
Abstract
Osteoporosis is a leading cause of morbidity in patients with inflammatory bowel disease (IBD). Bone loss is an early systemic process and occurs even before clinical disease manifests. Bone disease is attributed to vitamin D deficiency, steroid use, and/or systemic inflammation. In this review, we discuss the molecular pathways of bone loss mediated by inflammatory cytokines and other mediators. Further research will hopefully clarify the mechanisms of inflammation-induced bone loss in IBD and guide effective treatment modalities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Osteoporosis is the microarchitectural deterioration of bone and loss of bone mineral density (BMD) that increases fracture risk. Osteoporosis is a leading cause of morbidity and affects over 75 million people worldwide [1]. There were over 2 million osteoporosis-associated fractures in 2005 costing the health care system over $17 billion, estimated to increase to $25 billion by 2025 [2]. Glucocorticosteroid (GCS) use is the most common secondary cause of bone loss; 30% to 50% of those on GCS are likely to develop fractures [3]. Several other risk factors have been identified for bone loss, including post menopause, age, family history, sedentary lifestyle, malnutrition, smoking, and gastrointestinal factors (GI; inflammatory bowel disease [IBD], celiac disease, pancreatitis, and bypass surgery) [4]. The purpose of this review is to describe the mechanisms by which IBD affects signaling pathways involved with osteoblast (OB)–osteoclast (OC) crosstalk that leads to decreased BMD and osteoporosis.
Epidemiology of Bone Loss in GI Diseases
The incidence of osteopenia in IBD is 32% to 36% and osteoporosis is 7% to 15% [5]. In patients with ulcerative colitis (UC) following ileal pouch-anal anastomosis, the prevalence of low BMD is 32.1%. The risk of fragility fractures is 10.5% in the low BMD group and 5.9% in the normal BMD group [6]. The prevalence of fragility fractures in patients with chronic pancreatitis is 4.8%, Crohn’s disease (CD) 3.0%, celiac disease 5.0%, post gastrectomy 5.4%, versus controls 1.1%. The odds ratio (OR) of fractures in each group is significantly higher than in the control group (P < 0.0001) [7]
Children with IBD, even at the time of diagnosis, have mild cortical bone loss [8]. Similarly, children with celiac disease have a high prevalence of low bone mass at diagnosis [9]. On histomorphometric analysis, subjects with clinically dormant CD are found to have low BMD, trabecular thinning, and decreased mineral apposition rate, OC number, and surface area [10••]. Undiagnosed celiac disease is also associated with bone loss [11]. Even after being on a long-term gluten-free diet and calcium supplements, osteopenia associated with celiac disease is not completely reversed [12].
Based on our current understanding of osteoimmunology, it is evident that the mechanism of osteoporosis in inflammatory GI diseases is similar with other inflammatory systemic diseases, such as rheumatoid arthritis (RA), ankylosing spondylitis, systemic lupus erythematosus, dermatomyositis, and systemic sclerosis. In a population-based case–control study with a cohort comprising of 53,108 patients and 370,602 age-matched controls, the fracture risk was highest for patients with RA (OR, 3) and IBD (OR, 1.6) [13].
Mechanisms of Bone Loss in GI Diseases
Bone is a dynamic organ in a state of continuous bone formation and resorption responding to mechanical stress, microfractures, and various circulating factors. The main bone cell types are the OB, the bone-forming cell, and the OC, the bone-resorbing cell. OBs are derived from mesenchymal stem cells and OCs from hematopoietic stem cells. The bone loss in GI disease is multifactorial and complex, requiring an integrated management approach that targets the primary disease and mechanisms of bone loss. With respect to IBD, this strategy addresses systemic inflammation, malnutrition, malabsorption, and steroid therapy. This review focuses on the inflammatory mechanisms of bone loss in IBD.
