
Abstract
Patients with systemic sclerosis (SSc) can develop pulmonary hypertension (PH; mean pulmonary artery pressure ≥ 25 mm Hg) caused by pulmonary arterial hypertension (PAH), left ventricular disease, or pulmonary fibrosis. PAH is a pulmonary vascular disease, the diagnosis of which requires pulmonary capillary wedge pressure less than 15 mm Hg, pulmonary vascular resistance greater than 3 Wood Units, and exclusion of thromboembolism and parenchymal lung disease. Molecular mechanisms underlying PAH-SSc include activation of inflammatory and fibrogenic pathways in the vasculature and right ventricle. Circulating autoantibodies trigger endothelial damage and fibroblast activation. PAH most commonly occurs as a late complication in patients with limited cutaneous disease and anticentromere antibodies. Although echocardiography is a useful screening tool, heart catheterization is required to diagnose PAH before initiating therapy. Prognosis and therapeutic response are worse in PAH-SSc than in other PAH categories (median survival, 1–3 y). Approved therapies include prostacyclins, endothelin antagonists, and phosphodiesterase type 5 inhibitors. Research is needed to define disease mechanisms and develop effective therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Systemic sclerosis (SSc) is a systemic, inflammatory, autoimmune disease characterized by circulating autoantibodies and, clinically, by progressive and often severe fibrotic pathology of the skin, joints, and internal organs, notably the lungs, esophagus, and kidneys. SSc is divided into diffuse cutaneous (dcSSc) and limited cutaneous syndromes (lcSSc), depending on the extent of skin thickening. Skin thickening confined to the elbows, knees, or face characterizes lcSSc, whereas dcSSc reflects more extensive disease. Most dcSSc patients (about 60%) are Scl-70 positive (antitopoisomerase I), whereas 88.7% of lcSSc patients are anticentromere antibody positive [1••].
Pulmonary complications, including pulmonary arterial hypertension (PAH), pulmonary fibrosis (PF), or interstitial lung disease (ILD) are now the leading causes of death in SSc [2]. SSc patients with PAH usually present with fatigue, dyspnea, and exertional syncope; however, there are many causes of these nonspecific symptoms, including ILD, diastolic heart failure with preserved ejection fraction, aspiration pneumonitis, and deconditioning.
There are no proven disease-modifying treatments for SSc. Numerous immunosuppressive and antifibrotic agents have been investigated, including D-penicillamine, interferon, colchicine, methotrexate, relaxin, chlorambucil, photophoresis, and cyclophosphamide; however, none of these agents has been proven effective based on a large, randomized controlled trial and it is unclear whether any improve PAH-SSc. This review offers guidance on the diagnosis and management of PAH associated with SSc (PAH-SSc), highlights the need to better define disease mechanisms, and identifies the need for more effective therapies.
Prognosis
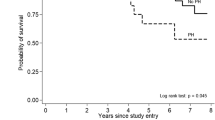
The 10-year survival rate for SSc has improved to 70%, up from 50% in the 1970s [3]. Although mortality from renal crisis has diminished in the past 30 years (from 42% to 6%) because of the use of angiotensin-converting enzyme inhibitors, the percentage of SSc patients dying from PAH and PF has increased (from 6% to 33%) [4]. Paradoxically, this has made PAH and PF the most common causes of death in SSc. Patients with PAH-SSc have a 1-year survival rate ranging from 50% to 87% [5••] and appear to have about a threefold increase in risk of mortality compared with idiopathic PAH (IPAH) [6]. ILD appears to be the major cause of mortality in dcSSc, with PAH the major cause of death in lcSSc.
Classification of Pulmonary Hypertension
Pulmonary hypertension (PH) is defined as a resting mean pulmonary artery pressure (mPAP) ≥ 25 mm Hg. The World Health Organization (WHO) classification recognizes five categories of PH: 1) PAH; 2) PH associated with left heart disease; 3) PH associated with lung disease/hypoxia; 4) thromboembolic PH; and 5) PH associated with multisystem disease (Table 1) [7•]. SSc patients may have category 1, 2, or 3 PH. Targeted therapies for category 1 PAH are not indicated for SSc patients with simple elevations of mPAP because of ILD or left ventricular disease. There is a common misunderstanding of the requirements to diagnose PAH, which in turn leads to misdiagnosis of PAH, especially in SSc patients. Rheumatologists should be familiar with the diagnostic algorithm and/or have a close referral relationship with a cardiologist or pulmonologist in a PH program.
