
Abstract
Systemic osteoporosis and increased fracture rates have been described in chronic inflammatory diseases such as rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, inflammatory bowel diseases, and chronic obstructive pulmonary disease. Most of these patients receive glucocorticoids, which have their own deleterious effects on bone. However, the other main determinant of bone fragility is the inflammation itself, as shown by the interactions between the inflammatory mediators, the actors of the immune system, and the bone remodelling. The inflammatory disease activity is thus on top of the other well-known osteoporotic risk factors in these patients. Optimal control of inflammation is part of the prevention of osteoporosis, and potent anti-inflammatory drugs have positive effects on surrogate markers of bone fragility. More data are needed to assess the anti-fracture efficacy of a tight control of inflammation in patients with a chronic inflammatory disorder. This review aimed at presenting different clinical aspects of inflammatory diseases which illustrate the relationships between inflammation and bone fragility.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inflammation is the body’s response to different pathogens and tissue damages; it involves the activation of the cells of both the innate and adaptive immune response and the production of cytokines (TNFα, interleukins, chemokines, interferons, etc.). The inflammatory process can affect several organs, including bone, where it is linked to an imbalance in bone remodelling. Systemic osteoporosis and increased fracture rates have been described in chronic inflammatory diseases involving joints (rheumatoid arthritis, spondyloarthropathies, psoriatic arthritis), gut (inflammatory bowel diseases), lung (chronic obstructive pulmonary disease), and several organs (systemic lupus erythematosus, systemic vasculitis, etc.). The role of inflammation in bone fragility is insufficiently recognized as most of the patients receive glucocorticoids, which have their own deleterious effects on bone. Inflammatory bone loss is a model of the interactions between the immune system and the bone remodelling [1]. In this review, after a brief summary of mechanisms mediating inflammatory bone loss, we present clinical data in a selection of five inflammatory diseases, which illustrate different aspects of the relationships between inflammation and bone fragility and allows the discussion of the role of anti-inflammatory drugs on bone protection.
Relationships between systemic inflammation and bone fragility
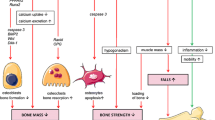
The inflammatory process is associated with an altered systemic bone remodelling, with increased bone resorption, and with impaired bone formation with the effect of inflammatory mediators on the differentiation and activity of osteoclasts and osteoblasts. Pro-inflammatory cytokines can influence osteoclastogenesis and osteoclasts’ activity (Fig. 1). Macrophage–colony-stimulating factor 1 (M-CSF) and receptor activators of NF-KB ligand (RANKL) are necessary and sufficient for osteoclastogenesis, which is enhanced by cytokines such as TNF, IL1, and IL6. RANK L, a member of the tumour necrosis factor (TNF)-α family, is one of the osteoclastogenic factors that directly act on osteoclast formation and activity like other cytokines and pro-inflammatory mediators: interleukins (IL)1, IL6, IL17, oncostatin M, leukaemia inhibitory factor, etc. [2, 3].

Schematic view of bone resorption regulation and immune cells
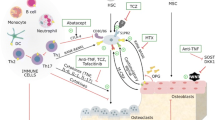
The evidence that B and T cells have a role in bone remodelling comes from a large amount of studies, including the observation of osteoporotic phenotype in B or T cell-deficient animals [4, 5]. Osteoclasts derive from the monocyte-macrophage lineage and thus share common progenitors and receptors with cells of the immune system [2, 3]. T cells play a role in the regulation of bone resorption and bone formation through the involvement of specialized cell lineages such as Th17 cells and Tregs. T cells (Th1, Th2, Th17 cells) are the main secretors of regulatory cytokines. A key cytokine during inflammatory process is TNFα, produced by activated T cells and macrophages. TNF alone cannot induce osteoclastogenesis, but does so under permissive concentrations of RANKL, targeting monocytes and macrophages to pre osteoclasts [6]. It has a role in recruiting osteoclast precursors and is a regulator of the pro inflammatory cascade. IL1 can enhance osteoclast formation and lifespan and mediate TNF-induced structural damages. IL6 is produced by T cells and macrophages, but also by osteoblasts in response to TNFα and IL1 [7]; it is a target of parathormone to recruit osteoclasts [8]. Th17 cells are the most osteoclastogenic subsets of T cells and play a pivotal role in the bone loss of inflammatory conditions (including psoriasis). Th17 cells potently induce osteoclastogenesis by secreting IL17, RANKL, TNF, IL1, and IL6, along with low levels of IFNγ. IL17 stimulates the release of RANKL by osteoblasts and osteocytes and potentiates the osteoclastogenic activity of RANKL by upregulating RANK [9]. Recent data suggest that PTH expands Th17 cells and increases IL17 levels in mice and humans, as IL17 acts as an “upstream cytokine” that increases the sensitivity of osteoblasts and osteocytes to parathormone [10]. In turn, regulatory T cells (Treg) protect against TNFα induced bone loss [11]. Tregs may act through different ways, including the direct cell-cell signalling via CTLA4 [12]. The balance between Th17 and Treg cells could be important for bone remodelling during the inflammatory process.
Attention has been paid recently on the role of B cells. There was no evidence so far of the presence of auto antibodies to mediate bone loss, till the observation that the presence of antibodies against citrullinated proteins (ACPAs) in rheumatoid arthritis (RA) is associated with a poor bone outcome, with more periarticular bone erosions and more systemic bone loss. This was explained by the presence of enzymes for citrullination of proteins, in osteoclasts’ precursors, and the expression of citrullinated vimentin on osteoclasts’ surface that ACPAs can target [13]. The binding of ACPA induces local production of TNFα which enhances osteoclasts’ formation and activity. This explains the intriguing observation that healthy patients with ACPAs, without any joint disease, have structural bone damage, with bone loss and reduced cortical thickness [14]. Immune complexes containing citrullinated components can increase local TNF production via the binding of antigen to Toll-like receptors [15]. In patients with RA being in remission, bone loss is arrested, although the level of circulating ACPAs does not change, suggesting that the local loop of autocrine effects of TNF or IL8 [16] is altered. Finally, activated B cells can express osteoclastogenic factors as RANKL, in models of periodontal infection, while CD8+ T cells inhibit this pathway [17].
Dendritic cells (DCs) are antigen-presenting cells that were not known to have a role in bone homeostasis. Indeed, DC-deficient animals have no altered bone phenotype. However, immature DCs can transdifferentiate into osteoclasts [18], leading to the observation that osteoclasts can be also antigen-presenting cells and activate in turn CD4+ and CD8+ T cells [19]. Whether the role of osteoclasts may differ according to the precursors they come from (either monocytes or DCs) is a hypothesis. In rheumatoid arthritis, the osteoclast-associated receptor (OSCAR) is induced in monocytes, predisposing these cells to commit to the osteoclast lineage [20].
The role of inflammatory cytokines in physiological bone remodelling is not fully understood, but there is evidence that they play a role in the pathogenesis of hormonal deficiency-induced bone loss. Nude mice deficient in T cells are protected against bone loss induced by ovariectomy; this bone loss occurs if these mice are restored with T cells from mild-type mice. Ovariectomy is also able to increase production of IL7 by T cells which in turn enhance the production of TNF by these cells. IL6-deficient mice are protected from oestrogen deficiency-induced bone loss. IL6 and associated IL17 production play a major role in osteoclast generation [21]. Deficiency of CCR2, a molecule involved in inflammation, protects animals from oestrogen-dependent resorption [22]. In postmenopausal healthy women, anti-IL1 or anti-TNF therapy can prevent part of the increase in bone resorption marker which occurs after discontinuation of oestrogen replacement therapy [23].