Interleukin/RANK-RANKL-OPG Axis
Receptor activator of nuclear factor-κB ligand (RANKL) is a circulating cytokine secreted by mature OB and T cells, interacts on the receptor activator of nuclear factor-κB (RANK) receptor located on the mature OC cell membrane, and activates bone resorption. RANKL is an absolute requirement of the OC to initiate bone resorption. The OB also secretes osteoprotegerin (OPG), a decoy receptor of the RANKL. Thus, the balance between RANKL and OPG determines if there will be net bone formation or resorption. Ashcroft et al. [14] demonstrated that in the interleukin (IL)-2–deficient mouse model of autoimmunity, bone loss was mediated via activated T cells secreting RANKL. Interestingly, they found that both the transcription and plasma levels of the decoy OPG were also increased but were not sufficient to reverse the increased RANKL-mediated bone loss [14]. This effect was supported by the increase in BMD seen with exogenous OPG administration, which also reversed colitis [14]. Moschen et al. [15] studied 180 patients with IBD and found that OPG plasma levels were elevated 2.4-fold in CD and 1.9-fold in UC, whereas soluble RANKL (sRANKL) levels were not significantly different in IBD patients compared with healthy controls. These findings corroborated the results from the IL-2–deficient murine models of colitis and suggested the role of OPG-RANK-RANKL axis in the decreased BMD associated with IBD [15]. Miheller et al. [16] showed that in patients treated with infliximab (a tumor necrosis factor [TNF]-α inhibitor) there was a statistically significant decrease in OPG levels and increase in osteocalcin and sRANKL, thus supporting the hypothesis that there is a counter-regulatory increase in serum OPG levels in patients with IBD [16••].
Several studies have shown elevated levels of circulating proinflammatory cytokines in IBD including TNF-α, IL-1β, IL-6, and IL-17. These cytokines have been implicated in OC-mediated bone resorption in experimental models through p38 mitogen-activated protein kinase (MAPK) pathways [17–20]. IL-1β is a key mediator of bone resorption in inflammatory conditions, such as RA and IBD. IL-1β promotes osteoclastogenesis by inducing RANKL expression on stromal cells and synergizing with RANKL to promote later stages of OC differentiation. IL-1β is also a potent inducer of nuclear factor (NF)-κB. High concentrations of IL-1β are found in both CD and UC [21]. Nemetz et al. [22] showed that in IBD patients, carriers of allele 2 of the AvaI gene (IL-1β-511*2 polymorphism), characterized by IL-1β hypersecretion, have a higher risk of diminished BMD (OR of 3.63 at the femoral neck) than healthy controls [22]. Apart from IL-1β, studies have shown that IBD patients exhibit significantly elevated IL-6 activity both locally in the lamina propria mononuclear cells and colonic epithelial cells [23] as well as in circulating monocytes [24].
CD patients with antibodies to Cbir1 (a flagellin that mediates mucosal inflammation) have a more complicated course [25]. Although IL-6 is significantly increased in monocytes from IBD patients, anti-CBir1(+) and IL-6 are inversely correlated. Anti-CBir+ antibodies are associated with decreased activation of NF-κB, which inhibits OC activation. [24]. IL-6 receptor blockade strongly reduces OC formation and bone erosion [26]. Humanized antibody against the IL-6 receptor (tocilizumab) has shown benefit in inflammatory arthritis seen in RA [27].
TNF-α appears to be the master regulator of bone loss in patients with IBD. It promotes osteoclastogenesis in conditions such as inflammatory osteolysis, by inducing OC differentiation, a function that requires the presence of RANKL, as evident from studies on RANK-deficient mice [28, 29]. Once differentiated, TNF-α can activate the OCs and this action is independent of RANK signaling [29]. TNF-α and RANKL markedly potentiate NF-κB and stress-activated protein kinase/c-Jun NH2-terminal kinase activity, two signaling pathways essential for osteoclastogenesis [30].
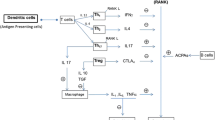
In addition to osteoclastogenesis, the effect of TNF-α on OBs is critical in the pathogenesis of reduced BMD seen with IBD. In the noninflammatory state, OB maturation is dependent on Wingless-Int signaling pathway (Wnt) and the genes regulated through the pathway such as Dmp1 (dentin matrix protein 1), Phex (phosphate-regulating gene with homologies to endopeptidases in the X chromosome), and Bsp (bone sialoprotein). The key regulator of this pathway appears to be a secreted factor termed Rspo2 (R-spondin 2) (Fig. 1). Knockout of Rspo2 abrogates Wnt-mediated OB maturation [31].