Prevalence and Epidemiology of SSc
The annual incidence and prevalence of SSc in Europe is lower (about 10 and 50 cases per million adults, respectively) [1••] than in the United States (19.3 and 242 cases per million adults, respectively) [8] and South Australia [9]. The prevalence of SSc also varies by region, gender, and ethnicity. African Americans have earlier disease onset and are more likely to have dcSSc. SSc more commonly afflicts women than men (3:1 in the United Kingdom; 6:1 in Europe; and 14:1 in Japan) [1••]. This is reminiscent of the 3:1 female–male preponderance in IPAH [10]. PH referral centers vary in percentage of SSc patients, with more seen in Chicago compared with the French National Registry [10, 11].
Subsets of SSc at Risk for PAH
Multiple registries describe SSc populations worldwide. The South Australian Scleroderma Register, a population-based cohort of 374 living and 234 deceased SSc patients revealed that isolated PH occurs as a late-stage complication, about 20 years after scleroderma onset. Isolated PH was most common in those with lcSSc and was predicted by multiple telangiectasia, reduced nailfold capillary density, digital ulceration, and gross reduction of the diffusing capacity of carbon monoxide (DLCO) [9]. No dcSSc patient in this cohort developed PAH.
A registry created by the European League Against Rheumatism Scleroderma Trials and Research (EUSTAR) group suggests that the autoantibody profile has better predictive value for development of PAH than traditional clinical risk factors. They compared risks of lung complications based on clinical findings versus the presence of autoantibodies in 3656 patients (87% of women, 1349 dcSSc, 2101 lcSSc) from 102 centers in 30 countries [1••]. The frequency of PH, diagnosed by echocardiography, was similar between dcSSc and lcSSc (22.3% and 20.5% of patients, respectively). However, the PAH surrogate in this study (PH with no PF) occurred more in lcSSc than dcSSc (9.2% vs 5.9%); conversely, PF, which was more common, occurred more frequently in dcSSc versus lcSSC (53.4 vs 34.7%). PH without PF was more prevalent in those with anticentromere versus anti-Scl70 antibodies (13% vs 5%) [1••]. Female patients were almost twice as likely to be anticentromere antibody positive as male patients (26.3% vs 50.3% in the lcSSc group). An important caveat is that this study includes no catheterization data, did not clearly define their diagnostic criteria for PH, and imprecisely referred to all PH as PAH. Nonetheless, PAH-SSc is more common in patients with lcSSc (formerly called CREST [calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia] syndrome) and in those with anticentromere antibodies [12]. ILD, the other major lung complication of SSc, is more common in patients with dcSSc who are Scl70 positive and anticentromere negative [13, 14]. Perhaps screening for PAH should focus on lcSSc patients with anticentromere antibody positivity. The female preponderance (relative to males) amongst lcSSc patient that have PH (6.5:1) is greater than the female preponderance amongst the general lcSSc population, suggesting females with lcSSc are at increased risk of the disease and may merit screening.
Histology and Molecular Features of PAH-SSc
There is a growing recognition that SSc is both a vascular disease and a fibrosing, inflammatory disease. Key features of its pathophysiology include endothelial cell injury induced by infection, immune-mediated cytotoxicity, and the presence of antiendothelial antibodies [15•]. Currently, there is no clear genetic basis for PAH-SSc. Bone morphogenetic protein receptor mutations, which are common in familial PAH, are rare in PAH-SSc [16]. Single nucleotide polymorphisms (SNPs) in the Fas promoter may predict susceptibility to SSc (perhaps reflecting altered susceptibility to apoptosis) [17], but no SNPs that predict PAH-SSc have been identified [5••]. SSc patients also have cellular and humoral immunologic abnormalities, which include chronic mononuclear cell infiltration of affected tissues, dysregulation of the growth factor production, and lymphokines.
The pulmonary vascular pathology in PAH-SSc includes plexiform lesions, medial hypertrophy of small pulmonary arteries, intimal fibrosis, adventitial thickening, and obliteration of small arteries (Fig. 1). It is indistinguishable from IPAH, explaining inclusion of both syndromes in category 1 PH. Nonetheless, certain pathologic abnormalities (eg, greater contribution from autoantibodies and inflammation) are more prevalent in PAH-SSc. These insults likely trigger the endothelial dysfunction [18] and fibroblast activation [19] that typify PAH-SSc.