A decrease in bone-forming cells (osteoblasts and osteocytes) activity is also observed during the inflammatory process. Blocking PTH signalling in T cells blunts the capacity of intermittent PTH to increase bone formation through a decrease in Wnt10b production by T cells [24]. This means that T cells are one of the targets of PTH for its bone anabolic effect. TNF can inhibit osteoblasts via inhibition of RUNX 2 [25]; IL1, IL6, and other cytokines can also inhibit osteoblastogenesis. Finally, TNF could control bone formation indirectly, as suggested by studies in TNF-transgenic mice in which osteocytes have an enhanced expression of sclerostin. TNF can induce DKK1, another inhibitor of the Wnt signalling pathway [26]. Sclerostin-inhibition effect has been studied on bone loss in a model of TNF transgenic mice with established inflammatory-induced osteoporosis [27].
All these data suggest that inflammation players can act on bone remodelling, with an unfavourable effect, and are the basis of the use of potent anti-inflammatory drugs to protect bone. In prospective studies, complete control of inflammation allows clinical improvement and thus increased mobility, and is accompanied by the absence of bone loss [28]. This is expected in SpA treated witn anti-TNF. In the BeSt study, conducted in patients with recent-onset, active RA with a tight control of inflammation, bone loss was limited in all treated groups, including in the one initially treated with high-dose prednisone [29]. Thus, there is a rationale for using anti-inflammatory drugs (including glucocorticoids) to protect bone from inflammatory-induced bone fragility. Bone fragility has been reported in a huge number of clinical studies conducted in patients with inflammatory diseases. Figure 2 demonstrates general and disease-specific risk factors for bone fragility in inflammatory diseases. The classical risk factors must be evaluated in such patients, bearing in mind the key role of systemic inflammation, and that some specific disease-related risks are added to the background risk.

Bone fragility in patients with inflammatory diseases
Inflammation and bone fragility in rheumatoid arthritis
Epidemiology of bone loss and fractures
Bone involvement in RA is one of the main extra-articular complications of RA: local bone destruction (bone erosions), localized peri-articular bone loss, and generalized bone loss. Bone erosions are the result of increased peri-inflammatory osteoclastic bone resorption in combination with suppressed bone formation, and rarely heal. Bone erosions in RA predict future bone destruction and generalized bone loss [30]. Bone degradation can occur as the result of synovitis, but in patients with ACPAs, it can already be present before RA becomes clinical and thus before clinical signs of synovitis [31]. Peri-articular bone loss is already occurring before RA becomes clinical and before bone erosions on conventional radiographs of hand and feet can be identified. It predicts future local bone destruction and generalized bone loss [32].
Generalized bone loss is found in RA, already early in the disease process. It is related to the general background risk (such as age, female gender, and body mass index) and to RA-specific factors, including systemic inflammation, disease activity and duration, immobility, and effect of the treatment of RA (Fig. 3).

Bone fragility in patients with rheumatoid arthritis
In RA, there is an increased risk of fractures of the vertebrae, the hip, and non-vertebral, non-hip fractures [33, 34]. Fracture risk is doubled compared to healthy age-matched controls, and fractures are a major component of comorbidities in RA. The risk of vertebral fractures can be increased early in the disease process. The aetiology of increased fracture risk in RA is multifactorial and includes the general background risk (clinical risk factors, low BMD) on top of RA-specific risk factors, which are related to disease activity, rheumatoid factor positivity, duration of disease, functional restrictions, sarcopenia, fall risk, the use of disease-modifying anti-rheumatic drugs (DMARDs) and biologicals, and the dose and duration of glucocorticoid (GC) therapy [35, 36] (Fig. 3).
Morbidity can be even more invalidating in RA patients with active disease or already pre-existing functional restriction. Furthermore, RA patients with a recent fracture have an increase in disease activity, and premature mortality is increased after hip fracture.
Risk factors and fracture assessment
Case finding
In view of the increased fracture risk, patients with RA older than 50 years, younger patients with persisting active disease, and patients with RA on GC therapy should be considered at risk for fractures and evaluated for fracture risk.
Risk evaluation
Several tools are available to evaluate fracture risk in RA. The general clinical risk evaluation includes increasing age, female gender, low body weight, personal and family history of fractures, fall risk, and lifestyle (smoking, excessive alcohol intake) (Fig. 3). These risk factors are to a variable degree included in fracture-risk algorithms such as FRAX, Garvan, and QFracture. However, the value of these algorithms for therapeutic decisions in RA is unclear. FRAX has been validated in many countries, but has been shown to overestimate fracture risk in RA [37], maybe because of the competing increased mortality in RA patients and in glucocorticoids users. Furthermore, it is unclear whether fixed or age-dependent fracture thresholds should be used for therapeutic decisions when using FRAX [37]. Compared to age-dependent thresholds, the use of fixed thresholds would under-diagnose/under-treat younger patients and over-diagnose/over-treat elderly patients [37].
Dual-energy X-ray absorptiometry (DXA) is the method of choice to measure bone mineral density (BMD) in the spine and hip. Imaging of the vertebrae is indicated as the prevalence of vertebral fractures is high in RA, and most of them are not accompanied with the signs and symptoms of an acute fracture. Indeed, RA patients receive GCs and analgesics because of painful joints, which can explain the high number of so-called asymptomatic vertebral fractures. However, the presence, number, and severity of vertebral fractures are associated with an increased fracture risk [33]. A prevalent non-traumatic vertebral fracture is therefore an indication for further fracture prevention, independent of BMD. Imaging of the spine is performed by radiography, and can also be performed using DXA with much lower radiation exposure, and a high negative predictive value for detecting vertebral fractures on radiography. It is therefore the preferred screening technique for diagnosing subclinical vertebral fractures, and can be performed at the same time as bone densitometry in RA patients.
Fracture prevention
In a patient with RA who has been identified at high risk for fracture, counselling starts with advices about lifestyle (stop smoking, limit alcohol intake), fall prevention, sufficient total calcium intake (1000–1200 mg/day in total), and vitamin D supplements (800 IU/day). The prescription of immunosuppressive medication, to reduce inflammation-induced bone loss, and minimizing dose and duration of GC treatment are essential measures. In patients receiving GCs, guidelines of prevention of glucocorticoid-induced osteoporosis (GIO) should be applied [38]. No randomized clinical trials (RCTs) with fracture reduction as primary endpoint are available in patients with RA. In patients on GCs, which included patients with RA, RCTs have shown that bisphosphonates (alendronate, risedronate, zoledronate) increase BMD, but no studies included fracture prevention as a primary endpoint. In a direct comparison study, the osteo-anabolic drug teriparatide significantly decreased the risk of vertebral fractures compared to alendronate in GC users [39], which is in line with the huge decrease of bone formation with GCs. Bisphosphonates (alendronate, risedronate, and zoledronate) and denosumab have been shown to reduce the risk of vertebral, non-vertebral, and hip fractures in postmenopausal women with osteoporosis; although this result was not obtained in patients selected on the basis of RA diagnosis. Denosumab and zoledronate have also been shown to inhibit progression of bone erosions in MTX-treated patients with RA [40].
Effect of biologicals on bone metabolism
The use of biologicals is associated with a decrease in osteoclastic activity, and in an increase in (markers of) bone formation, resulting in positive effects on BMD in the spine and hip in most, but not all, studies. However, to date no data show that DMARDs or biologicals are sufficient for fracture prevention in RA [41].
Relationships between inflammation and bone remodelling in spondyloarthropathies
Osteoporosis is a well-recognized feature of axial spondyloarthropathy (SpA). The primary disease localization in SpA is thought to be the enthesis, the zone in which tendons and ligaments insert into the bone. Although bone formation seems to be the cornerstone of the disease, SpA is also associated with a systemic osteoporosis, which has been reported as an early event and thus cannot be related only to spine ankylosis and immobilization. Moreover, this osteoporosis is unexpected as SpA typically affects young men, and glucocorticoids are not used in this disease, illustrating the link between inflammation and bone fragility [34].