Mechanism of bone loss in IBD. Under normal conditions, osteoblasts secrete RANKL, which binds to RANK receptor and activates NF-κB pathway, and in turn it controls DNA transcription necessary for osteoclastogenesis. OPG, a decoy receptor, blocks the interaction between RANKL and RANK causing decreased osteoclast activation. During inflammatory conditions, release of various cytokines such as IL-1, IL-6, and TNF-α and binding to its respective receptors cause increased activation of p38 MAPK, NF-κB, and JNK pathways causing excessive bone loss reducing BMD, seen in IBD. Osteoblast maturation is dependent on RSPO2, which controls several pathways responsible for osteoblast differentiation such as Wnt, Phex, Bsp, and Dmp1. TNF-α inhibits the activation of RSPO2 causing decreased bone mineralization. BMD—bone mineral density; Bsp—bone sialoprotein; Dmp1—dentin matrix acidic phosphoprotein 1; ECM—extracellular matrix; IBD—inflammatory bowel disease; IL—interleukin; IL-1R—interleukin-1 receptor; IL-6R—interleukin-6 receptor; JNK—c-Jun N-terminal kinases; NF-κB—nuclear factor-κB; OGP—osteoprotegerin; p38 MAPK—p38 mitogen-activated protein kinase; Phex—phosphate-regulating neutral endopeptidase; RANK—receptor activator of nuclear factor-κB; RANKL—receptor activator of nuclear factor-κB ligand; RSPO2—R-spondin-2; TNF-α—tumor necrosis factor-α; TNFR—tumor necrosis factor receptor; Wnt—Wingless-Int signaling pathway
The cause and effect relationship with TNF-α is established by several studies, which demonstrated the increase in BMD seen with infliximab, the monoclonal antibody against TNF-α. The REACH (A Pediatric Trial in Moderate to Severe Crohn’s Disease) study group showed that in 112 pediatric patients with CD (ages 6–17 years), infliximab induction at 5 mg/kg/dose and maintenance every 8 to 12 weeks showed statistically significant improvement in serum bone-specific alkaline phosphatase, N-terminal propeptide of type 1 collagen (P1NP), urine C-telopeptide of collagen cross-links (CTX-1), and deoxypyridinoline. The increases in CTX-1 and deoxypyridinoline likely reflect coupling of bone formation and resorption as increases in linear growth were seen (54-week increases in height z-score; P < 0.001) [32]. Results from prospective studies in adults have also shown the benefits of infliximab in improving BMD in patients with CD. Bernstein et al. [33•] showed improvement in BMD of the lumbar spine (L2–L4) and proximal left femur (neck and trochanter) in 46 CD patients treated with infliximab (5 mg/kg) at 6- to 8-week intervals for 1 year. Thirteen patients received concurrent prednisone at a mean dose of 10 mg/day (range: 5–15) [33•]. BMD increased at the lumbar spine by 2.4% ± 0.7% (P = 0.002), at the femoral trochanter by 2.8% ± 1.2% (P = 0.03), and at the femoral neck by 2.6% ± 0.7% (P = 0.001) [33•]. Interestingly, in a retrospective analysis on 61 patients with CD [34], patients with concurrent infliximab and bisphosphonate treatment exhibited a greater increase in BMD compared with those on bisphosphonates alone (+6.7%/year vs +4.46%/year; P = 0.045).
Because GCS remain the mainstay therapy for patients with IBD, does infliximab increase BMD through neutralization of steroid-mediated bone loss? This question was addressed in a study on 56 pediatric patients with IBD (35 CD, 21 UC) and an inverse correlation was found between bone marrow apparent density (BMAD) and IL-6 in patients with UC (r = −0.65); no correlation was found between BMAD and serum levels of TNF-α, IL-10, and IL-12 [17]. Disease activity indices inversely correlated with BMAD (r = −0.62 in patients with CD; r = −0.64 in patients with UC). Cumulative dose of GCS and duration of therapy did not correlate with BMAD. Patients treated with infliximab had a higher BMAD. It appears that at least in pediatric IBD, GCS appear to play a lesser role in the pathogenesis of decreased BMD [17].
