. Capturing the heterogeneity in systemic sclerosis with genome-wide expression profiling. Expert Rev Clin Immunol. 2011;7(4):463–73.
LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15(2):202–5.
Milano A, Pendergrass SA, Sargent JL, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS One. 2008;3(7):e2696.
•• Pendergrass SA, Lemaire R, Francis IP, et al. Intrinsic gene expression subsets of diffuse cutaneous systemic sclerosis are stable in serial skin biopsies. J Invest Dermatol. 2012;132(5):1363–73. This work extends the results of Milano et al., furnishing evidence that the SSc subsets identified are stable and may not simply reflect different stages during the development of SSc.
Yamamoto T. Animal model of systemic sclerosis. J Dermatol. 2010;37(1):26–41.
Artlett CM. Animal models of scleroderma: fresh insights. Curr Opin Rheumatol. 2010;22(6):677–82.
Jaffee BD, Claman HN. Chronic graft-versus-host disease (GVHD) as a model for scleroderma. I. Description of model systems. Cell Immunol. 1983;77(1):1–12.
Fleming JN, Shulman HM, Nash RA, et al. Cutaneous chronic graft-versus-host disease does not have the abnormal endothelial phenotype or vascular rarefaction characteristic of systemic sclerosis. PLoS One. 2009;4(7):e6203.
Ruzek MC, Jha S, Ledbetter S, et al. A modified model of graft-versus-host-induced systemic sclerosis (scleroderma) exhibits all major aspects of the human disease. Arthritis Rheum. 2004;50(4):1319–31.
Kaplan DH, Anderson BE, McNiff JM, et al. Target antigens determine graft-versus-host disease phenotype. J Immunol. 2004;173(9):5467–75.
•• Greenblatt MB, Sargent JL, Farina G, et al. Interspecies comparison of human and murine scleroderma reveals IL-13 and CCL2 as disease subset-specific targets. Am J Pathol. 2012;180(3):1080–94. Here we describe the contribution of IL-13 to the SclGVHD model, including the cellular sources and effects of IL-13. Also, molecular profiling of the SclGVHD model is used to map it to the inflammatory subset of SSc patients.
McCormick LL, Zhang Y, Tootell E, Gilliam AC. Anti-TGF-b treatment prevents skin and lung fibrosis in murine sclerodermatous graft-versus-host disease: a model for human scleroderma. J Immunol. 1999;163:5693–9.
Christmann RB, Wells AU, Capelozzi VL, Silver RM. Gastroesophageal reflux incites interstitial lung disease in systemic sclerosis: clinical, radiologic, histopathologic, and treatment evidence. Semin Arthritis Rheum. 2010;40(3):241–9.
Varga J, Pasche B. Transforming growth factor beta as a therapeutic target in systemic sclerosis. Nat Rev Rheumatol. 2009;5(4):200–6.
Abraham D. Connective tissue growth factor: growth factor, matricellular organizer, fibrotic biomarker or molecular target for anti-fibrotic therapy in SSc? Rheumatology (Oxford). 2008;47 Suppl 5:v8–9.
Trojanowska M. Role of PDGF in fibrotic diseases and systemic sclerosis. Rheumatology (Oxford). 2008;47 Suppl 5:v2–4.
Silver RM. Endothelin and scleroderma lung disease. Rheumatology (Oxford). 2008;47 Suppl 5:v25–6.
Minty A, Chalon P, Derocq JM, et al. Interleukin-13 is a new human lymphokine regulating inflammatory and immune responses. Nature. 1993;362(6417):248–50.
Supajatura V, Ushio H, Nakao A, et al. Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. J Clin Invest. 2002;109(10):1351–9.
Bellinghausen I, Brand P, Bottcher I, et al. Production of interleukin-13 by human dendritic cells after stimulation with protein allergens is a key factor for induction of T helper 2 cytokines and is associated with activation of signal transducer and activator of transcription-6. Immunology. 2003;108(2):167–76.
Kim EY, Battaile JT, Patel AC, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med. 2008;14(6):633–40.
Barlow JL, McKenzie AN. Nuocytes: expanding the innate cell repertoire in type-2 immunity. J Leukoc Biol. 2011;90(5):867–74.
• Neill DR, Wong SH, Bellosi A, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464(7293):1367–70. With Refs. [38] and [39], this paper identifies nuocytes as a novel IL-13-producing, IL-25 and IL33-responsive, innate immune cell.
• Moro K, Yamada T, Tanabe M, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. 2010;463(7280):540–4. With Refs. [37] and [39], this paper identifies a novel IL13-producing population that responds to IL-25.
• Price AE, Liang HE, Sullivan BM, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci USA. 2010;107(25):11489–94. With Refs. [37] and [38], this paper identifies systemically dispersed lineage negative cell that secretes IL-13 in response to IL-25 and IL33.
Nelms K, Keegan AD, Zamorano J, et al. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol. 1999;17:701–38.
Kelly-Welch AE, Hanson EM, Boothby MR, Keegan AD. Interleukin-4 and interleukin-13 signaling connections maps. Science. 2003;300(5625):1527–8.