Algorithm for early diagnosis of pulmonary arterial hypertension in patients with scleroderma. Concentric medial hypertrophy a and intimal fibrosis b can be seen in small pulmonary artery from a scleroderma lung. BNP B-type natriuretic peptide, CCBs calcium channel blockers, DLCO diffusing capacity of carbon monoxide, ECG electrocardiogram, ETRA endothelin receptor antagonist, FVC forced expiratory vital capacity, lcSSc limited cutaneous systemic sclerosis, mPAP mean pulmonary artery pressure, PDE-5 inhibitor phosphodiesterase type 5 inhibitor, PE pulmonary embolism, PF pulmonary fibrosis, RV right ventricular, RVH RV hypertrophy, TAPSE tricuspid annular plane systolic excursion, TR tricuspid regurgitation, V/Q ventilation/perfusion, WHO World Health Organization, WU Wood Units
PAH-SSc is a vascular disease. This likely results from increased endothelial apoptosis, increased inflammation and disordered angiogenesis. Endothelial cell apoptosis damages arteries and capillaries and reflects antibody-dependent, cell-mediated cytotoxicity, occurring via the Fas pathway [20]. Inflammation is also increased in PAH-SSc. This may be caused by activation of the calcium-activated, transcription factor, nuclear factor of activated T cells (NFAT) [21], although this has not been specifically tested in PAH-SSc. Vascular changes in PAH-SSc also reflect dysregulated angiogenesis. Clinically, poor angiogenesis is reflected by loss of nail bed vessels, reduced DLCO, and the presence of Raynaud phenomenon and telangiectasia. Perhaps as a compensatory mechanism, levels of vascular endothelial growth factor (VEGF) are elevated fourfold in SSc [22]. Choi et al. [22] suggest that high VEGF levels may serve as a surrogate indicator of capillary damage in SSc.
Autoantibodies can damage the endothelium and activate fibroblasts. The prevalence of antiendothelial cell autoantibodies (AECAs) in SSc is about 40% [18]. They are directed against many vascular cell targets, including endothelial cells [23] and fibroblasts [19]. These complex immunologic abnormalities are associated with severe and often progressive cutaneous and visceral fibrosis and obliteration of the lumen of small arteries and arterioles. There are increased levels of growth factors and cytokines in SSc, which may be responsible for 1) recruiting and activating fibroblast precursors and fibrocytes; 2) the profibrotic activation of fibroblasts; and 3) the proliferation of subendothelial and vascular smooth muscle cells, ultimately leading to fibroproliferative vasculopathy and tissue fibrosis [5••].
Clinical Features of PAH-SSc
PAH is a syndrome of dyspnea, fatigue, exertional syncope, and occasional hemoptysis that results in impaired right ventricular (RV) function caused by obstruction of the pulmonary vasculature. The clinical severity is assessed based on the WHO functional classification of PH (Table 2). PAH is a diagnosis of exclusion and the many other causes of PH must be shown to be absent. This usually requires (at minimum) electrocardiography (ECG), echocardiography, a CT scan, and ventilation/perfusion lung scan (to exclude pulmonary embolism and ILD), pulmonary function tests, and oximetry with a sleep study to exclude sleep apnea (Fig. 1). Cardiac catheterization with vasodilatory testing is required to confirm the diagnosis [24].
Echocardiographic studies have consistently overestimated the prevalence of PH and PAH-SSc. In a prospective study of Doppler echocardiography in 599 patients with SSc (including 29 patients with known PAH and 33 patients with suspected PAH) only 18 of the 33 that were suspected to have actually had PAH confirmed by catheterization. Three of the “suspected PAH” patients had left ventricular dysfunction and 12 had no PH [25]. In a prospective, 4-year study, 794 patients with SSc were screened for PAH using echocardiography, DLCO, and clinical assessment [26]. Catheterization was performed in patients either because of a suspected PA systolic pressure greater than 35 mm Hg or because of a low baseline DLCO or a decrease of more than 20% of expected DLCO in patients that did not have PF. The prevalence of confirmed PAH was only 12% (89 of 722 patients). The survival of this PAH-SSc cohort was poor; after 1, 2, and 3 years of follow-up, survival was 81%, 63%, and 56%, with adverse outcomes predicted by measure of impaired RV function (low cardiac index and high right atrial pressure) [26].
Screening for PAH-SSc
We propose a diagnostic algorithm for assessing SSc to exclude PAH (Fig. 1), although admittedly it is not evidence-based. It is consistent with approaches used to assess other PAH patients outlined in a consensus document from the American Heart Association and American College of Cardiology [27•]. The goal of the evaluation of SSc patients is to determine PA pressure and, if it is significantly elevated, exclude causes of PH such as chronic venous thromboembolism, left ventricular dysfunction, and ILD/hypoxia. The “cardiocentric” nature of our algorithm is important because category 2 and 3 PH are common in SSc. In SSc, mild left ventricular dysfunction, left atrial enlargement, and impaired diastolic relaxation are common [28]. The hemodynamic severity of the calculated pulmonary vascular resistance (PVR) may be less severe in PAH-SSc versus IPAH; however, RV function is often disproportionately depressed.