Insulin-Like Growth Factor Axis
The catabolic state and growth retardation seen in patients with IBD led to investigations exploring the growth hormone (GH) axis. In a retrospective analysis in 28 pediatric patients with IBD (25 CD, 3 UC), four children showed functional GH deficiency (decreased GH and decreased insulin-like growth factor-1 [IGF-1]) and 11 children showed GH resistance (increased GH and increased IGF-1). One child showed an impaired hepatic response to GH (increased GH and decreased IGF-1) [35]. Results were slightly more skewed in an analysis from 37 patients with IBD (N = 17 with CD and 20 with UC) in which total IGF-1 was reduced in 36% of CD patients and 41% of UC patients. Moreover, free IGF-1 increased significantly in patients with UC when treated with GCS; no change was seen in patients with CD [36].
Alteration of the IGF axis by inflammation was suggested by studies showing IGF-1 levels are inversely proportional to systemic inflammatory markers such as erythrocyte sedimentation rate, and C-reactive protein and that the alteration of IGF-binding protein-3/IGF-binding protein-2 ratio is altered in a manner that would reduce IGF-1 action [37, 38]. In vivo data from experimental colitis murine models (trinitrobenzenesulfonic acid [TNBS] colitis) showed that with normal stimulated GH secretion, the IGF-1 levels were reduced with reduced IGF mRNA expression, and this effect was reversed by neutralizing IL-6 [39]. TNF-α has been shown to inhibit GH-stimulated IGF-1 secretion in cultured rat liver cells [40]. There is evidence to suggest that anti TNF-α therapy (adalimumab) improves IGF-1 levels at least in RA with an improvement in the muscle wasting component of IBD [41].
The Role of Phex
Phex encodes a neutral (zinc) endopeptidase that regulates phosphate metabolism. Inactivating mutations of Phex leads to X-linked hypophosphatemic rickets with mineralization defects and renal phosphate wasting independent of vitamin D and phosphate levels [42]. Phex is expressed in the OB (and odontoblast) and is downregulated by TNF-α in IBD [43••, 44]. Uno et al. [45] demonstrated that animal models of chemically induced colitis demonstrated a 40% to 50% decrease in the Phex mRNA expression that was reversed with dietary curcumin, which has a potent anti-inflammatory effect in animal models of colitis [46•, 47–49] and human studies [44]. Anti-TNF-α antibody also restored Phex expression, further substantiating the role of TNF-α in its modulation. In vitro, OB-like osteosarcoma cells demonstrated a decrease in Phex mRNA on exogenous TNF-α administration in a concentration-dependent manner, associated with significantly decreased mineralization. OBs, derived from Hyp (Phex knockout) mice cultured in vitro, failed to mineralize [50]. Children with CD were found to have deranged OB function [51]; in vitro OBs also showed abnormal function when cultured in sera from patients with CD [52]. Thus, bone loss due to inflammation in IBD is mediated in part by the suppression of Phex, giving us a clearer insight into inflammatory bone disease.
TNF-α downregulates Phex transcriptionally with no effect on mRNA stability [45]. Majewski et al. [43••] identified that TNF-α induces poly (ADP-ribose) polymerase 1 (PARP-1), for enzymatic PARylation of the p65 (Rel A) subunit, increasing the affinity of NF-κB for the Phex promoter region and leading to decreased expression of Phex. Furthermore, in PARP-1–deficient mice and when PARP-1 is inhibited, downregulation of Phex is not seen.
In animal studies, inhibition of inhibitor of κB kinase (IκK) and NF-κB results in increased bone mass and density with no change in bone resorption [53••]. The increase in bone formation is mediated by an increase in the expression of Fos-related antigen-1 (Fra-1) [51], an indispensible transcription factor in bone formation [54, 55]. NF-κB is upregulated by inflammatory cytokines including TNF-α, IL-1, IL-6, and IL-7 [51], which are chronically elevated in systemic inflammatory conditions, including IBD.
Mapping of the IκK–NF-κB and PARP-1 pathways in bone loss characterizes them as potential targets for therapeutic intervention in bone loss of inflammatory diseases.