Epidemiology of bone loss and fractures
Prevalence of osteoporosis is 14–27 and 4–14% at the spine and hip, respectively, which is unexpected in these young patients aged 30–40 years [42, 43]. Prospective studies have shown that spine and hip BMD decreases predominantly in patients with active disease, with permanently increased CRP [44, 45]. A significant reduction of BMD of 5 and 3% at lumbar spine and femoral neck, respectively, has been shown after 19 months of follow-up in patients with active disease and an increase of CRP and ESR; in this study, serum IL6 levels were significantly higher in patients with active AS than in those with inactive disease [45].
In a cohort of 332 (52% males), young patients with early inflammatory back pain suggestive of spondyloarthropathies (disease duration of symptoms, 1.6 years), 13% of patients had a low BMD (Z-score ≤ − 2). The main determinants of low BMD were bone and systemic inflammation as assessed by MRI and biological parameters [46]. The presence of bone marrow oedema (BMO) lesions on MRI increases 5-fold the risk of having a low spine BMD, and the presence of BMO lesions on spine MRI was the single determinant of low hip BMD, suggesting a systemic effect of inflammation. The close relationship between BMO lesions on MRI and low BMD has been confirmed in axial SpA without radiographical involvement [47].
Among 267 patients with symptoms suggestive of axial SpA, the positive likelihood ratio of low BMD for an axial SpA diagnosis was 2.60 and 3.12 at the spine and hip, respectively [48]. These results were confirmed in patients without any radiographic abnormalities suggestive of axial SpA. This finding suggests a relationship between inflammation and increase of bone resorption in SpA.
Diagnosis of vertebral fractures (VFs) is difficult as a minority of them come to clinical attention. A case control study performed in the large General Practice Research Database showed an increased risk of clinical VF (OR = 3.26 (1.5–7.02)) in patients with SpA [49]. Patients can have deformities of vertebral bodies particularly at the thoracic spine due to erosions of the anterior corners, wedging secondary to discitis and hyperkyphosis. These deformities, which can be captured by semi-automated methods of morphometry using automatic positioning of points on vertebral contours, should not be taken into account for estimation of osteoporotic VFs as all vertebral deformities are not necessarily vertebral fractures. These fractures should be distinguished from transdiscal or transvertebral fractures which occur even after a minor trauma, mainly at the cervical spine, involving posterior arch in patients with ankylosed spine. Patients with vertebral fractures have lower BMD than patients without, and femoral neck is the best discriminant site [50]. However, low BMD is not sufficient for the prediction of fracture in this population.
Physiopathology and risk factors of bone fragility
SpA is characterized by excessive local bone formation and concomitant systemic bone loss. Patients with SpA have low cortical BMD measured by high-resolution peripheral quantitative CT of the ultradistal radius and tibia, illustrating the role of systemic inflammation on bone in this disease. The HLA B27 transgenic rat, which is a relevant model of SpA, has decreased bone strength, related to a significant decrease in bone volume, trabecular number, and trabecular thickness. This HLA B27 rat has an increased RANKL to osteoprotegerin mRNA ratio, suggesting the implication of this system in the systemic bone loss. In patients with SpA, serum concentrations of RANKL, and expression of intracellular RANKL in CD4+ and CD8+ T cells, are increased [51]. As expected, the initiation of anti-TNF therapy induces a decrease in serum CTX, suggesting an anti-osteoclastic effect of the anti-inflammatory drug [52]. Vitamin D receptor gene may also contribute to BMD changes in patients with SpA, as some polymorphisms are linked to systemic inflammation [53]. Advances in pathogenesis have been provided by a new mouse model that highlights the role of IL23 in entheseal inflammation. Gut-derived IL23 (even in subclinical gut involvement) can act on a previously unidentified subpopulation of entheseal resident T cells, which, in reaction, produce cytokines such as IL22 and IL17, involved in osteoproliferation and bone loss, respectively [54].
Case finding
In view of the increased fracture risk, patients with SpA older than 50 years, but also younger patients with persisting active disease, long disease duration, ankylosis spine, low body mass index (BMI), and male gender should be considered at risk for fractures and evaluated for fracture risk (Fig. 4). Disease duration and wall-occiput distance have been reported as risk factors for vertebral fractures. Patients with bamboo spine (i.e. ankylosed), hyperkyphosis, and difficulties with peripheral vision have potential impairments in balance and coordination and a high risk of falls.

Bone fragility in patients with SpA
Risk evaluation
Clinical risk factors should be assessed: age, gender, BMI, physical activity level, currently smoking, disease activity, hyperkyphosis. Syndesmophytes are a cause of artefactual increase of lumbar spine BMD, and studies use either DXA or quantitative computed tomography (QCT) for spine evaluation. The prevalence of low lumbar spine BMD increases with the use of QCT. DXA of hips is the preferred method in patients with syndesmophytes. Imaging of the vertebrae is indicated as the prevalence of vertebral fractures is high in SpA and most of them are not accompanied with the signs and symptoms of an acute fracture. FRAX tool is not validated in this population.
Fracture prevention
Inflammation plays a key role in bone loss in SpA, and a beneficial effect of anti-inflammatory drugs on bone is expected through both the increased mobility related to pain relief and a direct effect on bone. In a primary care-based nested case control study, the risk of any clinical fracture was decreased in patients with SpA taking non-steroidal anti-inflammatory drugs (NSAIDs) (OR 0.65 (0.50, 0.84)) [49]. The increased risk of fractures in patients with SpA seems to be significant only in those not on regular NSAID treatment [55]. However, in a nationwide case control study, there was an excess risk of any clinical fracture in patients with SpA, even higher in NSAIDs users, probably because these patients have a more severe disease and thus, a higher utilization of NSAIDs [56].
Several prospective open studies in patients with SpA receiving TNF blockers show a positive effect on BMD. In a 2-year follow-up study of 106 patients, BMD significantly increased 5.8 and 2.3% at the lumbar spine and hip BMD, respectively [57]. These results were confirmed after 6 years of continuous administration of TNF blockers in 59 patients with an increase in BMD of 11.8 and 3.6% at lumbar spine and hip sites, even after exclusion of patients with syndesmophytes [58]. Meta-analysis and systematic reviews showed an increase in BMD in patients treated with anti-TNFα [59]. Among patients with symptoms suggestive of early axial SpA, 42% had significant bone loss over 2 years; in this study, use of anti-TNF therapy was protective against bone loss and baseline use of NSAIDs had a protective effect on hip bone loss [60].
There are no guidelines for treatment of osteoporosis in SpA. In patients treated with TNF blockers, without any prevalent non-traumatic fracture, it seems logical to assess first the benefit of this treatment. However, in patients with severe osteoporosis and prevalent fractures, available guidelines in osteoporotic patients and in male osteoporosis must be used.
Relationships between inflammation and bone fragility in systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is a chronic systemic autoimmune disorder, predominantly affecting women in their reproductive years. Since survival of patients with SLE has improved over the past decades, increasing attention is focused on complications of the disease, including osteoporosis and fractures which occurred in young women.
Epidemiology of osteoporosis and fractures
Recent studies have demonstrated a high frequency of low BMD and both peripheral and vertebral fractures in patients with SLE. A population-based study in 7332 SLE patients and in 28,079 age- and sex-matched controls from the UK showed that osteoporosis incidence is 2.53-fold increased in SLE [61]. Osteoporosis (T-score less than − 2.5) is observed in 1.4–68% of SLE patients [62].
Population-based studies [63] have demonstrated a 1.2–4.7-fold increased incidence of symptomatic fractures in patients with SLE compared with matched controls [64]. Fracture risk was particularly high in patients with seizures or a past cerebrovascular event [64]. Moreover, a recent study demonstrated use of anti-epileptic drugs and chronic use of oral anti-coagulants as independent risk factors for symptomatic fractures in patients with SLE [65].