Tabata Y, Khurana Hershey GK. IL-13 receptor isoforms: breaking through the complexity. Curr Allergy Asthma Rep. 2007;7(5):338–45.
Donaldson DD, Whitters MJ, Fitz LJ, et al. The murine IL-13 receptor alpha 2: molecular cloning, characterization, and comparison with murine IL-13 receptor alpha 1. J Immunol. 1998;161(5):2317–24.
Murata T, Husain SR, Mohri H, Puri RK. Two different IL-13 receptor chains are expressed in normal human skin fibroblasts, and IL-4 and IL-13 mediate signal transduction through a common pathway. Int Immunol. 1998;10(8):1103–10.
Daines MO, Tabata Y, Walker BA, et al. Level of expression of IL-13R alpha 2 impacts receptor distribution and IL-13 signaling. J Immunol. 2006;176(12):7495–501.
Wood N, Whitters MJ, Jacobson BA, et al. Enhanced interleukin (IL)-13 responses in mice lacking IL-13 receptor alpha 2. J Exp Med. 2003;197(6):703–9.
Chiaramonte MG, Mentink-Kane M, Jacobson BA, et al. Regulation and function of the interleukin 13 receptor alpha 2 during a T helper cell type 2-dominant immune response. J Exp Med. 2003;197(6):687–701.
Leigh R, Ellis R, Wattie J, et al. Is interleukin-13 critical in maintaining airway hyperresponsiveness in allergen-challenged mice? Am J Respir Crit Care Med. 2004;170(8):851–6.
Fichtner-Feigl S, Young CA, Kitani A et al.: IL-13 signaling via IL-13R alpha2 induces major downstream fibrogenic factors mediating fibrosis in chronic TNBS colitis. Gastroenterology 2008, 135(6):2003–2013, 2013 e2001-2007.
Fichtner-Feigl S, Strober W, Kawakami K, et al. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. 2006;12(1):99–106.
Hasegawa M, Fujimoto M, Kikuchi K, Takehara K. Elevated serum levels of interleukin 4 (IL-4), IL-10, and IL-13 in patients with systemic sclerosis. J Rheumatol. 1997;24(2):328–32.
Fuschiotti P, Medsger Jr TA, Morel PA. Effector CD8+ T cells in systemic sclerosis patients produce abnormally high levels of interleukin-13 associated with increased skin fibrosis. Arthritis Rheum. 2009;60(4):1119–28.
Medsger Jr TA, Ivanco DE, Kardava L, et al. GATA-3 up-regulation in CD8+ T cells as a biomarker of immune dysfunction in systemic sclerosis, resulting in excessive interleukin-13 production. Arthritis Rheum. 2011;63(6):1738–47.
Sweiss NJ, Hushaw L, Thenappan T, et al. Diagnosis and management of pulmonary hypertension in systemic sclerosis. Curr Rheumatol Rep. 2010;12(1):8–18.
Christmann RB, Hayes E, Pendergrass S, et al. Interferon and alternative activation of monocyte/macrophages in systemic sclerosis-associated pulmonary arterial hypertension. Arthritis Rheum. 2011;63(6):1718–28.
Riccieri V, Rinaldi T, Spadaro A, et al. Interleukin-13 in systemic sclerosis: relationship to nailfold capillaroscopy abnormalities. Clin Rheumatol. 2003;22(2):102–6.
Cutolo M, Sulli A, Smith V. Assessing microvascular changes in systemic sclerosis diagnosis and management. Nat Rev Rheumatol. 2010;6(10):578–87.
Granel B, Allanore Y, Chevillard C, et al. IL13RA2 gene polymorphisms are associated with systemic sclerosis. J Rheumatol. 2006;33(10):2015–9.
Granel B, Chevillard C, Allanore Y, et al. Evaluation of interleukin 13 polymorphisms in systemic sclerosis. Immunogenetics. 2006;58(8):693–9.
Broen JC, Dieude P, Vonk MC, et al. Polymorphisms in the interleukin 4, interleukin 13, and corresponding receptor genes are not associated with systemic sclerosis and do not influence gene expression. J Rheumatol. 2012;39(1):112–8.
Zhu Z, Homer RJ, Wang Z, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103(6):779–88.
Zheng T, Oh MH, Oh SY, et al. Transgenic expression of interleukin-13 in the skin induces a pruritic dermatitis and skin remodeling. J Invest Dermatol. 2009;129(3):742–51.
Matsushita M, Yamamoto T, Nishioka K. Upregulation of interleukin-13 and its receptor in a murine model of bleomycin-induced scleroderma. Int Arch Allergy Immunol. 2004;135(4):348–56.
Aliprantis AO, Wang J, Fathman JW, et al. Transcription factor T-bet regulates skin sclerosis through its function in innate immunity and via IL-13. Proc Natl Acad Sci USA. 2007;104(8):2827–30.
Belperio JA, Dy M, Burdick MD, et al. Interaction of IL-13 and C10 in the pathogenesis of bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2002;27(4):419–27.
Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83.
Gallina G, Dolcetti L, Serafini P, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. 2006;116(10):2777–90.
Zhu Z, Ma B, Zheng T, et al. IL-13-induced chemokine responses in the lung: role of CCR2 in the pathogenesis of IL-13-induced inflammation and remodeling. J Immunol. 2002;168(6):2953–62.
Palmer G, Gabay C. Interleukin-33 biology with potential insights into human diseases. Nat Rev Rheumatol. 2011;7(6):321–9.
Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23(5):479–90.
Chackerian AA, Oldham ER, Murphy EE, et al. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J Immunol. 2007;179(4):2551–5.
Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci USA. 2009;106(22):9021–6.
Lefrancais E, Roga S, Gautier V, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci USA. 2012;109(5):1673–8.
Pichery M, Mirey E, Mercier P, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J Immunol. 2012;188(7):3488–95.
Kurowska-Stolarska M, Hueber A, Stolarski B, McInnes IB. Interleukin-33: a novel mediator with a role in distinct disease pathologies. J Intern Med. 2010;269(1):29–35.
Manetti M, Ibba-Manneschi L, Liakouli V, et al. The IL1-like cytokine IL33 and its receptor ST2 are abnormally expressed in the affected skin and visceral organs of patients with systemic sclerosis. Ann Rheum Dis. 2010;69(3):598–605.
Yanaba K, Yoshizaki A, Asano Y, et al. Serum IL-33 levels are raised in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. Clin Rheumatol. 2011;30(6):825–30.
Manetti M, Guiducci S, Ceccarelli C, et al. Increased circulating levels of interleukin 33 in systemic sclerosis correlate with early disease stage and microvascular involvement. Ann Rheum Dis. 2011;70(10):1876–8.
Miller AM. Role of IL-33 in inflammation and disease. J Inflamm (Lond). 2011;8(1):22.
Ho LH, Ohno T, Oboki K, et al. IL-33 induces IL-13 production by mouse mast cells independently of IgE-FcepsilonRI signals. J Leukoc Biol. 2007;82(6):1481–90.
Smithgall MD, Comeau MR, Yoon BR, et al. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol. 2008;20(8):1019–30.
Rankin AL, Mumm JB, Murphy E, et al. IL-33 induces IL-13-dependent cutaneous fibrosis. J Immunol. 2009;184(3):1526–35.
Yamamoto T. The bleomycin-induced scleroderma model: what have we learned for scleroderma pathogenesis? Arch Dermatol Res. 2006;297(8):333–44.
Tan FK, Zhou X, Mayes MD, et al. Signatures of differentially regulated interferon gene expression and vasculotrophism in the peripheral blood cells of systemic sclerosis patients. Rheumatology (Oxford). 2006;45(6):694–702.
York MR, Nagai T, Mangini AJ, et al. A macrophage marker, Siglec-1, is increased on circulating monocytes in patients with systemic sclerosis and induced by type I interferons and toll-like receptor agonists. Arthritis Rheum. 2007;56(3):1010–20.
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413(6857):732–8.
•• Farina GA, York MR, Di Marzio M, et al. Poly(I:C) drives type I IFN- and TGFbeta-mediated inflammation and dermal fibrosis simulating altered gene expression in systemic sclerosis. J Invest Dermatol. 2010;130(11):2583–93. This paper identifies expression of type I interferon and TGFβ target genes in the skin of SSc patients and demonstrates that chronic subcutaneous infusion of type I interferon eliciting poly I:C stimulates inflammation and fibrosis.
Farina G, York M, Collins C, Lafyatis R. dsRNA activation of endothelin-1 and markers of vascular activation in endothelial cells and fibroblasts. Ann Rheum Dis. 2011;70(3):544–50.
Cavassani KA, Ishii M, Wen H, et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205(11):2609–21.
White B. Immunopathogenesis of systemic sclerosis. Rheum Dis Clin North Am. 1996;22(4):695–708.
Roumm AD, Whiteside TL, Medsger Jr TA, Rodnan GP. Lymphocytes in the skin of patients with progressive systemic sclerosis. Quantification, subtyping, and clinical correlations. Arthritis Rheum. 1984;27(6):645–53.
Acknowledgments
Dr Aliprantis is supported by grants from the Burroughs Wellcome Fund and the National Institutes of Health (K08 AR 054859 and R01 AR060363). Dr Aliprantis’ work relevant to SSc was previously supported by the Scleroderma Research Foundation and Sanofi pharmaceuticals. The authors thank Drs Julia Charles, Susan Ritter, and Joerg Ermann for their thoughtful review and discussion of this manuscript.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Scleroderma
Rights and permissions
About this article
Cite this article
Greenblatt, M.B., Aliprantis, A.O. The Immune Pathogenesis of Scleroderma: Context Is Everything. Curr Rheumatol Rep 15, 297 (2013). https://doi.org/10.1007/s11926-012-0297-8
Published:
DOI: https://doi.org/10.1007/s11926-012-0297-8