The Role of Klotho
Klotho encodes an anti-inflammatory protein with widespread actions, including regulation of calcium and phosphate metabolism. It has been demonstrated that Klotho expression is downregulated in animal models of colitis, mediated by TNF-α and interferon (IFN)-γ [56••]. The extent of Klotho suppression was directly related to the severity of colitis and was reversed by anti-TNF-α antibodies. IFN-γ amplifies the effect of TNF-α and mediates the downregulation of Klotho by transcriptional repression of its promoter [57]. As Klotho maintains calcium and phosphate homeostasis, loss of Klotho mediated by TNF-α and other modulators of inflammation aggravates bone disease. An in vitro study on human umbilical vein endothelial cells showed that Klotho protein attenuated TNF-α–induced expression of adhesion molecules and activation of NF-κB [57], substantiating the involvement of Klotho in bone loss due to systemic inflammation.
Conclusions
Bone loss remains a major extraintestinal cause of morbidity in IBD, leading to impaired quality of life and productivity. It is clear that systemic inflammation and chronically high levels of circulating cytokines interact with bone through multiple pathways, synergizing to produce profound net bone loss, independent of other theoretically modifiable causes of bone loss. Even in pediatric IBD and clinically dormant IBD, osteopenia is manifest. The pathways leading to inflammatory bone disease are still inadequately understood and we need more studies to fill in the gaps. However, many factors have been identified as possible points of therapeutic manipulation, including TNF-α, RANKL, Phex, NF-κB, and PARP-1, among others. Using a multipronged approach to prevent and treat bone disease will likely be more successful because it is evident that no one factor in isolation can be implicated.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
EFFO and NOF. Who are candidates for prevention and treatment for osteoporosis? Osteoporos Int. 1997;7(1):1–6.
Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res. 2007;22:465.
Reid IR. Glucocorticoid osteoporosis - mechanisms and management. Eur J Endocrinol. 1997;137:209.
Pazianas M, Zaidi M, Subhani JM, Finch PJ, Ang L, Maxwell JD. Efferent loop small intestinal vitamin D receptor concentration and bone mineral density after Billroth II (Polya) gastrectomy in humans. Calcif Tissue Int. 2003;72(4):485–90.
Schulte C, Dignass AU, Mann K, Goebell H. Reduced bone mineral density and unbalanced bone metabolism in patients with inflammatory bowel disease. Inflamm Bowel Dis. 1998;4(4):268–75.
Shen B, Remzi FH, Oikonomou IK, Lu H, Lashner BA, Hammel JP, et al. Risk factors for low bone mass in patients with ulcerative colitis following ileal pouch-anal anastomosis. Am J Gastroenterol. 2009;104(3):639–46.
Tignor AS, Wu BU, Whitlock TL, Lopez R, Repas K, Banks PA, et al. High prevalence of low-trauma fracture in chronic pancreatitis. Am J Gastroenterol. 2010;105(12):2680–6.
Ward LM, Rauch F, Matzinger MA, Benchimol EI, Boland M, Mack DR. Iliac bone histomorphometry in children with newly diagnosed inflammatory bowel disease. Osteoporos Int. 2010;21:331–7.
Turner J, Pellerin G, Mager D. Prevalence of metabolic bone disease in children with celiac disease is independent of symptoms at diagnosis. J Pediatr Gastroenterol Nutr. 2009;49(5):589–93.
•• Oostlander AE et al. Dutch Initiative on Crohn and Colitis (ICC). Histomorphometric analysis reveals reduced bone mass and bone formation in patients with quiescent Crohn’s disease. Gastroenterology. 2011;140(1): 116–23. This study identifies that bone loss occurs even before clinically significant bowel disease has occurred, implicating chronic inflammation as the earliest cause for metabolic bone disease in IBD.
Godfrey JD et al. Morbidity and mortality among older individuals with undiagnosed celiac disease. Gastroenterology. 2010;139(3):763–9.
Pazianas M, Butcher GP, Subhani JM, Finch PJ, Ang L, Collins C, et al. Calcium absorption and bone mineral density in celiacs after long term treatment with gluten-free diet and adequate calcium intake. Osteoporos Int. 2005;16(1):56–63.
Weiss RJ, Wick MC, Ackermann PW, Montgomery SM. Increased fracture risk in patients with rheumatic disorders and other inflammatory diseases—a case-control study with 53,108 patients with fracture. J Rheumatol. 2010;37(11):2247–50.