Prevalent vertebral fractures have been demonstrated in 13.7–50% of the SLE patients with a mean age of 32–48 years [63, 66]. The majority of fractures is localized in the thoracic spine and is mild fractures. Age, low BMD, duration of menopause, male sex, previous use of intravenous methylprednisolone, high body mass index, and previous fractures were identified as risk factors for prevalent vertebral fractures (Fig. 5).

Factors contributing to bone fragility in patients with systemic lupus erythematosus. BMI body mass index, DHEA dehydroepiandrosterone, SLE systemic lupus erythematosus
The relationship between BMD and fracture incidence in SLE is not fully elucidated. Despite the high prevalence of reduced BMD and prevalent vertebral deformities in SLE, 29–35% of patients with at least one vertebral fracture has normal BMD [66, 67], which illustrates the limited value of BMD measurement in the assessment of fracture risk and the multifactorial aetiology of fractures in SLE. Therefore, assessment of vertebral fractures in addition to BMD measurement is recommended in all patients with SLE in whom bone density measurement is indicated because of the presence of clinical risk factors for fractures [63].
Physiopathology and risk factors of bone fragility
The aetiology of the increased bone loss in SLE is multifactorial, involving clinical osteoporosis risk factors, systemic inflammation, metabolic factors, serological factors, hormonal factors, possibly genetic factors, and medication-induced adverse effects [62] (Fig. 5). Age, postmenopausal status, low body weight, or low body mass index were recognized as independent risk factors for osteoporosis in patients with SLE [68, 69]. GCs are usually prescribed to patients with moderate or severe disease and, therefore, the association between GC use and fracture occurrence may point to an increased fracture risk in patients with active disease. The majority of SLE patients are on chronic GC treatment, and a 6-year follow-up study in Dutch SLE patients demonstrated a dose-dependent relationship between GC use and bone loss in the lumbar spine [70]. In SLE patients, serum levels of inflammatory parameters CRP and ESR are usually not markedly elevated. However, chronic inflammation may nevertheless contribute to bone loss in SLE. In patients with active SLE, increased serum levels of TNF [71] and oxidized low-density lipoprotein (LDL) [72] were demonstrated. Oxidized lipids induce activation of T cells, which in turn induce increased production of RANKL and TNF. Both TNF and RANKL enhance the maturation and activity of osteoclasts [71]. In addition, oxidized LDL may negatively influence bone formation by reducing osteoblast maturation [73]. Low-complement C4 levels (a measure of active SLE) were identified as a predictor of low spine BMD [68]. Moreover, a longitudinal study demonstrated significantly larger decrease in hip BMD in SLE patients with ≥ 3 disease flares over 5 years as compared to patients without disease flares [74], which illustrates the negative influence of disease activity on bone mass in SLE.
Vitamin D status might be negatively influenced by several factors in SLE: photosensitivity (leading to avoidance of sun exposure), use of sunscreens, renal failure, disease activity, GC therapy, and, probably, hydroxychloroquine use. A 6-year follow-up study in Dutch SLE patients revealed that low 25-hydroxyvitamin D (25(OH)D) levels at baseline were associated with increased bone loss in lumbar spine and hip [70].
Hydroxychloroquine, which is a drug frequently used for the treatment of SLE, is supposed to inhibit the conversion of 25(OH)D to 1,25(OH)2D (calcitriol) by inhibiting hydroxylase α1 [75]. Because calcitriol enhances intestinal calcium absorption, inhibition of 25(OH)D conversion to calcitriol may reduce uptake. A cross-sectional study in SLE patients demonstrated lower 1,25(OH)2D levels in hydroxychloroquine users as compared to non-users, while 25(OH)D levels were not different between both groups. In addition, a 6-year longitudinal study in SLE patients demonstrated significant bone loss in the hip in hydroxychloroquine users as compared to non-users [70], but this finding needs confirmation in other longitudinal studies in SLE populations.
Data on the role of autoimmunity in bone resorption in SLE are very limited. In a cross-sectional study on 34 patients with SLE, an association between the presence of anti-Sm and higher hip BMD was observed, while presence of anti-Ro/SSA was negatively associated with hip BMD [76]. The association between absence of anti-Ro/SSA and higher bone mass may be explained by the fact that anti-Ro/SSA positive patients with SLE are usually advised to avoid sun exposure. Bone density in SLE may be influenced by FOK-I vitamin D receptor (VDR) gene polymorphisms. A Dutch study revealed higher spine BMD in patients carrying the ff genotype of the FOK-I VDR compared to patients with FF and Ff genotypes, which might be in part explained by higher mean 25(OH)D serum levels in patients with the ff genotype [77].
Fracture prevention
Lifestyle measures are important, which include avoiding smoking, limiting alcohol intake, maintaining a normal body weight, avoiding falling, and performing regular weight-bearing exercise. Second, attention must be paid to an adequate calcium intake and sufficient serum 25(OH)D levels. For SLE patients with osteoporosis, with a previous fragility fracture, and/or receiving GCs, it is important to consider the prescription of an anti-resorptive agent. The bisphosphonates alendronate, risedronate, and zoledronic acid are recommended for the prevention and treatment of osteoporosis in GC-treated individuals without renal impairment. Bisphosphonates should be discussed in premenopausal women planning a pregnancy, since these agents are associated with fetal abnormalities in animal studies. They should only be prescribed in premenopausal women with severe osteoporosis or at a high risk for severe bone loss (e.g. those who are likely to be treated with high GC doses for a prolonged period of time).
Relationships between inflammation and bone fragility in inflammatory bowel diseases
Patients with inflammatory bowel diseases (IBD) (Crohn’s disease (CD) and ulcerative colitis (UC)) have an increased risk of osteoporosis and fractures, due to several risk factors (inflammation, low BMI, nutritional deficiencies, glucocorticoids (GCs), etc.).
Epidemiology of osteoporosis and fractures
In a case-control study on 6027 subjects with IBD (mean age 36 and 42 years for CD and UC, respectively) and 60,270 controls, the risk of osteoporotic fracture was increased by 40% [78]. However, this increased risk may be related to specific populations whose characteristics expose them to a higher risk. A study showed an increased risk of fracture only in postmenopausal women with CD, without any increase in patients with ulcerative colitis; in this study, there was a significant relationship between use of corticosteroids and fracture risk [79]. A case-control analysis conducted in the British General Practice Research Database showed a 40% increased risk of hip fracture after adjustment for the use of corticosteroids [80]. Overall, though patients with IBD may be at increased risk of fracture, the magnitude of risk appears to be small and is likely to be more important in those with other risk factors for fracture, those with more severe disease, and in the elderly.
The prevalence of osteoporosis is between 30 and 70% of IBD patients [81] in samples drawn from tertiary care settings. In studies where subjects are drawn from population-based sources, the rates of osteoporosis are lower, with estimates ranging from 5 to 15% [82]. There are conflicting results on the influence of the type of IBD (CD or UC) on the risk of osteoporosis, which depends more on the use of corticosteroids than on the type of IBD. A study conducted in 86 patients, followed over 4.3 years, showed that bone loss was on average similar to the expected bone loss in the general population [83]. In this study, the risk factors associated with bone loss were the decrease in BMI, age over 50 years, and the use of GCs (and thus indirectly the activity of the inflammatory disease).