Physical Examination, ECG, and Chest Radiography
Physical examination, ECG, and chest radiography can identify patients with significant PAH-SSc and RV hypertrophy (RVH), but are insensitive for the detection of early disease. An ECG with right-axis deviation, right atrial enlargement, and RVH suggests PAH, whereas left-axis deviation, left atrial enlargement, and left ventricular hypertrophy suggests diastolic heart failure. During physical examination, one should listen for a loud pulmonary component of the second heart sound and palpate the precordium for an RV lift. Tricuspid regurgitation murmurs are occasionally audible in advanced disease. The jugular venous pressure will be elevated only when the patient progresses to RV failure. When PH is severe, the right descending PA is enlarged (> 1.1 cm) on PA chest radiograph and there may be pruning (loss of peripheral vascular markings). On the lateral view, there is filling in of the retrosternal space when the RV is enlarged. These are specific, but insensitive, signs of PH, and the early detection of PAH-SSc largely depends on noninvasive testing.
Echocardiography
Echocardiography is the most commonly used screening tool for PH. Yearly echocardiography screening for patients with SSc is safe but there is no evidence that early diagnosis and treatment improve long-term outcomes [5••]. Doppler has reasonable sensitivity (90%) and specificity (75%) for detecting moderate-to-severe elevations in PAP in symptomatic SSc patients [29], but its utility in asymptomatic SSc patients is undetermined [30].
CMR Imaging
Cardiac magnetic resonance (CMR) yields exquisite image quality and provides accurate quantification of biventricular chamber size and function, characterization of the myocardial tissue, and evaluation of the pulmonary artery anatomy; however, it is not clear whether CMR is sensitive enough to detect the mild PH often seen in SSc. In a recent trial comparing noninvasive tests to right heart catheterization for identifying PH in SSc, the size of the main pulmonary artery as measured by CMR had a sensitivity of 68% and a specificity of 71% and was outperformed by echocardiography, which had a sensitivity and specificity of 68% and 96%, respectively [31]. It remains to be seen whether other CMR measurements, such as RV volume and hypertrophy, pulmonary artery flow patterns, interventricular dependence, pulmonary artery distensibility, and myocardial fibrosis are useful in PAH-SSc. Unique to CMR is its ability to directly visualize the presence of fibrosis or scar using a technique called late gadolinium enhancement. Many SSc patients have late gadolinium enhancement on CMR [32]; however, the clinical relevance of its ability to detect cardiac involvement awaits definition.
DLCO
The DLCO (in patients without ILD) likely reflects the integrity of the pulmonary vascular bed. A low and decreasing DLCO predicts the subsequent development of PAH-SSc [33]. Indeed, DLCO is predictive of poor outcomes in all types of PAH [34]. A retrospective case-control study of patients enrolled in the Pittsburgh Scleroderma Databank showed that individuals with PAH-SSc had a significantly lower DLCO (52% of predicted) versus matched SSc controls without PH (DLCO, 81% predicted) [33]. A subgroup analysis of patients who underwent multiple DLCO measurements for 15 years found that decreasing DLCO identified SSc patients who developed PAH.
Biomarkers
Antinuclear antibodies (ANAs) are positive in more than 90% of SSc; but their presence does not predict development of PAH. Antifibrillarin antibodies (anti-U3-RNP) are frequently found in PAH-SSc patients [23, 35]. Antiendothelial cell antibodies have been associated with PAH-SSc; however, there are no standard approaches for their measurement. B-type natriuretic peptide (BNP) is a cardiac hormone secreted by the ventricles in response to volume or pressure overload. BNP levels were elevated in SSc patients with cardiac disease [36]. There is a significant correlation between N-terminal (NT)-proBNP values and mPAP and PVR [26]. Good correlation between BNP and functional state/pulmonary hemodynamics has been confirmed by others (albeit with different threshold BNP values) [37], suggesting a similar role for monitoring BNP in PAH-SSc, as is the case in other forms of PAH. However, BNP or NT-proBNP, though predictive of prognosis at baseline and with therapy, have not yet been validated as a useful serial measure in PAH management.
Exercise Testing
Many centers use 6-minute walk distance (6MWD) as a primary outcome in the assessment of new PAH therapies and to track disease progression. Decreased oxygen saturation and walk distances are predictive of increased mortality in patients with IPAH [38]. Although this is true in PAH-SSc, the 6MWD cannot distinguish among SSc patients with PAH, ILD, and PF. Exercise treadmill testing, using metabolic equivalents, is a strong predictor of mortality in other forms of PAH and, if a patient with SSc can exercise adequately, it is a reproducible means to quantify functional status and monitor systemic blood pressure and oxygenation with exercise [39].
Cardiac Catheterization
A right and left heart catheterization with measurement of cardiac output is mandatory to avoid diagnostic misclassification and the use of expensive and potentially ineffective/harmful therapies in patients with category 2 and 3 PH [40].