Ashcroft AJ et al. Colonic dendritic cells, intestinal inflammation, and T cell-mediated bone destruction are modulated by recombinant osteoprotegerin. Immunity. 2003;19(6):849–61.
Moschen AR et al. The RANKL/OPG system is activated in inflammatory bowel disease and relates to the state of bone loss. Gut. 2005;54(4):479–87.
•• Miheller P et al. Changes of OPG and RANKL concentrations in Crohn’s disease after infliximab therapy. Inflamm Bowel Dis. 2007;13(11):1379–84. This study demonstrates that IBD treatment targeted at inflammatory cytokines can alter bone metabolism favorably, thereby establishing inflammation as a cause for bone loss.
Paganelli M et al. Inflammation is the main determinant of low bone mineral density in pediatric inflammatory bowel disease. Inflamm Bowel Dis. 2007;13(4):416–23.
Kumar S, Votta BJ, Rieman DJ, Badger AM, Gowen M, Lee JC. IL-1- and TNF-induced bone resorption is mediated by p38 mitogen activated protein kinase. J Cell Physiol. 2001;187(3):294–303.
Rovedatti L et al. Differential regulation of interleukin 17 and interferon gamma production in inflammatory bowel disease. Gut. 2009;58(12):1629–36.
Zhang YH, Heulsmann A, Tondravi MM, Mukherjee A, Abu-Amer Y. Tumor necrosis factor-alpha (TNF) stimulates RANKL-induced osteoclastogenesis via coupling of TNF type 1 receptor and RANK signaling pathways. J Biol Chem. 2001;276(1):563–8.
Mahida YR, Wu K, Jewell DP. Enhanced production of interleukin 1-beta by mononuclear cells isolated from mucosa with active ulcerative colitis of Crohn’s disease. Gut. 1989;30:835–8.
Nemetz A, Tóth M, García-González MA, Zágoni T, Fehér J, Peña AS, et al. Allelic variation at the interleukin 1beta gene is associated with decreased bone mass in patients with inflammatory bowel diseases. Gut. 2001;49(5):644–9.
Kusugami K et al. Elevation of interleukin-6 in inflammatory bowel disease is macrophage- and epithelial cell-dependent. Dig Dis Sci. 1995;40(5):949–59.
Shen C, Landers CJ, Derkowski C, Elson CO, Targan SR. Enhanced CBir1- specific innate and adaptive immune responses in Crohn’s disease. Inflamm Bowel Dis. 2008;14(12):1641–51.
Targan SR, Landers CJ, Yang H, et al. Antibodies to CBir1 flagellin define a unique response that is associated independently with complicated Crohn’s disease. Gastroenterology. 2005;128:2020–8.
Axmann R, Böhm C, Krönke G, Zwerina J, Smolen J, Schett G. Inhibition of interleukin-6 receptor directly blocks OC formation in vitro and in vivo. Arthritis Rheum. 2009;60(9):2747–56.
Garnero P, Thompson E, Woodworth T, Smolen JS. Rapid and sustained improvement in bone and cartilage turnover markers with the anti-interleukin-6 receptor inhibitor tocilizumab plus methotrexate in rheumatoid arthritis patients with an inadequate response to methotrexate: results from a substudy of the multicenter double-blind, placebo-controlled trial of tocilizumab in inadequate responders to methotrexate alone. Arthritis Rheum. 2010;62(1):33–43.
Li J et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci USA. 2000;97:1566–71.
Fuller K, Murphy C, Kirstein B, Fox SW, Chambers TJ. TNFα potently activates osteoclasts, through a direct action independent of and strongly synergistic with RANKL. Endocrinology. 2002;143:1108–18.
Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106:1481–8.
Friedman MS, Oyserman SM, Hankenson KD. Wnt11 promotes osteoblast maturation and mineralization through R-spondin 2. J Biol Chem. 2009;284(21):14117–25.
Thayu M, Leonard MB, Hyams JS, Crandall WV, Kugathasan S, Otley AR, et al. Improvement in biomarkers of bone formation during infliximab therapy in pediatric Crohn’s disease: results of the REACH study. Clin Gastroenterol Hepatol. 2008;6(12):1378–84.