Physiopathology and risk factors of bone fragility
Intestinal chronic inflammation leads to activation of T lymphocytes and production of pro-inflammatory cytokines including TNFα that activate the RANK-RANK-ligand pathway. Inflammatory colitis models have shown that early bone loss is related to the activation of RANK-RANKL system and is reversible upon administration of osteoprotegerin (OPG) [84]. The alterations of the OPG/RANKL system is associated with decreased BMD. In patients with IBD, the increase of serum OPG levels and the release of OPG by the gastrointestinal mucosa cells are inversely correlated with the decrease in bone density [85]. Inflammatory cytokines, including TNFα, can stimulate the production of sclerostin and thus decrease bone formation; the impaired bone formation has been described in animals, in chemical-induced colitis models. CD is considered as a more systemic inflammatory disease than UC. In patients with CD, there is a decrease in BMD before the use of glucocorticoids, suggesting that untreated systemic inflammation is responsible for low BMD [86]. In these patients, who are newly diagnosed and not yet treated for CD, serum TNF, IL6, IL1, and RANKL and OPG levels are high.
Some studies showed an increase in BMD after total colectomy with ileal pouch-anal anastomosis in UC patients, possibly due to the discontinuation of GCs, improvement in nutritional status, and decreased production of cytokines by the gut inflammation [87]. However, data are not consistent in this matter. In a study of 327 UC patients who underwent this surgery, 32% had low BMD 4 years after surgery, which suggests that bone loss continues after colectomy, or that spontaneous increase is not sufficient. The ileostomy performed in 126 patients with Crohn’s disease is associated with increased risk of osteoporosis, mainly explained by weight loss [88].
Use of glucocorticoids plays a major role on bone loss as nearly 35% of patients with IBD receive GCs within 1 year of diagnosis, and half of IBD patients with recent fracture use GCs [89]. The risk of osteoporosis is twice as high in patients who use corticosteroids (OR = 2.4 (1.5–3.4)) [90]. A higher annual bone loss of 5 to 10% is related to the use of high doses of corticosteroids prescribed during a relapse of IBD [91], which means that both uncontrolled inflammation and the need of corticosteroids are associated with bone loss. Another potential contributor to bone loss in IBD is low intake of calcium and vitamin D; CD frequently involves the small intestine which interferes with absorption of these elements. Moreover, active inflammation of the terminal ileum leads to a decreased reabsorption of secreted bile salts and reduces the absorption of vitamin D. Low vitamin D levels have also been shown to be associated with lower BMD among patients with IBD [92].
Fracture prevention
Osteoporosis screening by DXA is recommended for patients with IBD with a prior history of fracture, those with GCs intake > 3 months, and in patients over age 50 [93]. Calcium (1000–1200 mg/day) and vitamin D supplementation (600–800 UI/day) are recommended in patients with IBD. Strategies include using the lowest effective corticosteroid dose, administering corticosteroid therapy for the shortest duration, using corticosteroids with fewer systemic effects (e.g. budesonide), and using alternate medications (e.g. azathioprine, TNF blockers). In patients with osteoporosis, or with a previous fracture, anti-resorptive treatments should be considered. Bisphosphonates orally or intravenous have been shown to improve BMD in patients with IBD and could have a benefit on the risk of vertebral fractures [94] with a good tolerance. Prospective studies suggest that the TNF blockers have a positive effect on bone markers but there is no data on BMD changes in IBD patients receiving such treatments [95].
Relationship between inflammation and bone fragility in chronic obstructive pulmonary disease
Patients with chronic obstructive pulmonary disease (COPD) have a high prevalence of osteoporosis and vertebral fractures which results from chronic and systemic inflammation, the use of corticosteroids, low body mass, and a sedentary lifestyle. COPD is a typical situation in which the consequences of thoracic vertebral fractures can be severe.
Epidemiology of osteoporosis and fractures
Thoracic kyphosis related to vertebral fractures can impair lung function, and every single vertebral fracture decreases the vital capacity by 9% [96]. Patients with rib fractures have decreased ability to expectorate and may develop exacerbation of the lung disease. Osteoporosis and muscle weakness are important systemic complications of COPD. The prevalence of osteoporosis in COPD is 17% [97] in NHANES but up to 35% in a systematic review of 13 studies [98]. Osteoporosis is largely undertreated in patients with such condition; vertebral fractures are under-reported, although they are present on chest radiographies [98]. The prevalence of vertebral fracture is up to 40% in 2981 COPD patients [99]. The prevalence of hip fractures is unknown, but the presence of COPD carries a poor prognosis and an increased risk of mortality after hip fracture [100].
Physiopathology and risk factors of bone fragility
Corticosteroid use is obviously one of the determinants of bone fragility in COPD. However, the potential beneficial role of low doses of inhaled corticosteroids (as compared to high doses) has been suggested [101]. Indeed, inhaled corticosteroids can decrease lung inflammation, without or with a lower systemic bone effects as compared to oral corticosteroids. Frequent exacerbations of COPD may be risk factors for bone loss, including through the decreased physical activity and the use of higher doses of corticosteroids. Systemic inflammation is a major determinant of bone fragility. Studies have shown that matrix metalloproteinase-9 (MMP-9) and its cognate inhibitor TIMP-1, inflammatory cytokines TNF-α, IL1, and IL6, and the OPG/RANK/RANKL system may each play individual roles in the pathogenesis of osteoporosis in patients with COPD [102, 103].
Fracture prevention
A comprehensive management of COPD must incorporate assessment of osteoporosis and of associated risk factors such as smoking, sarcopenia, low BMI, reduced physical activity, and vitamin D deficiency [104]. The higher risk of bone complications is observed in older COPD patients with a low BMI and/or increased PTH level [104]. The assessment of vertebral fractures on chest X-rays should be a routine practice in patients with COPD (Fig. 6). Osteoporosis screening by DXA is recommended for patients with COPD with a prior history of fracture, those with GCs intake > 3 months, and in patients over age 50. There are no guidelines for treatment of osteoporosis in COPD. However, because of the use of systemic glucocorticoids, GIOP recommendations should be applied, including for anti-osteoporotic pharmaceutical drugs. Prevention of falls is an important issue, in particular in elderly patients.

Bone fragility in patients with COPD
Conclusion
Patients with chronic inflammatory disorders are at high risk of osteoporosis and fragility fractures due to disease severity (related to both the activity and the duration of the disease) on top of the well-known demographic risk factors, such as age, low BMI, familial osteoporosis, etc. Glucocorticoids use is not the single determinant of osteoporosis in these patients. Complete control of inflammation is crucial in the prevention of osteoporosis. Data are needed to study the anti-fracture efficacy of potent anti-inflammatory drugs, which allow glucocorticoid sparing effect, and on the indications and duration of the anti-osteoporotic drugs in patients with chronic inflammatory disorders.