Treatment
New therapies for PAH-SSc improve hemodynamics, symptoms, quality of life, and functional capacity; however, none has demonstrated a survival benefit (in part because studies are usually small and last only a few months). In general, the outcome of patients with PAH-SSc is worse than patients with IPAH, which perhaps is not surprising in light of systemic comorbidities in scleroderma [6, 41, 42]. The clinical trials that have studied various interventions in PAH-SSc are summarized in Table 3. The most effective agent studied to date for treatment of PAH-SSc, based on limited subgroup analyses, appears to be continuous intravenous epoprostenol.
General Measures
Although there are no trial data, consensus supports the use of supplemental oxygen to maintain saturation levels above 90%, use of diuretics to minimize edema, and digoxin for management right heart failure or atrial arrhythmias. Consensus guidelines recommend anticoagulating patients with advanced IPAH and advanced PAH associated with connective tissue [27•]. However, less than 50% of PAH-SSc remain on long-term warfarin, largely because of occult lower gastrointestinal bleeding [5••].
Approved PAH Therapies
PAH therapy includes phosphodiesterase type 5 (PDE-5) inhibitors (sildenafil and tadalafil, which cost about $15,000/year), endothelin receptor antagonists (bosentan and ambrisentan, which cost about $48,000/year) and prostacyclins (continuous infusion epoprostenol, treprostinil, subcutaneous treprostinil, or inhaled iloprost, which cost > $80,000/year). Treatment is based on two criteria: WHO functional classification and responsiveness to acute vasodilator testing. Patients who are WHO class IV should receive epoprostenol as initial therapy. Although it is expensive and cumbersome it is effective in PAH-SSc. Patients who respond to an acute vasodilator, such as inhaled nitric oxide can be safely treated with a calcium channel blocker [27•]. Although there are strong feelings (and a little science) regarding calcium channel blockers in PAH-SSC, these inexpensive drugs are appealing, because of the almost universal presence of Raynaud phenomenon, for which they are first-line therapy. In most cases, we recommend nifedipine or diltiazem, rather than verapamil because of the latter’s negative effects on RV contractility. For vasodilator unresponsive patients who are WHO class II-III, oral therapy can be used. Either a PDE-5 inhibitor (sildenafil or tadalafil) or an endothelin antagonist (bosentan, ambrisentan, or sitaxsentan) is appropriate.
Continuous intravenous epoprostenol delivered by pump through a permanent indwelling catheter improves exercise capacity, functional class, cardiopulmonary hemodynamics, and symptoms in patients with IPAH [43]. Epoprostenol is the only PAH-specific therapy that has demonstrated a survival benefit in a randomized, prospective clinical trial, although this was in IPAH [43]. In 102 patients with PAH-SSc, epoprostenol also significantly improved exercise capacity and hemodynamics compared with conventional therapy, but failed to improve survival at 12 weeks [44]. In an open-label extension, the 1- and 2-year probabilities of survival for all treated subjects were 0.71 and 0.52, respectively [45••].
Continuous subcutaneous treprostinil, significantly increases exercise capacity, and improves symptoms and hemodynamics in connective tissue disease associated with PAH [46]. The utility of prostacyclins is limited by the need for meticulous catheter care, continuous infusion, and daily preparation of the medication by patients whose hand function may be impaired.
Endothelin Receptor Antagonists
The first oral therapy approved for therapy of PAH was bosentan. The initial efficacy study included a subgroup analysis of patients with PAH-SSc and revealed a nonsignificant trend toward improvement in 6MWD relative to placebo [47]. A 48-week open-label study of 52 WHO class III patients with various connective tissue diseases (mostly PAH-SSc) noted functional improvements in 27% of patients but no improvement in quality of life. The 1-year survival rate was 92% [48]; however, the results of open-label, nonrandomized trials must be interpreted with caution.
In marked contrast, in a trial comparing bosentan in IPAH versus PAH-SSc, Girgis et al. [49] found that, although first-line bosentan monotherapy was associated with long-term improvement in functional class and good overall survival in WHO class III IPAH, PAH-SSc patients were either stable or declined and tended to have worse survival than IPAH (79% vs 100% at 2 years) [49]. There was 17% drug discontinuation because of hepatotoxicity.
Selective endothelin receptor A antagonists (sitaxsentan approved in Europe; ambrisentan approved in the United States) preserve the vasodilatory action of the endothelin B receptor. It has not been demonstrated that this potential benefit translates into an important benefit in PAH. Hepatic toxicity is a class effect. The use of any endothelin antagonist is contraindicated in pregnancy because of teratogenicity.