• Bernstein M, Irwin S, Greenberg GR. Maintenance infliximab treatment is associated with improved bone mineral density in Crohn's disease. Am J Gastroenterol. 2005;100(9):2031–5. This study corroborates inflammation as a major cause of bone disease in IBD, and its reversal with blocking TNF-α with infliximab.
Pazianas M, Rhim AD, Weinberg AM, Su C, Lichtenstein GR. The effect of anti-TNF-alpha therapy on spinal bone mineral density in patients with Crohn’s disease. Ann N Y Acad Sci. 2006;1068:543–56.
Wong SC, Smyth A, McNeill E, Galloway PJ, Hassan K, McGrogan P, et al. The growth hormone insulin-like growth factor 1 axis in children and adolescents with inflammatory bowel disease and growth retardation. Clin Endocrinol (Oxf). 2010;73(2):220–8.
Eivindson M, Grønbaek H, Flyvbjerg A, Frystyk J, Zimmermann-Nielsen E, Dahlerup JF. The insulin-like growth factor (IGF)-system in active ulcerative colitis and Crohn’s disease: relations to disease activity and corticosteroid treatment. Growth Horm IGF Res. 2007;17(1):33–40.
Street ME et al. Relationships between serum IGF-1, IGFBP-2, interleukin-1beta and interleukin-6 in inflammatory bowel disease. Horm Res. 2004;61(4):159–64.
Corkins MR, Gohil AD, Fitzgerald JF. The insulin-like growth factor axis in children with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2003;36(2):228–34.
Ballinger A. Fundamental mechanisms of growth failure in inflammatory bowel disease. Horm Res. 2002;58 Suppl 1:7–10.
Wolf M, Bohm S, Brand M, Kreymann G. Proinflammatory cytokines interleukin 1 beta and tumor necrosis factor alpha inhibit growth hormone stimulation of Insulin-like growth factor I synthesis and growth hormone receptor mRNA levels in cultured rat liver cells. Eur J Endocrinol. 1996;135729–737.
Sarzi-Puttini P, Atzeni F, Schölmerich J, Cutolo M, Straub RH. Anti-TNF antibody treatment improves glucocorticoid induced insulin-like growth factor 1 (IGF1) resistance without influencing myoglobin and IGF1 binding proteins 1 and 3. Ann Rheum Dis. 2006;65(3):301–5.
A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet. 1995;11(2):130–6.
•• Majewski PM, Thurston RD, Ramalingam R, Kiela PR, Ghishan FK. Cooperative role of NF-κB and poly(ADP-ribose) polymerase 1 (PARP-1) in the TNF-induced inhibition of PHEX expression in osteoblasts. J Biol Chem. 2010;285(45):34828–38. This study identifies another mechanism by which TNF mediates bone loss in IBD, and implicates NF-κB and PARP-1 in the same, raising the possibility of therapeutic intervention.
Hanai H, Iida T, Takeuchi K, Watanabe F, Maruyama Y, Andoh A, et al. Curcumin maintenance therapy for ulcerative colitis: randomized, multicenter, double-blind, placebo-controlled trial. Clin Gastroenterol Hepatol. 2006;4(12):1502–6.
Uno JK, Kolek OI, Hines ER, Xu H, Timmermann BN, Kiela PR, et al. The role of tumor necrosis factor alpha in down-regulation of osteoblast Phex gene expression in experimental murine colitis. Gastroenterology. 2006;131(2):497–509.
• Ung VY, Foshaug RR, MacFarlane SM, Churchill TA, Doyle JS, Sydora BC, Fedorak RN. Oral administration of curcumin emulsified in carboxymethyl cellulose has a potent anti-inflammatory effect in the IL-10 gene-deficient mouse model of IBD. Dig Dis Sci. 2010;55(5):1272–7. This study identifies curcumin as anti-inflammatory in colitis, and further elucidates pathways of inflammation-mediated bone loss.
Lubbad A, Oriowo MA, Khan I. Curcumin attenuates inflammation through inhibition of TLR-4 receptor in experimental colitis. Mol Cell Biochem. 2009;322(1–2):127–35.