References
Geusens P, Goldring SR, Briot K, Roux C (2016) The role of the immune system in the development of osteoporosis and fracture risk. In: Lorenzo J (ed) Osteoimmunology: interactions of the immune and skeletal systems, 2nd edn. Elsevier, pp 187–214
Tetelbaum SL (2000) Bone resorption by osteoclasts. Science 289:1504–1508
Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Boyle WJ, Riggs BL (2000) The roles of osteoprotegerin and osteoprotegerin ligand in the paracrine regulation of bone resorption. J Bone Miner Res 15:2–12
Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S et al (1999) Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoproteogerin ligand. Nature 402:304–309
Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP et al (2007) B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood 109:3839–3849
Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL (2000) TNFα induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest 106:1481–1488
Axmann R, Böhm C, Krönke G, Zwerina, Smolen J, Schett G (2009) Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum 60:2747–2756
Greenfield EM, Gornik SA, Horowitz MC, Donahue HJ, Shaw SM (1993) Regulation of cytokine expression in osteoblasts by parathyroid hormone: rapid stimulation of interleukin-6 and leukemia inhibitory factor mRNA. J Bone Miner Res 8:1163–1171
Adamopoulos IE, Chao CC, Geissler R, Laface D, Blumenschein W, Iwakura Y et al (2010) Interleukin-17A upregulates receptor activator of NF-kappaB on osteoclast precursors. Arthritis Res Ther 12:R29
Pacifici R (2016) The role of IL-17 and TH17 cells in the bone catabolic activity of PTH. Front Immunol 7:57
Zaiss MM, Axmann R, Zwerina J, Polzer K, Gückel E, Skapenko A, Schulze-Koops H, Horwood N, Cope A, Schett G (2007) Treg cells suppress ostoclast formation. A new link between the immune system and bone. Arthritis Rheum 56:4104–4112
Axmann R, Herman S, Zaiss M, Franz S, Polzer K, Zwerina J, Hermann M, Smolen J, Schett G (2008) CTLA-4 directly inhibits osteoclast formation. Ann Rheum Dis 67:1603–1609
Harre U, Georgess D, Bang H, Bozec A, Axamann R, Ossipova E et al (2012) Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J Clin Invest 122:1791–1802
Kleyer A, Finzel S, Rech J, Manger B, Krieter M, Faustini F et al (2014) Bone loss before the clinical onset of rheumatoid arthritis in subjects with anticitrullinated protein antibodies. Ann Rheum Dis 73:854–860
Sokolove J, Zhao X, Chandra PE, Robinson WH (2011) Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcγ receptor. Arthritis Rheum 63:53–62
Krishnamurthy A, Joshua V, Haj Hensvold A, Jin T, Sun M, Vivar N, Ytterberg AJ et al (2016) Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann Rheum Dis 75:721–729
Choi Y, Mi Woo K, Ko SH, Lee YJ, Park SJ, Kim HM et al (2001) Osteoclastogenesis is enhanced by activated B cells but suppressed by activated CD8+ T cells. Eur J Immunol 31:2179–2188
Rivollier A, Mazzorana M, Tebib J, Piperno M, Aitsiselmi T, Rabourdin-Combe C et al (2004) Immature dentritic cell transdifferentiation into osteoclasts: a novel pathway sustained by the rheumatoid arthritis microenvironment. Blood 104:4029–4037
Haiyan L, Hong S, Qian J, Zheng Y, Yang J, Yi Q (2010) Cross talk between the bone and immune systems: osteoclasts function as antigen-presenting cells and activate CD4+ and CD8+ T cells. Blood 116:210–217
Herman S, Mueller R, Kronke G, Zwerina J, Redlich K, Hueber A, Gelse H, Neumann E, Mueller-Ladner U, Schett G (2008) OSCAR, a key co-stimulation molecule for osteoclasts, is induced in patients with rheumatoid arthritis. Arthritis Rheum 58:3041–3050
Wong PKK, Quin JMW, Sims NA, van Nieuwenhuijze A, Campbell IK, Wicks IP (2006) Interleudin-6 modulates production of T lymphocyte-derived cytokines in antigen-induced arthritis and drives inflammation-induced ostoclastogenesis. Arthritis Rheum 54:158–168
Binder NB, Niederreiter B, Hoffmann O, Stange R, Pap T, Stulnig TM et al (2009) Estrogen-dependent and C-C chemokine receptor-2 dependent pathways determine osteoclast behavior in osteoporosis. Nat Med 15:417–424
Charactcharoenwitthaya N, Khosla S, Atkinson EJ, McCready LK, Riggs BL (2007) Effect of blockade of TNF-α and interleukin-1 action on bone resorption in early postmenopausal women. J Bone Miner Res 22:724–729
Bedi B, Li JY, Tawfeek H, Baek KH, Adams J, Vangara SS et al (2012) Silencing of parathyroid hormone (PTH) receptor 1 in T cells blunts the bone anabolic activity of PTH. Proc Natl Acad Sci U S A 109:E725–E733
Gilbert L, He X, Farmer P, Rubin J, Drissi H, van Wijnen AJ et al (2002) Expression of the osteoblast differentiation factor RUNX2 (Cbfa1/AML3/Pebp2aA) is inhibited by tumor necrosis factor-α. J Biol Chem 277:2695–2701
Heiland GR, Zwerina K, Baum W, Kireva T, Distler JH, Grisanti M et al (2010) Neutralisation of Dkk-1 protects from systemic bone loss during inflammation and reduces sclerostin expression. Ann Rheum Dis 69:2152–2159
Chen XX, Baum W, Dwyer D, Stock M, Schwabe K, Ke HZ et al (2013) Sclerostin inhibition reverses systemic, periarticular and local bone loss in arthritis. Ann Rheum Dis 72:1732–1736
Wijbrandts CA, Klaasen R, Dijkgraaf MGW, Gerlag DM, van Eck-Smit BLF, Tak PP (2009) Bone mineral density in rheumatoid arthritis patients 1 year after adalimumab therapy: arrest of bone loss. Ann Rheum Dis 68:373–376.57
Güler-Yüksel M, Bijsterbosch J, Goekoop-Ruiterman YP et al (2008) Changes in bone mineral density in patients with recent onset, active rheumatoid arthritis. Ann Rheum Dis 67:823–828
Güler-Yüksel M, Allaart CF, Goekoop-Ruiterman YP et al (2009) Changes in hand and generalised bone mineral density in patients with recent-onset rheumatoid arthritis. Ann Rheum Dis 68:330–336
Schett G, Firestein GS (2010) Mr Outside and Mr Inside: classic and alternative views on the pathogenesis of rheumatoid arthritis. Ann Rheum Dis 69:787–789
Haugeberg G, Green MJ, Quinn MA et al (2006) Hand bone loss in early undifferentiated arthritis: evaluating bone mineral density loss before the development of rheumatoid arthritis. Ann Rheum Dis 65:736–740
van Staa TP, Geusens P, Bijlsma JW et al (2006) Clinical assessment of the long-term risk of fracture in patients with rheumatoid arthritis. Arthritis Rheum 54:3104–3112
Weiss RJ, Wick MC, Ackermann PW, Montgomery SM (2010) Increased fracture risk in patients with rheumatic disorders and other inflammatory diseases—a case-control study with 53,108 patients with fracture. J Rheumatol 37:2247–2250
Amin S, Gabriel SE, Achenbach SJ, Atkinson EJ, Melton LJ 3rd (2013) Are young women and men with rheumatoid arthritis at risk for fragility fractures? A population-based study. J Rheumatol 40:1669–1676
Ochi K, Inoue E, Furuya T, Ikari K, Toyama Y, Taniguchi A, Yamanaka H, Momohara S (2015) Ten-year incidences of self-reported non-vertebral fractures in Japanese patients with rheumatoid arthritis: discrepancy between disease activity control and the incidence of non-vertebral fracture. Osteoporos Int 26:961–968
Klop C, de Vries F, Bijlsma JW, Leufkens HG, Welsing PM (2016) Predicting the 10-year risk of hip and major osteoporotic fracture in rheumatoid arthritis and in the general population: an independent validation and update of UK FRAX without bone mineral density. Ann Rheum Dis 75:2095–2100