PDE-5 Inhibitors
Sildenafil, a PDE-5 inhibitor, inhibits destruction of the vasodilator cyclic guanosine monophosphate (cGMP) [50]. Because PDE-5 is induced in the pulmonary circulation and right ventricle in PAH, PDE-5 inhibitors that counteract the cGMP downregulation have beneficial effects, lowering PVR and increasing cardiac output. Sildenafil (three times daily) and tadalafil (once daily) are approved for use in PAH (including PAH-SSc) in the United States. Sildenafil improves 6MWD in individuals with connective tissue disease (CTD)-related PAH. A post-hoc analysis of PAH-CTD found improvements in functional class, 6MWD, and hemodynamics after 12 weeks of therapy [51]. However, less than half had PAH-SSc and a proper prospective assessment is required (Table 3).
Tadalafil, a once-daily PDE-5 inhibitor, significantly improved 6MWD in the PAH-CTD group; however, the proportion of patients with SSc was not reported [52]. Furthermore, more than half of the participants were on additional therapy with bosentan at enrollment and the effect of tadalafil was reduced in those patients simultaneously receiving bosentan. A recent systematic review of all randomized controlled trials evaluating the efficacy of sildenafil, sitaxsentan, and bosentan showed that the effect size (ie, the ratio of the treatment effect; mean differences in 6MWD between treatment and placebo groups) was not statistically significant for either sildenafil or endothelin receptor antagonists [53]. This meta-analysis is provocative and highlights the limited response in 6MWD to oral therapies in PAH-SSc and signifies the urgent need for larger and longer, appropriately powered trials in PAH-SSc.
Combination Therapy
Combination therapies have become common practice in pulmonary hypertension centers despite limited data supporting their benefit. Data in PAH-SSc are even more limited and therefore recommendations cannot be made at this time.
Lung Transplantation
Lung transplantation remains a treatment of last resort for PAH. Many SSc patients are excluded because of multiorgan disease or esophageal dysmotility, which may increase the risk of aspiration. Nonetheless, with proper screening, 2-year survival rates similar to those achieved as in IPF or IPAH was achieved in a cohort of 29 SSc patients (albeit with higher 6-month mortality) [54].
Treatment Summary
A recent consensus document from the European League Against Rheumatism recommends bosentan as initial therapy for PAH-SSc [55]. In our view, this is questionable based on the warning requiring long-term monitoring of liver function, the interactions with warfarin, the risk of exacerbation of edema/development of heart failure, and the high cost. The optimal first-line therapy for PAH-SSc remains controversial and more data are needed before a formal recommendation of one of the three classes can be made.
Conclusions
PAH-SSc is an important cause of mortality in SSc. Careful diagnosis is required to exclude PF and other causes of PH, which are prevalent. Risk of developing PAH is highest in females with lcSSc and anticentromere antibodies. More research is required to understand the mechanisms of this condition and to identify useful biomarkers and therapies. For now, PAH-SSc patients are treated much the same as other WHO category 1 PH patients. Oral therapy is used for functional class II-III patients (sildenafil or bosentan) and intravenous epoprostenol (delivered by continuous infusion, possibly with addition of sildenafil) is reserved for WHO class IIIb-IV patients. Research is required to help PH physicians better treat SSc patients both in early and late disease.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
•• Walker UA, Tyndall A, Czirjak L, et al.: Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group database. Ann Rheum Dis 2007, 66:754–763. This large European registry provides information on the predictive value of classical risk factors versus antibody profile for organ involvement in scleroderma. It concludes that anticentromere antibody patients are particularly at risk for PAH.
Ioannidis JP, Vlachoyiannopoulos PG, Haidich AB, et al.: Mortality in systemic sclerosis: an international meta-analysis of individual patient data. Am J Med 2005, 118:2–10.
Ranque B, Mouthon L: Geoepidemiology of systemic sclerosis. Autoimmun Rev 2009 Nov 10 (Epub ahead of print).
Steen VD, Medsger TA: Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis 2007, 66:940–944.
•• Hassoun PM: Pulmonary arterial hypertension complicating connective tissue diseases. Semin Respir Crit Care Med 2009, 30:429–439. This article is a well-written review by an expert in pulmonary medicine who has extensive experience with the care and management of patients with SSc and various forms of pulmonary hypertension.
Kawut SM, Taichman DB, Archer-Chicko CL, et al.: Hemodynamics and survival in patients with pulmonary arterial hypertension related to systemic sclerosis. Chest 2003, 123:344–350.
• Simonneau G, Robbins IM, Beghetti M, et al.: Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2009, 54:S43–S54. This article reviews the new WHO classification for pulmonary hypertension. The importance of distinguishing category 1 (PAH) from the more common forms of PH is evident.
Mayes MD, Lacey JV Jr, Beebe-Dimmer J, et al.: Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum 2003, 48:2246–2255.
Cox SR, Walker JG, Coleman M, et al.: Isolated pulmonary hypertension in scleroderma. Intern Med J 2005, 35:28–33.
Thenappan T, Shah SJ, Rich S, Gomberg-Maitland M: A USA-based registry for pulmonary arterial hypertension: 1982–2006. Eur Respir J 2007, 30:1103–1110.