Venkataranganna MV, Rafiq M, Gopumadhavan S, Peer G, Babu UV, Mitra SK. NCB-02 (standardized Curcumin preparation) protects dinitrochlorobenzene- induced colitis through down-regulation of NFkappa-B and iNOS. World J Gastroenterol. 2007;13(7):1103–7.
Camacho-Barquero L, Villegas I, Sánchez-Calvo JM, Talero E, Sánchez-Fidalgo S, Motilva V, et al. Curcumin, a Curcuma longa constituent, acts on MAPK p38 pathway modulating COX-2 and iNOS expression in chronic experimental colitis. Int Immunopharmacol. 2007;7(3):333–42.
Xiao ZS, Crenshaw M, Guo R, Nesbitt T, Drezner MK, Quarles LD. Intrinsic mineralization defect in Hyp mouse osteoblasts. Am J Physiol. 1998;275(4 Pt 1):E700–8.
Hyams JS, Wyzga N, Kreutzer DL, Justinich CJ, Gronowicz GA. Alterations in bone metabolism in children with inflammatory bowel disease: an in vitro study. J Pediatr Gastroenterol Nutr. 1997;24(3):289–95.
Varghese S, Wyzga N, Griffiths AM, Sylvester FA. Effects of serum from children with newly diagnosed Crohn disease on primary cultures of rat osteoblasts. J Pediatr Gastroenterol Nutr. 2002;35(5):641–8.
•• Chang J, Wang Z, Tang E, Fan Z, McCauley L, Franceschi R, Guan K, Krebsbach PH, Wang CY. Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat Med. 2009;15(6):682–9. This study implicates NF-κB in bone loss and identifies it as a point of therapeutic intervention.
Jochum W, David JP, Elliott C, Wutz A, Plenk Jr H, Matsuo K, et al. Increased bone formation and osteosclerosis in mice overexpressing the transcription factor Fra-1. Nat Med. 2000;6(9):980–4.
Eferl R, Hoebertz A, Schilling AF, Rath M, Karreth F, Kenner L, et al. The Fos-related antigen Fra-1 is an activator of bone matrix formation. EMBO J. 2004;23(14):2789–99.
•• Thurston RD, Larmonier CB, Majewski PM, Ramalingam R, Midura-Kiela M, Laubitz D, Vandewalle A, Besselsen DG, Mühlbauer M, Jobin C, Kiela PR, Ghishan FK. Tumor necrosis factor and interferon-gamma down-regulate Klotho in mice with colitis. Gastroenterology. 2010;138(4):1384–94, 1394. This study identifies the role of Klotho, a novel pathway, in inflammation-mediated bone loss in IBD.
Maekawa Y, Ishikawa K, Yasuda O, Oguro R, Hanasaki H, Kida I, et al. Klotho suppresses TNF-alpha-induced expression of adhesion molecules in the endothelium and attenuates NF-kappaB activation. Endocrine. 2009;35(3):341–6.
Acknowledgments
MA and SA would like to thank Ankit Amin (MS) and Anastasia Floros (MS) for their inputs in the preparation of figure.
Disclosure
Conflicts of interest: M. Agrawal: none; S. Arora: none; J. Li: none; R. Rahmani: none; L. Sun: none; A.F. Steinlauf: none; J.I. Mechanick: has been a consultant for Abbott Nutrition and Nestle Nutrition; has received grant support from Select Medical Corp.; has received honoraria from Abbott Nutrtion and Nestle Nutrition; has received payment for development of educational presentations including service on speakers’ bureaus from Abbott Nutrition; and has received travel/accommodations expenses covered or reimbursed by Abbott Nutrition and Nestle Nutrition; M. Zaidi: has been a consultant for Amgen, Procter & Gamble, Roche and Genentech, GlaxoSmithKline, Warner Chilcott, and Novartis; has given expert testimony for Bowman and Brooke, Simes, and VEnables; has two patents on FSH and TSH, only if they mature into a drug, from Mount Sinai School of Medicine; has received payment for development of educational presentations including service on speakers’ bureaus from CME Education LLC and also various CME prams at academic institutions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Agrawal, M., Arora, S., Li, J. et al. Bone, Inflammation, and Inflammatory Bowel Disease. Curr Osteoporos Rep 9, 251–257 (2011). https://doi.org/10.1007/s11914-011-0077-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-011-0077-9