Lekamwasam S, Adachi JD, Agnusdei D et al (2012) A framework for the development of guidelines for the management of glucocorticoid-induced osteoporosis. Osteoporos Int 23:2257–2276
Saag KG, Zanchetta JR, Devogelaer JP, Adler RA, Eastell R, See K, Krege JH, Krohn K, Warner MR (2009) Effects of teriparatide versus alendronate for treating glucocorticoid-induced osteoporosis: thirty-six-month results of a randomized, double-blind, controlled trial. Arthritis Rheum 60:3346–3355
Vis M, Güler-Yüksel M, Lems WF (2012) Can bone loss be prevented in RA? Osteoporos Int 24:2541–2553
Manara M, Sinigaglia L (2015) Bone and TNF in rheumatoid arthritis: clinical implications. RMD Open 1(Suppl 1):e000065. doi:10.1136/rmdopen-2015-000065
Van der Weijden MAC, Claushuis TAM, Nazari T, Lems WF, Dijkmans BAC, Van der Horst-Bruinsma EI (2012) High prevalence of low bone mineral density in patients within 10 years of onset of ankylosing spondylitis: a systematic review. Clin Rheumatol 31:1529–1535
El Maghraoui A, Borderie D, Cherruau B, Edouard R, Dougados M, Roux C (1999) Osteoporosis, body composition, and bone turnover in ankylosing spondylitis. J Rheumatol 26:2205–2209
Maillefert JF, Aho S, El Maghraoui A, Dougados M, Roux C (2001) Changes in bone density in patients with ankylosing spondylitis: a 2 year follow-up study. Osteoporos Int 12:605–609
Gratacos J, Collado A, Pons F, Osaba M, Sammarti R, Roqué M et al (1999) Significant loss of bone mass in patients with early, active ankylosing spondylitis. A follow-up study. Arthritis Rheum 42:2319–2324
Briot K, Durnez A, Paternotte S, Miceli-Richard C, Dougados M, Roux C (2013) Bone oedema on MRI is highly associated with low bone mineral density in patients with early inflammatory back pain: results from the DESIR cohort. Ann Rheum Dis 72:1914–1919
Akgöl G, Kamanli A, Ozgocmen S (2014) Evidence for inflammation-induced bone loss in non-radiographic axial spondyloarthritis. Rheumatolojy (Oxford) 53:497–501
Forien M, Molto A, Etcheto A et al (2015) Bone mineral density in patients with symptoms suggestive of spondyloarthritis. Osteoporos Int 26:1647–1653
Vosse D, Landewé R, van der Heijde D, van der Linden S, van Staa TP, Geusens P (2009) Ankylosing spondylitis and the risk of fracture: results from a large primary care-based nested case-control study. Ann Rheum Dis 68:1839–1842
Geusens P, De Winter L, Quaden D, Vanhoof J, Vosse D, van den Bergh J, Somers V (2015) The prevalence of vertebral fractures in spondyloarthritis: relation to disease characteristics, bone mineral density, syndesmophytes and history of back pain and trauma. Arthritis Res Ther 17:294
Stupphann D, Ranner M, Krenbeck D et al (2008) Intracellular and surface RANK L are differentially regulated in patients with ankylosing spondylitis. Rheumatol Int 28:987–993
Briot K, Garnero P, Le Henanff A, Dougados M, Roux C (2005) Body weight, body composition and bone turnover changes in patients with spondyloarthropathy receiving anti-TNF alpha treatment. Ann Rheum Dis 64:1137–1140
Arends S, Spoorenberg A, Bruyn GAW et al (2011) The relation between bone mineral density, bone turnover markers, and vitamin D status in ankylosing spondylitis patients with active disease: a cross-sectional analysis. Osteoporos Int 22:1431–1439
Sherlock JP, Joyce-Shaikh B, Turner SP et al (2012) IL-23 induces spondyloarthropathy by acting on ROR-γt+CD3+CD4−CD8−entheseal resident T cells. Nat Med 18:1069–1076
Muñoz-Ortego J, Vestergaard P, Rubio JB, Wordsworth P, Judge A, Javaid MK, Arden NK, Cooper C, Díez-Pérez A, Prieto-Alhambra D (2014) Ankylosing spondylitis is associated with an increased risk of vertebral and nonvertebral clinical fractures: a population-based cohort study. J Bone Miner Res 29(8):1770–1776
Prieto-Alhambra D, Muñoz-Ortego J, De Vries F, Vosse D, Arden NK, Bowness P, Cooper C, Diez-Perez A, Vestergaard P (2015) Ankylosing spondylitis confers substantially increased risk of clinical spine fractures: a nationwide case-control study. Osteoporos Int 26(1):85–91
Briot K, Gossec L, Kolta S, Dougados M, Roux C (2008) Prospective assessment of body weight, body composition, and bone density changes in patients with spondyloarthropathy receiving anti-tumor necrosis factor-alpha treatment. J Rheumatol 35:855–861
Durnez A, Paternotte S, Fechtenbaum J, Landewé RB, Dougados M, Roux C, Briot K (2013) Increase in bone density in patients with spondyloarthritis during anti-tumor necrosis factor therapy: 6-year follow-up study. J Rheumatol 40:1712–1718
Haroon NN, Sriganthan J, Al Ghanim N et al (2014) Effect of TNF-alpha inhibitor treatment on bone mineral density in patients with ankylosing spondylitis: a systematic review and meta-analysis. Semin Arthritis Rheum 44:155–161
Briot K, Etcheto A, Miceli-Richard C, Dougados M, Roux C (2016) Bone loss in patients with early inflammatory back pain suggestive of spondyloarthritis: results from the prospective DESIR cohort. Rheumatology (Oxford) 55:335–422
Rees F, Doherty M, Grainge M, Lanyon P, Davenport G, Zhang W (2015) Burden of comorbidity in systemic lupus erythematosus in the UK, 1999-2012. Arthritis Care Res (Hoboken) 68:819–827
Bultink IEM (2012) Osteoporosis and fractures in systemic lupus erythematosus. Arthritis Care Res (Hoboken) 64:2–8
Bultink IEM, Lems WF (2016) Lupus and fractures. Curr Opin Rheumatol 28:426–432
Bultink IE, Harvey NC, Lalmohamed A, Cooper C, Lems WF, van Staa TP, de Vries F (2014) Elevated risk of clinical fractures and associated risk factors in patients with systemic lupus erythematosus versus matched controls: a population-based study in the United Kingdom. Osteoporos Int 25:1275–1283
Carli L, Tani C, Spera V, Vagelli R, Vagnani S, Mazzantini M, Di Munno O, Mosca M (2016) Risk factors for osteoporosis and fragility fractures in patients with systemic lupus erythematosus. Lupus Sci Med 3:e000098
Li EK, Tam LS, Griffith JF, Zhu TY, Li TK, Li M, Wong KC, Chan M, Lam CW, Chu FS, Wong KK, Leung PC, Kwok A (2009) High prevalence of asymptomatic vertebral fractures in Chinese women with systemic lupus erythematosus. J Rheumatol 36:1646–1652
Mendoza-Pinto C, García-Carrasco M, Sandoval-Cruz H, Muñoz-Guarneros M, Escárcega RO, Jiménez-Hernández M, Munguia-Realpozo P, Sandoval-Cruz M, Delezé-Hinojosa M, López-Colombo A, Cervera R (2009) Risk factors of vertebral fractures in women with systemic lupus erythematosus. Clin Rheumatol 28:579–585
Petri M (1995) Musculoskeletal complications of systemic lupus erythematosus in the Hopkins Lupus Cohort: an update. Arthritis Care Res 8:137–145
Bultink IE, Lems WF, Kostense PJ, Dijkmans BA, Voskuyl AE (2005) Prevalence of and risk factors for low bone mineral density and vertebral fractures in patients with systemic lupus erythematosus. Arthritis Rheum 54:2044–2050
Jacobs J, Korswagen LA, Schilder AM, van Tuyl LH, Dijkmans BAC, Lems WF, Voskuyl AE, Bultink IEM (2013) Six-year follow-up study of bone mineral density in patients with systemic lupus erythematosus. Osteoporos Int 24:1827–1833