Humbert M, Sitbon O, Chaouat A, et al.: Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 2006, 173:1023–1030.
Steen V, Medsger TA Jr: Predictors of isolated pulmonary hypertension in patients with systemic sclerosis and limited cutaneous involvement. Arthritis Rheum 2003, 48:516–522.
Catoggio LJ, Bernstein RM, Black CM, et al.: Serological markers in progressive systemic sclerosis: clinical correlations. Ann Rheum Dis 1983, 42:23–27.
Steen VD: The lung in systemic sclerosis. J Clin Rheumatol 2005, 11:40–46.
• Wigley FM: Vascular disease in scleroderma. Clin Rev Allergy Immunol 2009, 36:150–175. This article provides a useful review of the causes and consequences of scleroderma-associated vascular disease by a leader in the field.
Morse J, Barst R, Horn E, et al.: Pulmonary hypertension in scleroderma spectrum of disease: lack of bone morphogenetic protein receptor 2 mutations. J Rheumatol 2002, 29:2379–2381.
Liakouli V, Manetti M, Pacini A, et al.: The -670G>A polymorphism in the FAS gene promoter region influences the susceptibility to systemic sclerosis. Ann Rheum Dis 2009, 68:584–590.
Pignone A, Scaletti C, Matucci-Cerinic M, et al.: Anti-endothelial cell antibodies in systemic sclerosis: significant association with vascular involvement and alveolo-capillary impairment. Clin Exp Rheumatol 1998, 16:527–532.
Chizzolini C, Raschi E, Rezzonico R, et al.: Autoantibodies to fibroblasts induce a proadhesive and proinflammatory fibroblast phenotype in patients with systemic sclerosis. Arthritis Rheum 2002, 46:1602–1613.
Sgonc R, Gruschwitz MS, Boeck G, et al.: Endothelial cell apoptosis in systemic sclerosis is induced by antibody-dependent cell-mediated cytotoxicity via CD95. Arthritis Rheum 2000, 43:2550–2562.
Bonnet S, Rochefort G, Sutendra G, et al.: The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci U S A 2007, 104:11418–11423.
Choi JJ, Min DJ, Cho ML, et al.: Elevated vascular endothelial growth factor in systemic sclerosis. J Rheumatol 2003, 30:1529–1533.
Tamby MC, Chanseaud Y, Humbert M, et al.: Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial hypertension. Thorax 2005, 60:765–772.
Badesch DB, Champion HC, Sanchez MA, et al.: Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2009, 54:S55–S66.
Hachulla E, Gressin V, Guillevin L, et al.: Early detection of pulmonary arterial hypertension in systemic sclerosis: a French nationwide prospective multicenter study. Arthritis Rheum 2005, 52:3792–3800.
Mukerjee D, St George D, Coleiro B, et al.: Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: application of a registry approach. Ann Rheum Dis 2003, 62:1088–1093.
• McLaughlin VV, Archer SL, Badesch DB, et al.: ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation 2009, 119:2250–2294. (Published erratum appears in Circulation 2009, 120:e13.) This American Heart Association/American College of Cardiology consensus document on the diagnosis and management of PAH is a useful reference article.
Meune C, Avouac J, Wahbi K, et al.: Cardiac involvement in systemic sclerosis assessed by tissue-doppler echocardiography during routine care: a controlled study of 100 consecutive patients. Arthritis Rheum 2008, 58:1803–1809.
Denton CP, Cailes JB, Phillips GD, et al.: Comparison of Doppler echocardiography and right heart catheterization to assess pulmonary hypertension in systemic sclerosis. Br J Rheumatol 1997, 36:239–243.
Mukerjee D, St George D, Knight C, et al.: Echocardiography and pulmonary function as screening tests for pulmonary arterial hypertension in systemic sclerosis. Rheumatology (Oxford) 2004, 43:461–466.
Hsu VM, Moreyra AE, Wilson AC, et al.: Assessment of pulmonary arterial hypertension in patients with systemic sclerosis: comparison of noninvasive tests with results of right-heart catheterization. J Rheumatol 2008, 35:458–465.
Tzelepis GE, Kelekis NL, Plastiras SC, et al.: Pattern and distribution of myocardial fibrosis in systemic sclerosis: a delayed enhanced magnetic resonance imaging study. Arthritis Rheum 2007, 56:3827–3836.
Steen VD, Graham G, Conte C, et al.: Isolated diffusing capacity reduction in systemic sclerosis. Arthritis Rheum 1992, 35:765–770.
Chandra S, Shah SJ, Thenappan T, et al.: Carbon monoxide diffusing capacity and mortality in pulmonary arterial hypertension. J Heart Lung Transplant 2009 Sep 25 (Epub ahead of print).