Svenungsson E, Fei GZ, Jensen-Urstad K, de Faire U, Hamsten A, Frostegard J (2003) TNF-alpha: a link between hypertriglyceridaemia and inflammation in SLE patients with cardiovascular disease. Lupus 12:454–461
Frostegard J, Svenungsson E, Wu R, Gunnarsson I, Lundberg IE, Klareskog L, Hörkkö S, Witztum JL (2005) Lipid peroxidation is enhanced in patients with systemic lupus erythematosus and is associated with arterial and renal disease manifestations. Arthritis Rheum 52:192–200
Moerman EJ, Teng K, Lipschitz DA, Lecka-Czernik B (2004) Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell 6:379–389
Zhu TY, Griffith JF, Au SK, Tang XA, Kwok AW, Leung PC, Li EK, Tam LS (2014) Bone mineral density change in systemic lupus erytehmatosus: a 5-year followup study. J Rheumatol 41:1990–1997
Huisman AM, White KP, Algra A, Harth M, Vieth R, Jacobs JW, Bijlsma JW, Bell DA (2013) Vitamin D levels in women with systemic lupus erythematosus and fibromyalgia. J Rheumatol 11:2535–2539
Mok CC, Mak A, Ma KM (2005) Bone mineral density in postmenopausal Chinese patients with systemic lupus erythematosus. Lupus 14:106–112
Jacobs J, Voskuyl AE, Korswagen LA, Theunissen R, Cohen Tervaert JW, Bultink IEM (2015) The association between FOK-I vitamin D receptor gene polymorphisms and bone mineral density in patients with systemic lupus erythematosus. Clin Exp Rheumatol 33:0765
Bernstein CN, Blanchard JF, Leslie W, Wajda A, Yu BN (2000) The incidence of fracture among patients with inflammatory bowel disease. A population-based cohort study. Ann Intern Med 133:795–799
Vestergaard P, Mosekilde L (2002) Fracture risk in patients with celiac disease, Crohn’s disease, and ulcerative colitis: a nationwide follow-up study of 16,416 patients in Denmark. Am J Epidemiol 156:1–10
Card T, West J, Hubbard R, Logan RF (2004) Hip fractures in patients with inflammatory bowel disease and their relationship to corticosteroid use: a population based cohort study. Gut 53:251–255
Leslie WD, Miller N, Rogala L, Bernstein CN (2009) Body mass and composition effect bone density in recently diagnosed inflammatory bowel disease: the Manitoba IBD Cohort Study. Inflamm Bowel Dis 15:39–46
Tsai MS, Lin CL, Tu YK, Lee PH, Kao CH (2015) Risks and predictors of osteoporosis in patients with inflammatory bowel diseases in an Asian population: a nationwide population-based cohort study. Int J Clin Pract 69:235–241
Targownik LE, Leslie WD, Carr R, Clara I, Miller N, Rogala L, Graff LA, Walker JR, Bernstein CN (2012) Longitudinal change in bone mineral density in a population-based cohort of patients with inflammatory bowel disease. Calcif Tissue Int 91:356–363
Ashcroft AJ, Cruickshank SM, Croucher PI, Perry MJ, Rollinson S, Lippitt JM, Child JA, Dunstan C, Felsburg PJ, Morgan GJ, Carding SR (2003) Colonic dendritic cells, intestinal inflammation, and T cell-mediated bone destruction are modulated by recombinant osteoprotegerin. Immunity 19:849–861
Moschen AR, Kaser A, Enrich B, Ludwiczek O, Gabriel M, Obrist P, Wolf AM, Tilg H (2005) The RANKL/OPG system is activated in inflammatory bowel disease and relates to the state of bone loss. Gut 54:479–487
Ghosh S, Cowen S, Hannan WJ, Ferguson A (1994) Low bone mineral density in Crohn’s disease, but not in ulcerative colitis, at diagnosis. Gastroenterology 107:1031–1039
Abitbol V, Roux C, Gillemant S, Valleur P, Hautefeuille P, Dougados M, Couturier D, Chaussade S (1998) Bone assessment in patients with ileal pouch-anal anastomosis for inflammatory bowel disease. Br J Surg:1551–1554
Gupta S, Shen B (2013) Bone loss in patients with the ileostomy and ileal pouch for inflammatory bowel disease. Gastroenterol Rep (Oxf) 1:159–165
Bernstein CN, Blanchard JF, Metge C, Yogendran M (2003) The association between corticosteroid use and development of fractures among IBD patients in a population-based database. Am J Gastroenterol 98:1797–1801
Abraham BP, Prasad P, Malaty HM (2014) Vitamin D deficiency and corticosteroid use are risk factors for low bone mineral density in inflammatory bowel disease patients. Dig Dis Sci 59:1878–1884
Roux C, Abitbol V, Chaussade S, Kolta S, Guillemant S, Dougados M, Amor B, Couturier D (1995) Bone loss in patients with inflammatory bowel disease: a prospective study. Osteoporos Int 5:156–160
Leslie WD, Miller N, Rogala L, Bernstein CN (2008) Vitamin D status and bone density in recently diagnosed inflammatory bowel disease: the Manitoba IBD Cohort Study. Am J Gastroenterol 103:1451–1459
Bernstein CN, Leslie WD, Leboff MS (2003) AGA technical review on osteoporosis in gastrointestinal diseases. Gastroenterology 124:795–841
Melek J, Sakuraba A (2014) Efficacy and safety of medical therapy for low bone mineral density in patients with inflammatory bowel disease: a meta-analysis and systematic review. Clin Gastroenterol Hepatol 12:32–44.e5
Franchimont N, Putzeys V, Collette J, Vermeire S, Rutgeerts P, De Vos M, Van Gossum A, Franchimont D, Fiasse R, Pelckmans P, Malaise M, Belaiche J, Louis E (2004) Rapid improvement of bone metabolism after infliximab treatment in Crohn's disease. Aliment Pharmacol Ther 20(6):607–614
Majumdar SR, Villa-Roel C, Lyons KJ, Rowe BH (2010) Prevalence and predictors of vertebral fracture in patients with chronic obstructive pulmonary disease. Respir Med 104:260–266
Harrison RA, Siminoski K, Vetkanayagam D, Majumdar SR (2007) Osteoporosis-related kyphosis and impairments in pulmonary function: a systematic review. J Bone Miner Res 22:448–557
Graat-Verboom L, Spruit MA, van den Borne BE et al (2009) CIRO Network. Correlates of osteoporosis in chronic obstructive pulmonary disease: an underestimated systemic component. Respir Med 103:1143–1151
Nutti R, Siviero P, Maggi S et al (2009) Vertebral fractures in patients with chronic obstructive pulmonary disease: the EOLO study. Osteoporos Int 20:989–998
De Luise C, Brimacombe M, Pedersen L, Sorensen HT (2008) Chronic obstructive pulmonary disease and mortality following hip fracture: a population-based cohort study. Eur J Epidemiol 23:115–122
Mathioudakis AG, Amanetopoulou SG, Gialmanidis IP et al (2013) Impact of long-term treatement with low-dose inhaled corticosteroids on the bone mineral density of chronic obstructive pulmonary disease patients: aggravating or beneficial? Respirology 18:147–153
Bai P, Sun Y, Jin J, Hou J, Li R, Zhang Q, Wang Y (2011) Disturbance of the OPG/RANK/RANKL pathway and systemic inflammation in COPD patients with emphysema and osteoporosis. Respir Res 12:157
Zhang PF, Pan L, Luo ZY, Zhao HJ, Cai SX (2013) Interrelationship of circulating matrix metalloproteinase-9, TNF-α, and OPG/RANK/RANKL systems in COPD patients with osteoporosis. COPD 10:650–656
Graat-Verboom L, van den Borne BEEM, Smeenk FWJM, Spruit MA, Wouters EFM (2011) Osteoporosis in COPD outpatients based on bone mineral density and vertebral fractures. J Bone Miner Res 26:561–568
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Rights and permissions
About this article

Cite this article
Briot, K., Geusens, P., Em Bultink, I. et al. Inflammatory diseases and bone fragility. Osteoporos Int 28, 3301–3314 (2017). https://doi.org/10.1007/s00198-017-4189-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-017-4189-7