Okano Y, Steen VD, Medsger TA Jr: Autoantibody to U3 nucleolar ribonucleoprotein (fibrillarin) in patients with systemic sclerosis. Arthritis Rheum 1992, 35:95–100.
Allanore Y, Wahbi K, Borderie D, et al.: N-terminal brain natriuretic peptide in systemic sclerosis: a new cornerstone of cardiovascular assessment? Ann Rheum Dis 2009 68:1885–1889.
Ciurzynski M, Bienias P, Lichodziejewska B, et al.: Non-invasive diagnostic and functional evaluation of cardiac involvement in patients with systemic sclerosis. Clin Rheumatol 2008, 27:991–997.
Paciocco G, Martinez FJ, Bossone E, et al.: Oxygen desaturation on the six-minute walk test and mortality in untreated primary pulmonary hypertension. Eur Respir J 2001, 17:647–652.
Shah SJ, Thenappan T, Rich S, et al.: Value of exercise treadmill testing in the risk stratification of patients with pulmonary hypertension. Circ Heart Fail 2009, 2:278–286.
Halpern SD, Taichman DB: Misclassification of pulmonary hypertension due to reliance on pulmonary capillary wedge pressure rather than left ventricular end-diastolic pressure. Chest 2009, 136:37–43.
Fisher MR, Mathai SC, Champion HC, et al.: Clinical differences between idiopathic and scleroderma-related pulmonary hypertension. Arthritis Rheum 2006, 54:3043–3050.
McLaughlin VV, Presberg KW, Doyle RL, et al.: Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 2004, 126:78 S–92 S.
Barst RJ, Rubin LJ, Long WA, et al.: A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study Group. N Engl J Med 1996, 334:296–302.
Badesch DB, Tapson VF, McGoon MD, et al.: Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med 2000, 132:425–434.
•• Badesch DB, McGoon MD, Barst RJ, et al.: Longterm survival among patients with scleroderma-associated pulmonary arterial hypertension treated with intravenous epoprostenol. J Rheumatol 2009, 36:2244–2249.
Oudiz RJ, Schilz RJ, Barst RJ, et al.: Treprostinil, a prostacyclin analogue, in pulmonary arterial hypertension associated with connective tissue disease. Chest 2004, 126:420–427.
Rubin LJ, Badesch DB, Barst RJ, et al.: Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002, 346:896–903. (Published erratum appears in N Engl J Med 2002, 346:1258.)
Denton CP, Pope JE, Peter HH, et al.: Long-term effects of bosentan on quality of life, survival, safety and tolerability in pulmonary arterial hypertension related to connective tissue diseases. Ann Rheum Dis 2008, 67:1222–1228.
Girgis RE, Mathai SC, Krishnan JA, et al.: Long-term outcome of bosentan treatment in idiopathic pulmonary arterial hypertension and pulmonary arterial hypertension associated with the scleroderma spectrum of diseases. J Heart Lung Transplant 2005, 24:1626–1631.
Archer SL, Michelakis ED: Phosphodiesterase type 5 inhibitors for pulmonary arterial hypertension. N Engl J Med 2009, 361:1864–1871.
Badesch DB, Hill NS, Burgess G, et al.: Sildenafil for pulmonary arterial hypertension associated with connective tissue disease. J Rheumatol 2007, 34:2417–2422.
Galie N, Brundage BH, Ghofrani HA, et al.: Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009, 119:2894–2903.
Avouac J, Wipff J, Kahan A, Allanore Y: Effects of oral treatments on exercise capacity in systemic sclerosis related pulmonary arterial hypertension: a meta-analysis of randomised controlled trials. Ann Rheum Dis 2008, 67:808–814.
Schachna L, Medsger TA Jr, Dauber JH, et al.: Lung transplantation in scleroderma compared with idiopathic pulmonary fibrosis and idiopathic pulmonary arterial hypertension. Arthritis Rheum 2006, 54:3954–3961.
Kowal-Bielecka O, Landewe R, Avouac J, et al.: EULAR recommendations for the treatment of systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group (EUSTAR). Ann Rheum Dis 2009, 68:620–628.
Acknowledgement
The authors would like to thank Dr Stuart Rich for his thoughtful review and advice.
Disclosure
Dr. Archer reports no potential conflicts of interest relevant to this article.
[AU: ONLY ARTICLES FROM 2007 TO THE PRESENT CAN BE ANNOTATED -- ANNOTATIONS FOR ARTICLES BEFORE 2007 HAVE THEREFORE BEEN REMOVED]
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sweiss, N.J., Hushaw, L., Thenappan, T. et al. Diagnosis and Management of Pulmonary Hypertension in Systemic Sclerosis. Curr Rheumatol Rep 12, 8–18 (2010). https://doi.org/10.1007/s11926-009-0078-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11926-009-0078-1